Evaluating Face2Gene as a Tool to Identify Cornelia de Lange Syndrome by Facial Phenotypes

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
2.1. Clinical and Molecular Diagnosis of the Individuals Analysed
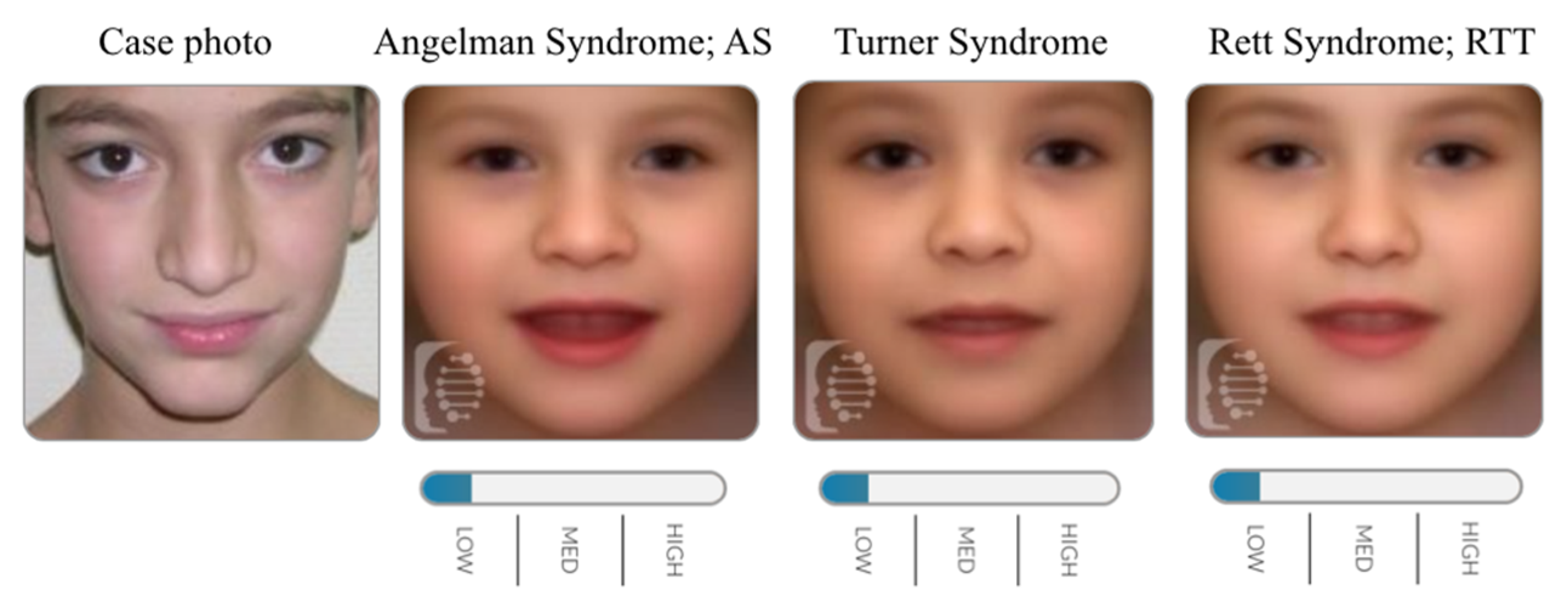
2.2. Identifying Cornelia de Lange Syndrome using Face2Gene
2.3. Face2Gene Evauation for Facial Images of CdLS Patients at Different Ages
2.4. Face2Gene Evaluation for Facial Image of CdLS Patients with Different Causative Genes
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CdLS | Cornelia de Lange Syndrome |
| RSTS | Rubinstein-Taybi Syndrome |
| PMID | PubMed Unique Identifier |
References
- Ramos, F.J.; Puisac, B.; Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Bueno, I.; Deardorff, M.A.; Hennekam, R.C.; Kaiser, F.J.; Krantz, I.D.; Musio, A.; et al. Clinical utility gene card for: Cornelia de Lange syndrome. Eur. J. Hum. Genet. 2015, 23, 1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huisman, S.A.; Redeker, E.J.W.; Maas, S.M.; Mannens, M.M.; Hennekam, R.C.M. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J. Med. Genet. 2013, 50, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Braunholz, D.; Teresa-Rodrigo, M.E.; Obieglo, C.; Gener, B.; Schwarzmayr, T.; Strom, T.M.; Gómez-Puertas, P.; Puisac, B.; et al. Somatic mosaicism in a Cornelia de Lange syndrome patient with NIPBL mutation identified by different next generation sequencing approaches. Clin. Genet. 2014, 86, 595–597. [Google Scholar] [CrossRef]
- Pozojevic, J.; Parenti, I.; Graul-Neumann, L.; Ruiz Gil, S.; Watrin, E.; Wendt, K.S.; Werner, R.; Strom, T.M.; Gillessen-Kaesbach, G.; Kaiser, F.J. Novel mosaic variants in two patients with Cornelia de Lange syndrome. Eur. J. Med. Genet. 2018, 61, 680–684. [Google Scholar] [CrossRef]
- Pié, J.; Puisac, B.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E.; Gil-Rodríguez, M.; Baquero-Montoya, C.; Ramos-Cáceres, M.; Bernal, M.; Ayerza-Casas, A.; Bueno, I.; et al. Special cases in Cornelia de Lange syndrome: The Spanish experience. Am. J. Med. Genet. Part C 2016, 172, 198–205. [Google Scholar] [CrossRef]
- Watrin, E.; Kaiser, F.J.; Wendt, K.S. Gene regulation and chromatin organization: Relevance of cohesin mutations to human disease. Curr. Opin. Genet. Dev. 2016, 37, 59–66. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, X. Roles of cohesin in chromosome architecture and gene expression. Semin. Cell Dev. Biol. 2019, 90, 187–193. [Google Scholar] [CrossRef]
- Mehta, G.D.; Kumar, R.; Srivastava, S.; Ghosh, S.K. Cohesin: Functions beyond sister chromatid cohesion. FEBS Lett. 2013, 587, 2299–2312. [Google Scholar] [CrossRef] [Green Version]
- Sarogni, P.; Pallotta, M.M.; Musio, A. Cornelia de Lange syndrome: From molecular diagnosis to therapeutic approach. J. Med. Genet. 2019. [Google Scholar] [CrossRef]
- Mannini, L.; Cucco, F.; Quarantotti, V.; Krantz, I.D.; Musio, A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum. Mutat. 2013, 34, 1589–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, I.; Gervasini, C.; Pozojevic, J.; Wendt, K.S.; Watrin, E.; Azzollini, J.; Braunholz, D.; Buiting, K.; Cereda, A.; Engels, H.; et al. Expanding the clinical spectrum of the “HDAC8-phenotype”—implications for molecular diagnostics, counseling and risk prediction. Clin. Genet. 2016, 89, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. Part A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avagliano, L.; Parenti, I.; Grazioli, P.; Di Fede, E.; Parodi, C.; Mariani, M.; Kaiser, F.J.; Selicorni, A.; Gervasini, C.; Massa, V. Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin. Genet. 2020, 97, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piché, J.; van Vliet, P.P.; Pucéat, M.; Andelfinger, G. The expanding phenotypes of cohesinopathies: One ring to rule them all! Cell Cycle 2019, 18, 2828–2848. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Teresa-Rodrigo, M.E.; Pozojevic, J.; Ruiz Gil, S.; Bader, I.; Braunholz, D.; Bramswig, N.C.; Gervasini, C.; Larizza, L.; Pfeiffer, L.; et al. Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum. Genet. 2017, 136, 307–320. [Google Scholar] [CrossRef]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef]
- Groza, T.; Köhler, S.; Moldenhauer, D.; Vasilevsky, N.; Baynam, G.; Zemojtel, T.; Schriml, L.M.; Kibbe, W.A.; Schofield, P.N.; Beck, T.; et al. The Human Phenotype Ontology: Semantic Unification of Common and Rare Disease. Am. J. Hum. Genet. 2015, 97, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef]
- Pié, J.; Gil-Rodríguez, M.C.; Ciero, M.; López-Viñas, E.; Ribate, M.P.; Arnedo, M.; Deardorff, M.A.; Puisac, B.; Legarreta, J.; de Karam, J.C.; et al. Mutations and variants in the cohesion factor genes NIPBL, SMC1A, and SMC3 in a cohort of 30 unrelated patients with Cornelia de Lange syndrome. Am. J. Med. Genet. Part A 2010, 152, 924–929. [Google Scholar] [CrossRef] [Green Version]
- Krawczynska, N.; Wierzba, J.; Jasiecki, J.; Wasag, B. Molecular characterization of two novel intronic variants of NIPBL gene detected in unrelated Cornelia de Lange syndrome patients. BMC Med. Genet. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Gillis, L.A.; McCallum, J.; Kaur, M.; DeScipio, C.; Yaeger, D.; Mariani, A.; Kline, A.D.; Li, H.H.; Devoto, M.; Jackson, L.G.; et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet. 2004, 75, 610–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, K.C.; Xu, B.; Krantz, I.D.; Gerton, J.L. NIPBL Controls RNA Biogenesis to Prevent Activation of the Stress Kinase PKR. Cell Rep. 2016, 14, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisac, B.; Teresa-Rodrigo, M.E.; Hernández-Marcos, M.; Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Visnes, T.; Bot, C.; Gómez-Puertas, P.; Kaiser, F.J.; Ramos, F.J.; et al. mRNA Quantification of NIPBL Isoforms A and B in Adult and Fetal Human Tissues, and a Potentially Pathological Variant Affecting Only Isoform A in Two Patients with Cornelia de Lange Syndrome. Int. J. Mol. Sci. 2017, 18, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teresa-Rodrigo, M.E.; Eckhold, J.; Puisac, B.; Dalski, A.; Gil-Rodríguez, M.C.; Braunholz, D.; Baquero, C.; Hernández-Marcos, M.; de Karam, J.C.; Ciero, M.; et al. Functional characterization of NIPBL physiological splice variants and eight splicing mutations in patients with Cornelia de Lange syndrome. Int. J. Mol. Sci. 2014, 15, 10350–10364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, L.; Liang, D.; Huang, Y.; Pan, Q.; Wu, L. Two novel NIPBL gene mutations in Chinese patients with Cornelia de Lange syndrome. Gene 2015, 555, 476–480. [Google Scholar] [CrossRef]
- Wierzba, J.; Gil-Rodríguez, M.C.; Polucha, A.; Puisac, B.; Arnedo, M.; Teresa-Rodrigo, M.E.; Winnicka, D.; Hegardt, F.G.; Ramos, F.J.; Limon, J.; et al. Cornelia de Lange syndrome with NIPBL mutation and mosaic Turner syndrome in the same individual. BMC Med. Genet. 2012, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E.; Vicente-Gabas, A.; Bernal, M.; Casale, C.H.; Bueno-Lozano, G.; Bueno-Martínez, I.; Queralt, E.; et al. Severe ipsilateral musculoskeletal involvement in a Cornelia de Lange patient with a novel NIPBL mutation. Eur. J. Med. Genet. 2014, 57, 503–509. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pié, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Teresa-Rodrigo, M.E.; Hernández-Marcos, M.; Bueno-Lozano, G.; Bueno-Martínez, I.; Remeseiro, S.; Fernández-Hernández, R.; Bassecourt-Serra, M.; Rodríguez de Alba, M.; et al. Could a patient with SMC1A duplication be classified as a human cohesinopathy? Clin. Genet. 2014, 85, 446–451. [Google Scholar] [CrossRef]
- Deep learning for genomics. Nat. Genet. 2019, 51, 1. [CrossRef] [PubMed]
- Rohatgi, S.; Clark, D.; Kline, A.D.; Jackson, L.G.; Pié, J.; Siu, V.; Ramos, F.J.; Krantz, I.D.; Deardorff, M.A. Facial diagnosis of mild and variant CdLS: Insights from a dysmorphologist survey. Am. J. Med. Genet. Part A 2010, 152, 1641–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basel-Vanagaite, L.; Wolf, L.; Orin, M.; Larizza, L.; Gervasini, C.; Krantz, I.D.; Deardorff, M.A. Recognition of the Cornelia de Lange syndrome phenotype with facial dysmorphology novel analysis. Clin. Genet. 2016, 89, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Pantel, J.T.; Zhao, M.; Mensah, M.A.; Hajjir, N.; Hsieh, T.; Hanani, Y.; Fleischer, N.; Kamphans, T.; Mundlos, S.; Gurovich, Y.; et al. Advances in computer-assisted syndrome recognition by the example of inborn errors of metabolism. J. Inherit. Metab. Dis. 2018, 41, 533–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishima, H.; Suzuki, H.; Doi, M.; Miyazaki, M.; Watanabe,, S.; Matsumoto, T.; Morifuji, K.; Moriuchi, H.; Yoshiura, K.I.; Kondoh, T.; et al. Evaluation of Face2Gene using facial images of patients with congenital dysmorphic syndromes recruited in Japan. J. Hum. Genet. 2019, 64, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Alcalde, I.; Mendieta-Moreno, J.I.; Puisac, B.; Gil-Rodríguez, M.C.; Hernández-Marcos, M.; Soler-Polo, D.; Ramos, F.J.; Ortega, J.; Pié, J.; Mendieta, J.; et al. Two-step ATP-driven opening of cohesin head. Sci. Rep. 2017, 7, 3266. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Age Photo | CdLS Score | Gene | Variant Type | Exon/Intron | Mutation (hg 19) | Protein | Inheritance | Novelty |
|---|---|---|---|---|---|---|---|---|---|---|
| #N01 | M | 5 | 14 | NIPBL | missense | 35 | c.6242G>T | p.Gly2081Val | de novo | Patient reported [20] |
| #N02 | M | 1 | 10 | NIPBL | splice variant | 3i | c.230+1G>A | - | de novo | Patient reported [20] |
| #N03 | F | 2 | 13 | NIPBL | nonsense | 10 | c.2146C>T | p.Gln716* | de novo | Patient reported [20] |
| #N04 | F | 1 | 15 | NIPBL | missense | 37 | c.6449T>C | p.Leu2150Pro | - | Patient Reported [2,20] |
| #N05 | M | 14 | 12 | NIPBL | nonframeshiftDeletion | 30 | c.5689_5691delAAT | p.Asn1897del | de novo | Patient Reported [2,20] |
| #N06 | F | 4 | 13 | NIPBL | frameshiftDeletion | 20 | c.4321G>T | p.Phe1442Lysfs*3 | de novo | Patient reported [20] |
| #N07 | F | 13 | 16 | NIPBL | frameshiftDeletion | 10 | c.2479_2480delAG | p.Arg827Glyfs*2 | de novo | Patient reported [20] |
| #N08 | M | 9 | 14 | NIPBL | nonsense | 3 | c.133C>T | p.Arg45* | de novo | New CdLS Variant |
| #N09 | F | 24 | 9 | NIPBL | missense | 36 | c.6316G>C | p.Val2106Leu | de novo | ClinVar |
| #N10 | F | 32 | 15 | NIPBL | missense | 41 | c.7012G>C | p.Ala2338Pro | de novo | ClinVar |
| #N11 | F | 37 | 15 | NIPBL | splice variant | 32i | c.5862+2insGAG | - | de novo | Similar Variant described in the literature c.5862 + 1delG [21] |
| #N12 | M | 16 | 15 | NIPBL | frameshiftDeletion | 10 | c.3060_3063delAGAG | p.Glu1021Thrfs*22 | - | Variant described in the literature [11] |
| #N13 | F | 27 | 13 | NIPBL | missense | 29 | c.5471C>T | p.Ser1824Leu | de novo | New CdLS Variant |
| #N14 | F | 40 | - | NIPBL | missense | 40 | c.6893G>A | p.Arg2298His | de novo | Variant described in the literature [11,22,23] |
| #N15 | M | 13 | 10 | NIPBL | missense | 47 | c.8387A>G | p.Tyr2796Cys | familial (m) | Patient reported [24] |
| #N16 | F | 2 | 14 | NIPBL | missense | 36 | c.6269G>T | p.Ser2090Ile | de novo | Patient reported [20] |
| #N17 | M | 3 | 13 | NIPBL | splice variant | 28i | c.5329-6T>G | - | familial (p) | Patient reported [25] |
| #N18 | F | 8 | 13 | NIPBL | frameshiftDeletion | 44 | c.7438_7439delAG | p.Arg2480Lysfs*5 | de novo | Patient reported [20] |
| #N19 | M | 16 | 13 | NIPBL | exon 4 deletion | 4 | - | - | - | New CdLS Variant |
| #N20 | F | 5 | 15 | NIPBL | missense | 39 | c.6647A>C | p.Tyr2216Ser | de novo | Patient reported [4] |
| #N21 | F | 7 | 14 | NIPBL | missense | 36 | c.6272G>A | p.Cys2091Tyr | - | Variant described in the literature [26] |
| #N22 | M | 1 | 16 | NIPBL | splice variant | 19i | c.4320+5G>C | - | de novo | Patient reported [20,25] |
| #N23 | F | 5 | 14 | NIPBL | nonsense | 39 | c.6880C>T | p.Gln2294* | de novo | Patient reported [20] |
| #N24 | F | 1 | 15 | NIPBL | nonsense | 9 | c.1445_1448delGAGA | p.Arg482Asnfs*20 | - | Patient reported [27] |
| #N25 | M | 3 | 16 | NIPBL | missense | 39 | c.6647A>G | p.Tyr2216Cys | de novo | Patient reported [28] |
| #N26 | F | 7 | 13 | NIPBL | missense | 40 | c.6860T>C | p.Leu2287Pro | - | New CdLS Variant |
| #N27 | M | 1 | 15 | NIPBL | nonsense | 29 | c.5455C>T | p.Arg1819* | de novo | New CdLS Variant |
| #N28 | F | 9 | 15 | NIPBL | frameshiftInsertion | 41 | c.6964_6965insATTTA | p.Ala2325* | - | New CdLS Variant |
| #N29 | F | 2 | 13 | NIPBL | splice variant | 21i | c.4560+4A>G | - | de novo | New CdLS Variant |
| #N30 | F | 1 | 15 | NIPBL | frameshiftDeletion | 38 | c.6549_6552delCTCA | p.His2183Glnfs*13 | de novo | New CdLS Variant |
| #N31 | M | 4 | 17 | NIPBL | splice variant | 20i | c.4422-1G>T | - | - | New CdLS Variant |
| #N32 | M | 34 | 14 | NIPBL | splice variant | 2i | c.65-5A>G | - | - | LOVD |
| #N33 | F | 16 | 15 | NIPBL | nonsense | 9 | c.992C>T | p.Arg308* | - | New CdLS Variant |
| #S34 | M | 5 | 12 | SMC1A | missense | 4 | c.587G>A | p.Arg196His | de novo | Patient reported [20,29] |
| #S35 | F | 27 | 14 | SMC1A | nonframeshiftInsertion | 5 | c.802_804delAAG | p.Lys268del | de novo | Patient reported [20] |
| #S36 | M | 4 | 13 | SMC1A | missense | 13 | c.2132 G>A | p.Arg711Gln | de novo | Patient reported [20] |
| #S37 | F | 7 | 14 | SMC1A | missense | 15 | c.2369G>A | p.Arg790Gln | - | Patient reported [13] |
| #S38 | F | 2 | - | SMC1A | nonframeshiftDeletion | 5 | c.802_804delAAG | p.Lys268del | - | Variant described in the literature [20] |
| #S39 | F | 2 | 13 | SMC1A | splice variant | 2 | c.44-1G>A | - | - | New CdLS Variant |
| #S40 | M | 11 | - | SMC1A | missense | 22 | c.3340A>T | p.Asn1114Tyr | familial (m) | New CdLS Variant |
| #S41 | F | 41 | 15 | SMC1A | nonframeshiftDeletion | 5 | c.802_804delAAG | p.Lys268del | - | Variant described in the literature [20] |
| #H42 | F | 4 | 8 | HDAC8 | missense | 6 | c.562G>A | p.Ala188Thr | de novo | Clin Var |
| #H43 | M | 3 | 12 | HDAC8 | missense | 9 | c.958G>A | p.Gly320Arg | - | ClinVar |
| #H44 | F | 6 | 9 | HDAC8 | missense | 7 | c.709G>T | p.Asp237Tyr | - | New CdLS Variant |
| #H45 | M | 5 | 11 | HDAC8 | missense | 4 | c.305G>A | p.Cys102Tyr | de novo | New CdLS Variant |
| #H46 | F | 11 | 8 | HDAC8 | missense | 5 | c.468T>G | p.Asn156Lys | de novo | Patient reported [12] |
| #R47 | F | 3 | 8 | RAD21 | missense | 11 | c.1382C>T | p.Thr461Ile | familial (p) | Patient reported (In press) |
| #R48 | F | 5 | - | RAD21 | 4.7 Mb deletion | whole gene | 8q24.11q24.12(117765326_122494596)x1 | - | New CdLS Variant | |
| #R49 | M | 8 | 10 | RAD21 | 504 Kb deletion | whole gene | 8q24.11 (117765326_118270323)x1 | - | New CdLS Variant | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latorre-Pellicer, A.; Ascaso, Á.; Trujillano, L.; Gil-Salvador, M.; Arnedo, M.; Lucia-Campos, C.; Antoñanzas-Pérez, R.; Marcos-Alcalde, I.; Parenti, I.; Bueno-Lozano, G.; et al. Evaluating Face2Gene as a Tool to Identify Cornelia de Lange Syndrome by Facial Phenotypes. Int. J. Mol. Sci. 2020, 21, 1042. https://doi.org/10.3390/ijms21031042
Latorre-Pellicer A, Ascaso Á, Trujillano L, Gil-Salvador M, Arnedo M, Lucia-Campos C, Antoñanzas-Pérez R, Marcos-Alcalde I, Parenti I, Bueno-Lozano G, et al. Evaluating Face2Gene as a Tool to Identify Cornelia de Lange Syndrome by Facial Phenotypes. International Journal of Molecular Sciences. 2020; 21(3):1042. https://doi.org/10.3390/ijms21031042
Chicago/Turabian StyleLatorre-Pellicer, Ana, Ángela Ascaso, Laura Trujillano, Marta Gil-Salvador, Maria Arnedo, Cristina Lucia-Campos, Rebeca Antoñanzas-Pérez, Iñigo Marcos-Alcalde, Ilaria Parenti, Gloria Bueno-Lozano, and et al. 2020. "Evaluating Face2Gene as a Tool to Identify Cornelia de Lange Syndrome by Facial Phenotypes" International Journal of Molecular Sciences 21, no. 3: 1042. https://doi.org/10.3390/ijms21031042