Associations between the Complement System and Choroidal Neovascularization in Wet Age-Related Macular Degeneration

 ,
,
Abstract
:1. Introduction
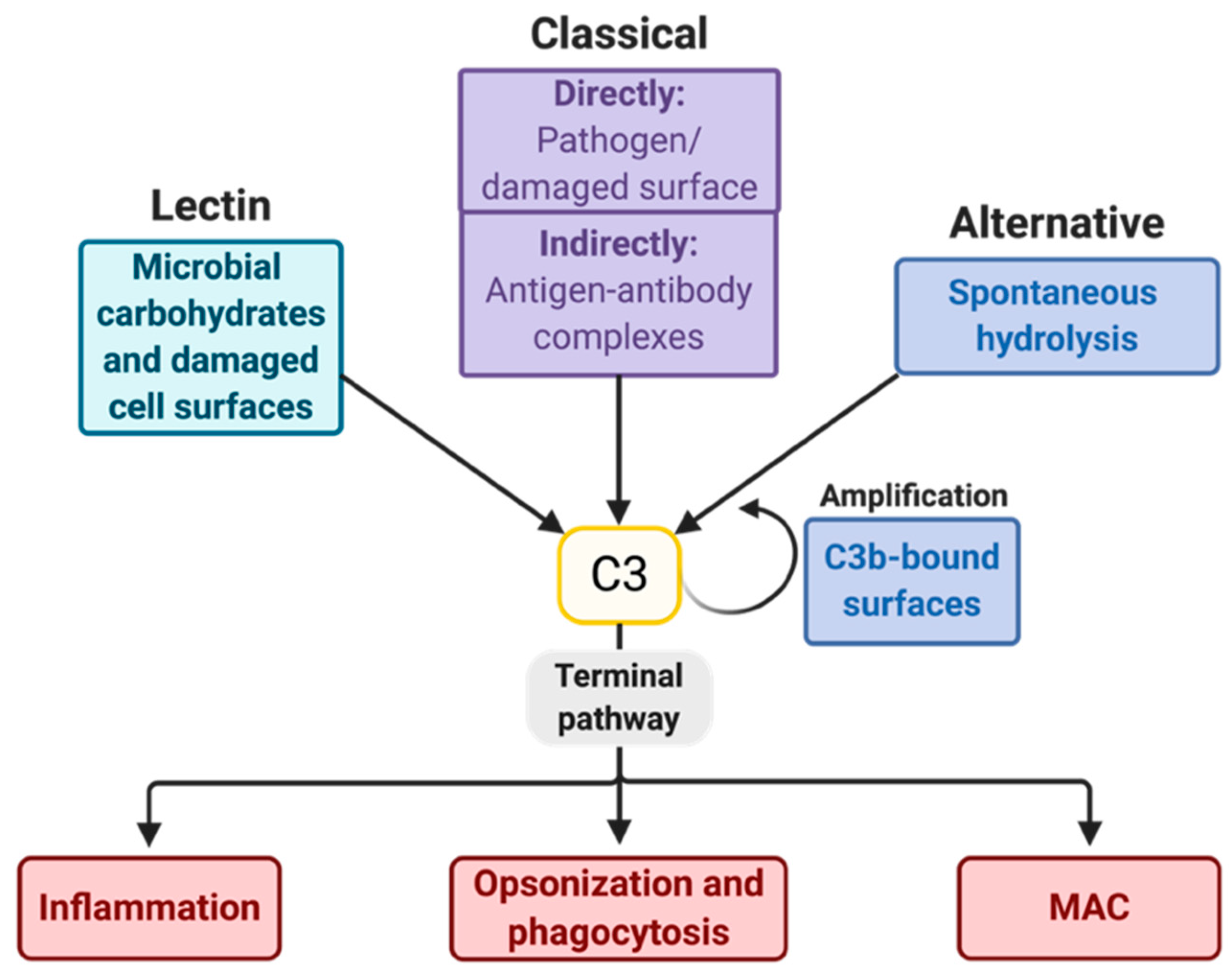
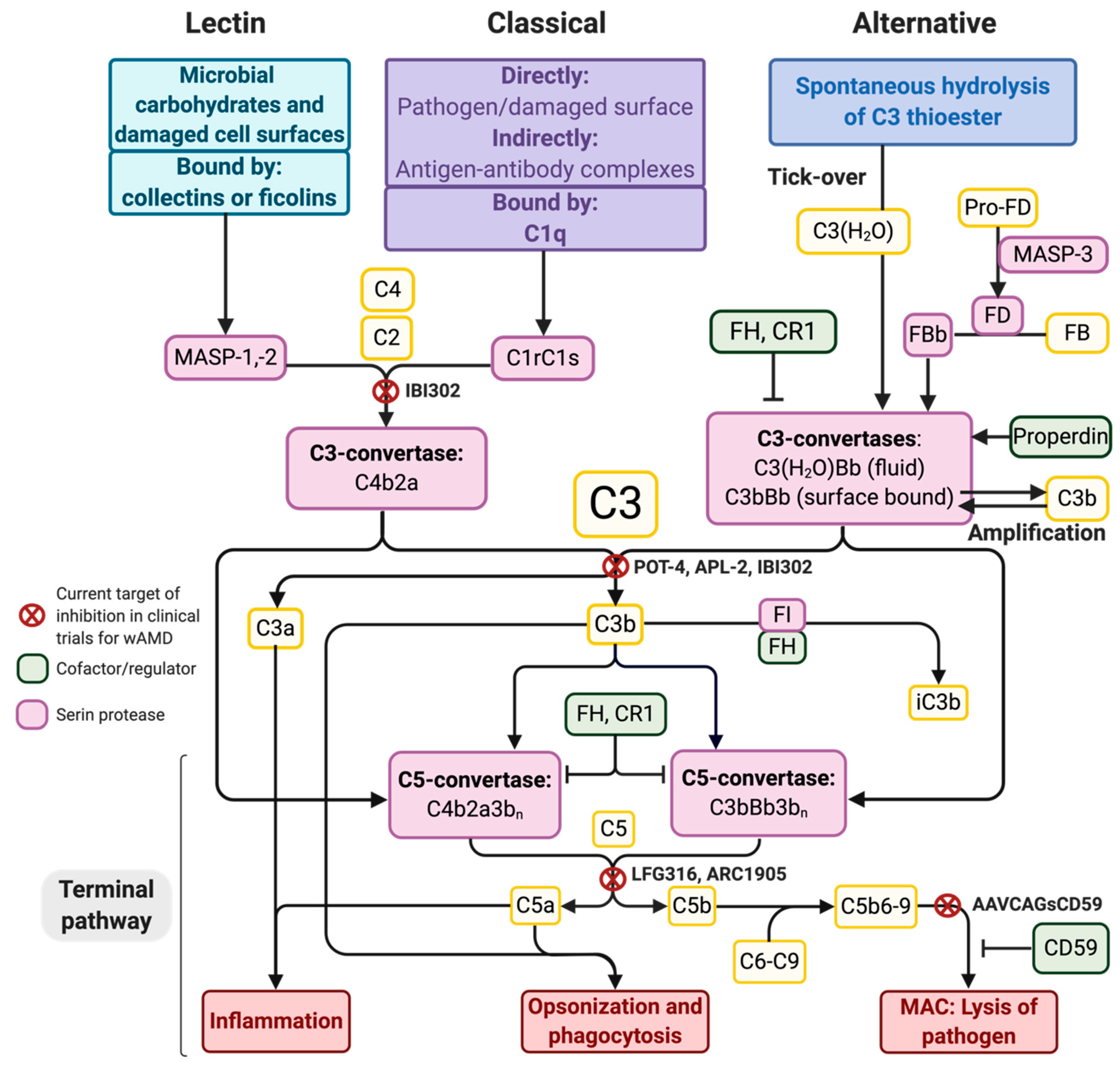
2. The Complement System and Potential Targets for Inhibition
3. Genetic Associations between AMD and the Complement System
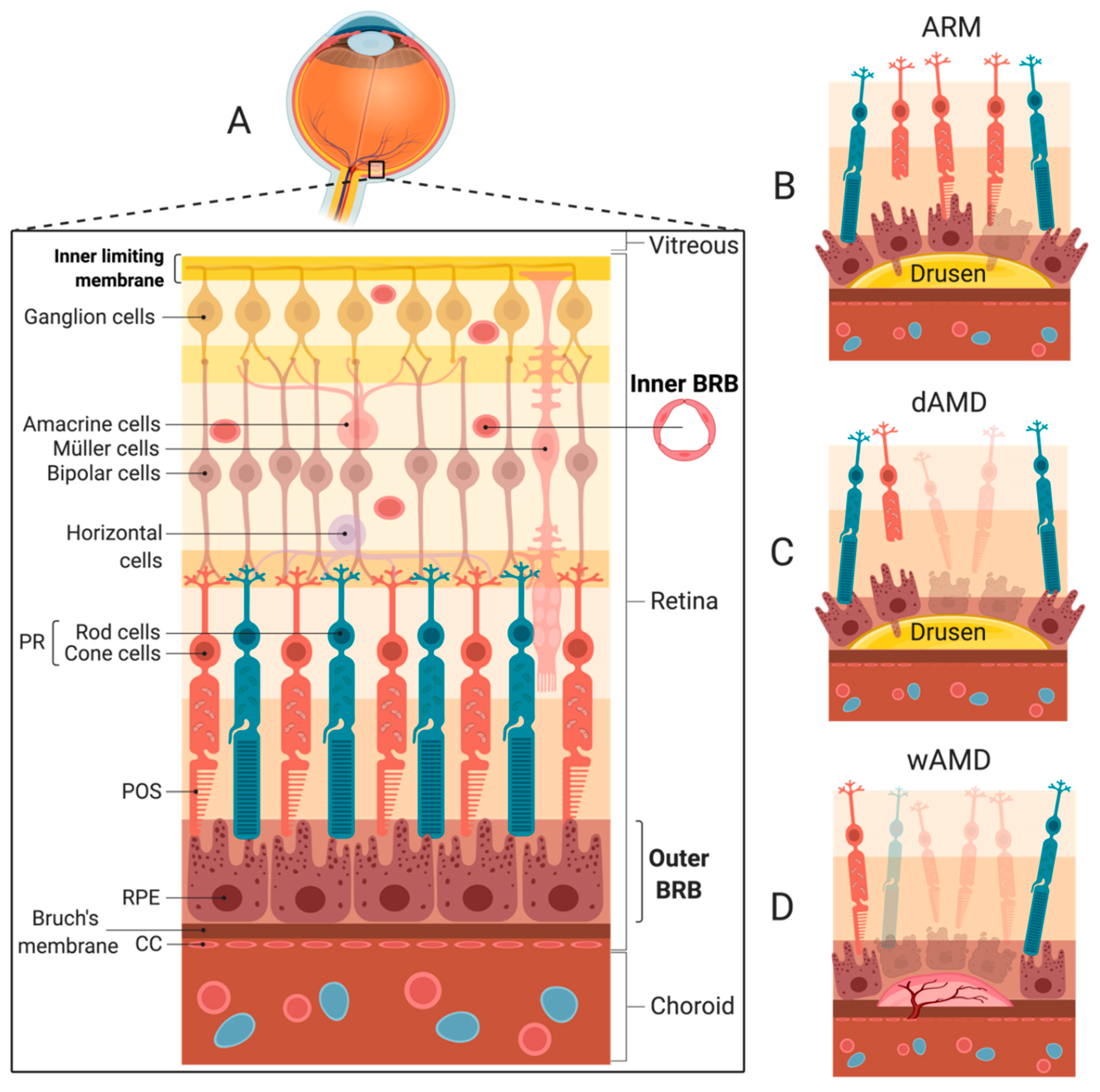
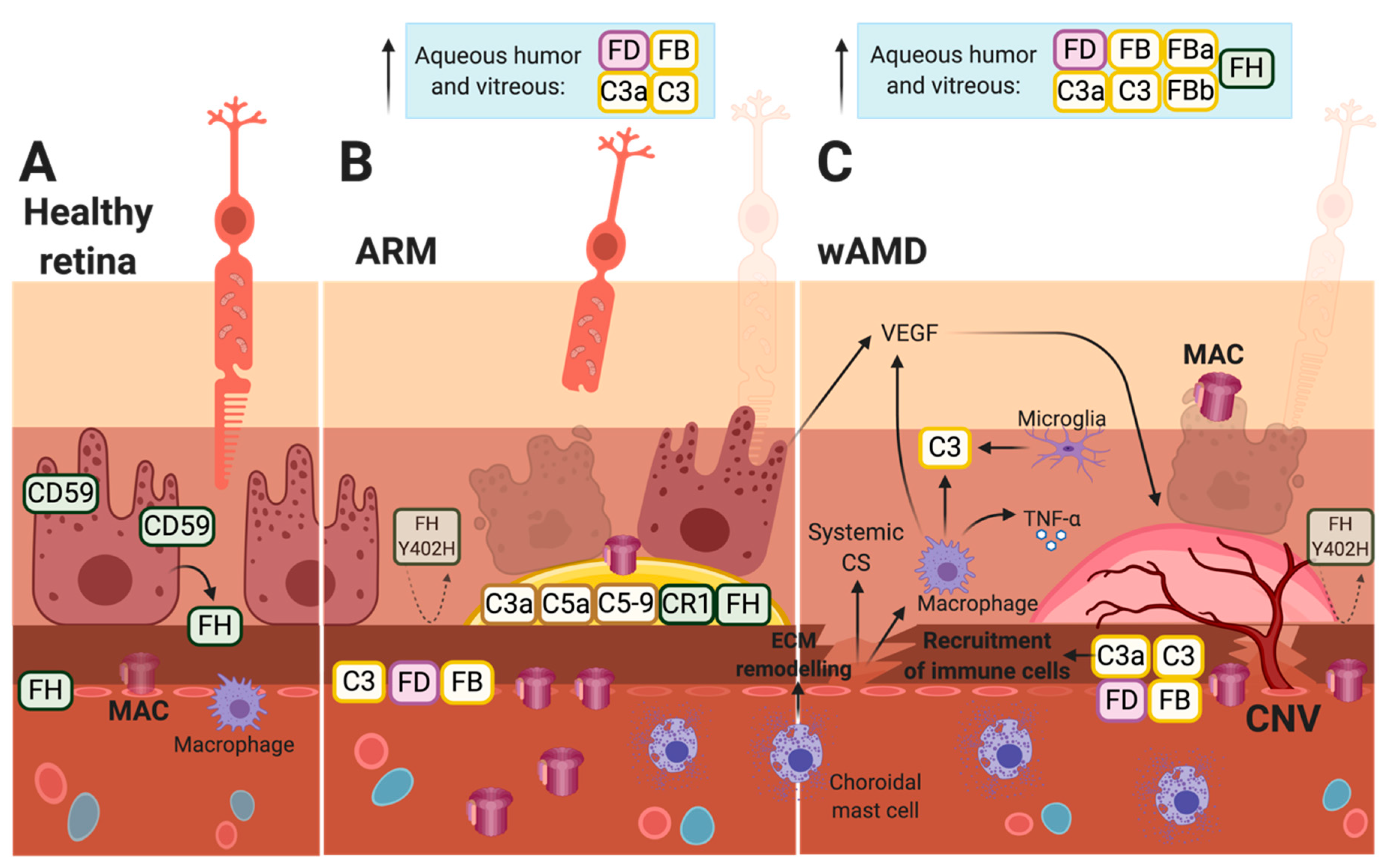
4. Choroidal Neovascularization and wAMD
5. Local Production of Complement Factors in the Retina
6. Complement Involved in Laser-Induced CNV Formation in Mice
7. Current Treatment and Clinical Trials for Complement Inhibition in wAMD
8. Future Therapeutic Approach
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| AMD | age-related macular degeneration |
| AP | alternative pathway |
| ARM | age-related maculopathy |
| BRB | blood-retina barrier |
| C3 | complement component 3 (same for C2, C4, etc.) |
| C3(H2O)Bb | fluid-phase C3-convertase |
| CC | choriocapillaris |
| CNV | choroidal neovascularization |
| CP | classical pathway |
| CR1 | complement receptor 1 |
| dAMD | dry AMD |
| EMA | European Medicines Agency |
| ECM | extracellular matrix |
| FDA | Food and Drug Administration |
| FB | factor B |
| FD | factor D |
| FH | factor H |
| FI | factor I |
| GA | geographic atrophy |
| GAG | glycosaminoglycans |
| GWAS | genome-wide association study |
| Ig | immunoglobulin |
| KO | knock-out |
| LP | lectin pathway |
| MAC | membrane-attack complex |
| MASP | MBL-associated serine protease |
| MBL | mannose-binding lectin |
| mRNA | messenger RNA |
| PEDF | pigment endothelial-derived factor |
| PR | photoreceptor |
| rCD59 | recombinant CD59 |
| RPE | retinal pigment epithelium |
| SNP | single nucleotide polymorphism |
| TNF-α | tumor necrosis factor-alpha |
| VEGF | vascular endothelial growth factor |
| wAMD | wet AMD |
References
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor h (hf1/cfh) predisposes individua1. hageman, g. s. et al. a common haplotype in the complement regulatory gene factor h (hf1/cfh) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hageman, G.S.; Luthert, P.J.; Victor Chong, N.H.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the rpe-bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732. [Google Scholar] [CrossRef]
- Parsons, N.; Annamalai, B.; Obert, E.; Schnabolk, G.; Tomlinson, S.; Rohrer, B. Inhibition of the alternative complement pathway accelerates repair processes in the murine model of choroidal neovascularization. Mol. Immunol. 2019, 108, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, M.J.; de Jong, E.K.; den Hollander, A.I. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Park, D.H.; Connor, K.M.; Lambris, J.D. The challenges and promise of complement therapeutics for ocular diseases. Front. Immunol. 2019, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.F.; Aptsiauri, N.; Hageman, G.S. Structure and composition of drusen associated with glomerulonephritis: Implications for the role of complement activation in drusen biogenesis. Eye 2001, 15, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Bora, N.S.; Matta, B.; Lyzogubov, V.V.; Bora, P.S. Relationship between the complement system, risk factors and prediction models in age-related macular degeneration. Mol. Immunol. 2015, 63, 176–183. [Google Scholar] [CrossRef]
- Van Lookeren Campagne, M.; Strauss, E.C.; Yaspan, B.L. Age-related macular degeneration: Complement in action. Immunobiology 2016, 221, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Korb, L.C.; Ahearn, J.M. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: Complement deficiency and systemic lupus erythematosus revisited. J. Immunol. 1997, 158, 4525–4528. [Google Scholar]
- Taylor, P.R.; Carugati, A.; Fadok, V.A.; Cook, H.T.; Andrews, M.; Carroll, M.C.; Savill, J.S.; Henson, P.M.; Botto, M.; Walport, M.J. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 2000, 192, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Kuraya, M.; Ming, Z.; Liu, X.; Matsushita, M.; Fujita, T. Specific binding of l-ficolin and h-ficolin to apoptotic cells leads to complement activation. Immunobiology 2005, 209, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Raaschou-Jensen, N.; Roos, A.; Daha, M.R.; Madsen, H.O.; Borrias-Essers, M.C.; Ryder, L.P.; Koch, C.; Garred, P. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur. J. Immunol. 2003, 33, 2853–2863. [Google Scholar] [CrossRef]
- McGrath, F.D.G.; Brouwer, M.C.; Arlaud, G.J.; Daha, M.R.; Hack, C.E.; Roos, A. Evidence that complement protein c1q interacts with c-reactive protein through its globular head region. J. Immunol. 2006, 176, 2950–2957. [Google Scholar] [CrossRef] [Green Version]
- Kumar-Singh, R. The role of complement membrane attack complex in dry and wet amd-from hypothesis to clinical trials. Exp. Eye Res. 2019, 184, 266–277. [Google Scholar] [CrossRef]
- Hänsch, G.M.; Seitz, M.; Betz, M. Effect of the late complement components c5b-9 on human monocytes: Release of prostanoids, oxygen radicals and of a factor inducing cell proliferation. Int. Arch. Allergy Appl. Immunol. 1987, 82, 317–320. [Google Scholar] [CrossRef]
- Lueck, K.; Wasmuth, S.; Williams, J.; Hughes, T.R.; Morgan, B.P.; Lommatzsch, A.; Greenwood, J.; Moss, S.E.; Pauleikhoff, D. Sub-lytic c5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye 2011, 25, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Kunchithapautham, K.; Rohrer, B. Sublytic membrane-attack-complex (mac) activation alters regulated rather than constitutive vascular endothelial growth factor (vegf) secretion in retinal pigment epithelium monolayers. J. Biol. Chem. 2011, 286, 23717–23724. [Google Scholar] [CrossRef] [Green Version]
- Ambati, J.; Ambati, B.K.; Yoo, S.H.; Ianchulev, S.; Adamis, A.P. Age-related macular degeneration: Etiology, pathogenesis, and therapeutic strategies. Surv. Ophthalmol. 2003, 48, 257–293. [Google Scholar] [CrossRef]
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of age-related macular degeneration in europe: The past and the future. Ophthalmology 2017, 124, 1753–1763. [Google Scholar] [CrossRef] [Green Version]
- Vingerling, J.R.; Dielemans, I.; Hofman, A.; Grobbee, D.E.; Hijmering, M.; Kramer, C.F.L.; de Jong, P.T.V.M. The prevalence of age-related maculopathy in the rotterdam study. Ophthalmology 1995, 102, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Heal. 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Kay, P.; Yang, Y.C.; Paraoan, L. Directional protein secretion by the retinal pigment epithelium: Roles in retinal health and the development of age-related macular degeneration. J. Cell. Mol. Med. 2013, 17, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.C.; Bressler, N.M.; Bressler, S.B.; Chisholm, I.H.; Coscas, G.; Davis, M.D.; de Jong, P.T.V.M.; Klaver, C.C.W.; Klein, B.E.K.; Klein, R.; et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv. Ophthalmol. 1995, 39, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Klaver, C.C.; Assink, J.J.; Van Leeuwen, R.; Wolfs, R.C.; Vingerling, J.R.; Stijnen, T.; Hofman, A.; de Jong, P.T. Incidence and progression rates of age-related maculopathy: The rotterdam study | iovs | arvo journals. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2237–2241. [Google Scholar]
- Davis, M.D.; Gangnon, R.E.; Lee, L.Y.; Hubbard, L.D.; Klein, B.E.K.; Klein, R.; Ferris, F.L.; Bressler, S.B.; Milton, R.C. The age-related eye disease study severity scale for age-related macular degeneration: Areds report no. 17. Arch. Ophthalmol. 2005, 123, 1484–1498. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.; Meuer, S.M.; Myers, C.E.; Buitendijk, G.H.S.; Rochtchina, E.; Choudhury, F.; De Jong, P.T.V.M.; McKean-Cowdin, R.; Iyengar, S.K.; Gao, X.; et al. Harmonizing the classification of age-related macular degeneration in the three-continent amd consortium. Ophthalmic Epidemiol. 2014, 21, 14–23. [Google Scholar] [CrossRef]
- Sallo, F.B.; Peto, T.; Dandekar, S.; Leung, I.; Bird, A.C. The international classification system and progression of amd. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1811. [Google Scholar]
- Ishibashi, T.; Patterson, R.; Ohnishi, Y.; Inomata, H.; Ryan, S.J. Formation of drusen in the human eye. Am. J. Ophthalmol. 1986, 101, 342–353. [Google Scholar] [CrossRef]
- Schuman, S.G.; Koreishi, A.F.; Farsiu, S.; Jung, S.H.; Izatt, J.A.; Toth, C.A. Photoreceptor layer thinning over drusen in eyes with age-related macular degeneration imaged in vivo with spectral-domain optical coherence tomography. Ophthalmology 2009, 116, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gass, J.D.M. Drusen and disciform macular detachment and degeneration. Arch. Ophthalmol. 1973, 90, 206–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labardini, C.P.; Blumenthal, E.Z. Causative pathogens in endophthalmitis after intravitreal injection of anti-vascular endothelial growth factor agents. Rambam Maimonides Med. J. 2018, 9, e0032. [Google Scholar] [CrossRef] [PubMed]
- Daien, V.; Nguyen, V.; Essex, R.; Morlet, N.; Barthelmes, D.; Gillies, M.; Hunt, A.; Dayajeewa, C.; Hunyor, A.; Fraser-Bell, S.; et al. Incidence and outcomes of infectious and noninfectious endophthalmitis after intravitreal injections for age-related macular degeneration. Ophthalmology 2018, 125, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.V.; Leitner, W.P.; Staples, M.K.; Anderson, D.H. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp. Eye Res. 2001, 73, 887–896. [Google Scholar] [CrossRef]
- Anderson, D.H.; Mullins, R.F.; Hageman, G.S.; Johnson, L. V A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 2002, 134, 411–431. [Google Scholar] [CrossRef]
- Van Der Schaft, T.L.; Mooy, C.M.; De Bruijn, W.C.; De Jong, P.T.V.M. Early stages of age-related macular degeneration: An immunofluorescence and electron microscopy study. Br. J. Ophthalmol. 1993, 77, 657–661. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.V.; Ozaki, S.; Staples, M.K.; Erickson, P.A.; Anderson, D.H. A potential role for immune complex pathogenesis in drusen formation. Exp. Eye Res. 2000, 70, 441–449. [Google Scholar] [CrossRef]
- Lorés-Motta, L.; Paun, C.C.; Corominas, J.; Pauper, M.; Geerlings, M.J.; Altay, L.; Schick, T.; Daha, M.R.; Fauser, S.; Hoyng, C.B.; et al. Genome-wide association study reveals variants in cfh and cfhr4 associated with systemic complement activation: Implications in age-related macular degeneration. Ophthalmology 2018, 125, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.F.; Schoo, D.P.; Sohn, E.H.; Flamme-Wiese, M.J.; Workamelahu, G.; Johnston, R.M.; Wang, K.; Tucker, B.A.; Stone, E.M. The membrane attack complex in aging human choriocapillaris: Relationship to macular degeneration and choroidal thinning. Am. J. Pathol. 2014, 184, 3142–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.F.; Dewald, A.D.; Streb, L.M.; Wang, K.; Kuehn, M.H.; Stone, E.M. Elevated membrane attack complex in human choroid with high risk complement factor h genotypes. Exp. Eye Res. 2011, 93, 565–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirco, K.R.; Flamme-Wiese, M.J.; Wiley, J.S.; Potempa, L.A.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Evaluation of serum and ocular levels of membrane attack complex and c-reactive protein in cfh-genotyped human donors. Eye 2018, 32, 1740–1742. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Perveen, R.; Hakobyan, S.; Morgan, B.P.; Sim, R.B.; Bishop, P.N.; Day, A.J. Impaired binding of the age-related macular degeneration-associated complement factor h 402h allotype to bruch’s membrane in human retina. J. Biol. Chem. 2010, 285, 30192–30202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, K.B.; Fijalkowski, N.; Cano, M.; Handa, J.T. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J. Pathol. 2013, 229, 729–742. [Google Scholar] [CrossRef] [Green Version]
- Hecker, L.A.; Edwards, A.O.; Ryu, E.; Tosakulwong, N.; Baratz, K.H.; Brown, W.L.; Issa, P.C.; Scholl, H.P.; Pollok-Kopp, B.; Schmid-Kubista, K.E.; et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum. Mol. Genet. 2010, 19, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, R.; Hartnett, M.E.; Atkinson, J.P.; Giclas, P.C.; Rosner, B.; Seddon, J.M. Plasma complement components and activation fragments: Associations with age-related macular degeneration genotypes and phenotypes. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5818–5827. [Google Scholar] [CrossRef]
- Heesterbeek, T.J.; Lechanteur, Y.T.E.; Lorés-Motta, L.; Schick, T.; Daha, M.R.; Altay, L.; Liakopoulos, S.; Smailhodzic, D.; den Hollander, A.I.; Hoyng, C.B.; et al. Complement activation levels are related to disease stage in amd. Investig. Ophthalmol. Vis. Sci. 2020, 61, 18. [Google Scholar] [CrossRef] [Green Version]
- Scholl, H.P.N.; Issa, P.C.; Walier, M.; Janzer, S.; Pollok-Kopp, B.; Börncke, F.; Fritsche, L.G.; Chong, N.V.; Fimmers, R.; Wienker, T.; et al. Systemic complement activation in age-related macular degeneration. PLoS ONE 2008, 3, e2593. [Google Scholar] [CrossRef]
- Altay, L.; Sitnilska, V.; Schick, T.; Widmer, G.; Duchateau-Nguyen, G.; Piraino, P.; Jayagopal, A.; Drawnel, F.M.; Fauser, S. Early local activation of complement in aqueous humour of patients with age-related macular degeneration. Eye 2019, 33, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Loyet, K.M.; DeForge, L.E.; Katschke, K.J.; Diehl, L.; Graham, R.R.; Pao, L.; Sturgeon, L.; Lewin-Koh, S.C.; Hollyfield, J.G.; van Lookeren Campagne, M. Activation of the alternative complement pathway in vitreous is controlled by genetics in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6628–6637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schick, T.; Steinhauer, M.; Aslanidis, A.; Altay, L.; Karlstetter, M.; Langmann, T.; Kirschfink, M.; Fauser, S. Local complement activation in aqueous humor in patients with age-related macular degeneration. Eye 2017, 31, 810–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Alper, C.A.; Myron Johnson, A.; Birtch, A.G.; Moore, F.D. Human c’3: Evidence for the liver as the primary site of synthesis. Science 1969, 163, 286–288. [Google Scholar] [CrossRef] [Green Version]
- Morgan, B.P.; Gasque, P. Extrahepatic complement biosynthesis: Where, when and why? Clin. Exp. Immunol. 1997, 107, 1–7. [Google Scholar] [CrossRef]
- Gulati, P.; Lemercier, C.; Guc, D.; Lappin, D.; Whaley, K. Regulation of the synthesis of c1 subcomponents and c1-inhibitor. Behring Inst. Mitt. 1993, 196–203. [Google Scholar]
- Wirthmueller, U.; Dewald, B.; Thelen, M.; Schäfer, M.K.; Stover, C.; Whaley, K.; North, J.; Eggleton, P.; Reid, K.B.; Schwaeble, W.J. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J. Immunol. 1997, 158, 4444–4451. [Google Scholar]
- Pauly, D.; Agarwal, D.; Dana, N.; Schäfer, N.; Biber, J.; Wunderlich, K.A.; Jabri, Y.; Straub, T.; Zhang, N.R.; Gautam, A.K.; et al. Cell-type-specific complement expression in the healthy and diseased retina. Cell Rep. 2019, 29, 2835–2848. [Google Scholar] [CrossRef]
- El-Lati, S.G.; Dahinden, C.A.; Church, M.K. Complement peptides c3a- and c5a-induced mediator release from dissociated human skin mast cells. J. Investig. Dermatol. 1994, 102, 803–806. [Google Scholar] [CrossRef] [Green Version]
- Hook, W.A.; Siraganian, R.P.; Wahl, S.M. Complement-induced histamine release from human basophils i. generation of activity in human serum. J. Immunol. 1975, 114, 1185–1190. [Google Scholar] [PubMed]
- Siraganian, R.P.; Hook, W.A. Complement-induced histamine release from human basophils ii. mechanism of the histamine release reaction. J. Immunol. 1976, 116, 639–646. [Google Scholar] [PubMed]
- Hayashi, M.; Machida, T.; Ishida, Y.; Ogata, Y.; Omori, T.; Takasumi, M.; Endo, Y.; Suzuki, T.; Sekimata, M.; Homma, Y.; et al. Cutting edge: Role of masp-3 in the physiological activation of factor d of the alternative complement pathway. J. Immunol. 2019, 203, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Hourcade, D.E. The role of properdin in the assembly of the alternative pathway c3 convertases of complement. J. Biol. Chem. 2006, 281, 2128–2132. [Google Scholar] [CrossRef] [Green Version]
- Bexborn, F.; Andersson, P.O.; Chen, H.; Nilsson, B.; Ekdahl, K.N. The tick-over theory revisited: Formation and regulation of the soluble alternative complement c3 convertase (c3(H2O)bb). Mol. Immunol. 2008, 45, 2370–2379. [Google Scholar] [CrossRef] [Green Version]
- Fearon, D.T.; Austen, K.F. Properdin: Binding to c3b and stabilization of the c3b dependent c3 convertase. J. Exp. Med. 1975, 142, 856–863. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Bishop, P.N.; Day, A.J. Complement factor h and age-related macular degeneration: The role of glycosaminoglycan recognition in disease pathology. Biochem. Soc. Trans. 2010, 38, 1342–1348. [Google Scholar] [CrossRef] [Green Version]
- Leffler, J.; Herbert, A.P.; Norström, E.; Schmidt, C.Q.; Barlow, P.N.; Blom, A.M.; Martin, M. Annexin-ii, dna, and histones serve as factor h ligands on the surface of apoptotic cells. J. Biol. Chem. 2010, 285, 3766–3776. [Google Scholar] [CrossRef] [Green Version]
- Weeks, D.E.; Conley, Y.P.; Tsai, H.J.; Mah, T.S.; Schmidt, S.; Postel, E.A.; Agarwal, A.; Haines, J.L.; Pericak-Vance, M.A.; Rosenfeld, P.J.; et al. Age-related maculopathy: A genomewide scan with continued evidence of susceptibility loci within the 1q31, 10q26, and 17q25 regions. Am. J. Hum. Genet. 2004, 75, 174–189. [Google Scholar] [CrossRef] [Green Version]
- Seddon, J.M.; Santangelo, S.L.; Book, K.; Chong, S.; Cote, J. A genomewide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am. J. Hum. Genet. 2003, 73, 780–790. [Google Scholar] [CrossRef] [Green Version]
- Majewski, J.; Schultz, D.W.; Weleber, R.G.; Schain, M.B.; Edwards, A.O.; Matise, T.C.; Acott, T.S.; Ott, J.; Klein, M.L. Age-related macular degeneration-a genome scan in extended families. Am. J. Hum. Genet. 2003, 73, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, S.K.; Song, D.; Klein, B.E.K.; Klein, R.; Schick, J.H.; Humphrey, J.; Millard, C.; Liptak, R.; Russo, K.; Jun, G.; et al. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am. J. Hum. Genet. 2004, 74, 20–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, G.R.; Yashar, B.M.; Zhao, Y.; Ghiasvand, N.M.; Zareparsi, S.; Branham, K.E.H.; Reddick, A.C.; Trager, E.H.; Yoshida, S.; Bahling, J.; et al. Age-related macular degeneration: A high-resolution genome scan for susceptibility loci in a population enriched for late-stage disease. Am. J. Hum. Genet. 2004, 74, 482–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor h polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef]
- Rivera, A.; Fisher, S.A.; Fritsche, L.G.; Keilhauer, C.N.; Lichtner, P.; Meitinger, T.; Weber, B.H.F. Hypothetical loc387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor h to disease risk. Hum. Mol. Genet. 2005, 14, 3227–3236. [Google Scholar] [CrossRef] [Green Version]
- Jakobsdottir, J.; Conley, Y.P.; Weeks, D.E.; Mah, T.S.; Ferrell, R.E.; Gorin, M.B. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am. J. Hum. Genet. 2005, 77, 389–407. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Camp, N.J.; Sun, H.; Tong, Z.; Gibbs, D.; Cameron, D.J.; Chen, H.; Zhao, Y.; Pearson, E.; Li, X.; et al. A variant of the htra1 gene increases susceptibility to age-related macular degeneration. Science 2006, 314, 992–993. [Google Scholar] [CrossRef]
- DeWan, A.; Liu, M.; Hartman, S.; Zhang, S.S.M.; Liu, D.T.L.; Zhao, C.; Tam, P.O.S.; Chan, W.M.; Lam, D.S.C.; Snyder, M.; et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 2006, 314, 989–992. [Google Scholar] [CrossRef]
- Black, J.R.M.; Clark, S.J. Age-related macular degeneration: Genome-wide association studies to translation. Genet. Med. 2016, 18, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Tan, P.L.; Bowes Rickman, C.; Katsanis, N. AMD and the alternative complement pathway: Genetics and functional implications. Hum. Genomics 2016, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, J.R.W.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement c3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerness, J.A.; Maller, J.B.; Neale, B.M.; Reynolds, R.C.; Daly, M.J.; Seddon, J.M. Variation near complement factor i is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef]
- Sofat, R.; Casas, J.P.; Webster, A.R.; Bird, A.C.; Mann, S.S.; Yates, J.R.W.; Moore, A.T.; Sepp, T.; Cipriani, V.; Bunce, C.; et al. Complement factor h genetic variant and age-related macular degeneration: Effect size, modifiers and relationship to disease subtype. Int. J. Epidemiol. 2012, 41, 250–262. [Google Scholar] [CrossRef]
- Wegscheider, B.J.; Weger, M.; Renner, W.; Steinbrugger, I.; März, W.; Mossböck, G.; Temmel, W.; El-Shabrawi, Y.; Schmut, O.; Jahrbacher, R.; et al. Association of complement factor h y402h gene polymorphism with different subtypes of exudative age-related macular degeneration. Ophthalmology 2007, 114, 738–742. [Google Scholar] [CrossRef]
- Borras, C.; Canonica, J.; Jorieux, S.; Abache, T.; El Sanharawi, M.; Klein, C.; Delaunay, K.; Jonet, L.; Salvodelli, M.; Naud, M.C.; et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.N.; Issa, P.C.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor h binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Herbert, A.P.; Deakin, J.A.; Schmidt, C.Q.; Blaum, B.S.; Egan, C.; Ferreira, V.P.; Pangburn, M.K.; Lyon, M.; Uhrín, D.; Barlow, P.N. Structure shows that a glycosaminoglycan and protein recognition site in factor h is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. J. Biol. Chem. 2007, 282, 18960–18968. [Google Scholar] [CrossRef] [Green Version]
- Nauta, A.J.; Daha, M.R.; Van Kooten, C.; Roos, A. Recognition and clearance of apoptotic cells: A role for complement and pentraxins. Trends Immunol. 2003, 24, 148–154. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Yasuma, T.R.; Tomida, D.; Nakamura, M.; Ishikawa, K.; Kikuchi, M.; Ohmi, Y.; Niwa, T.; Hamajima, N.; Furukawa, K.; et al. C9-r95x polymorphism in patients with neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 508–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penfold, P.L.; Provis, J.M.; Billson, F.A. Age-related macular degeneration: Ultrastructural studies of the relationship of leucocytes to angiogenesis. Graefe’s Arch. Clin. Exp. Ophthalmol. 1987, 225, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Green, W.R.; Wilson, D.J. Choroidal neovascularization. Ophthalmology 1986, 93, 1169–1176. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Campa, C.; Costagliola, C.; Incorvaia, C.; Sheridan, C.; Semeraro, F.; De Nadai, K.; Sebastiani, A. Inflammatory mediators and angiogenic factors in choroidal neovascularization: Pathogenetic interactions and therapeutic implications. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Pepper, M.S. Extracellular proteolysis and angiogenesis. Thromb. Haemost. 2001, 86, 346–355. [Google Scholar] [CrossRef]
- Grossniklaus, H.E.; Green, W.R. Choroidal neovascularization. Am. J. Ophthalmol. 2004, 137, 496–503. [Google Scholar] [CrossRef]
- Kvanta, A.; Algvere, P.V.; Berglin, L.; Seregard, S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1929–1934. [Google Scholar] [CrossRef]
- Hinton, D.R.; Lopez, P.F.; Sippy, B.D.; Lambert, H.M.; Thach, A.B. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1996, 37, 855–868. [Google Scholar]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal aflibercept (vegf trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef]
- Liu, K.; Song, Y.; Xu, G.; Ye, J.; Wu, Z.; Liu, X.; Dong, X.; Zhang, M.; Xing, Y.; Zhu, S.; et al. Conbercept for treatment of neovascular age-related macular degeneration: Results of the randomized phase 3 phoenix study. Am. J. Ophthalmol. 2019, 197, 156–167. [Google Scholar] [CrossRef]
- Heier, J.S.; Antoszyk, A.N.; Pavan, P.R.; Leff, S.R.; Rosenfeld, P.J.; Ciulla, T.A.; Dreyer, R.F.; Gentile, R.C.; Sy, J.P.; Hantsbarger, G.; et al. Ranibizumab for treatment of neovascular age-related macular degeneration. a phase i/ii multicenter, controlled, multidose study. Ophthalmology 2006, 113, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.J.; Leibole, M.A.; Tezel, T.; Ferguson, T.A. Fas ligand (cd95 ligand) controls angiogenesis beneath the retina. Nat. Med. 1999, 5, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ryan, A.M.; Rohan, R.; Amano, S.; Agular, S.; Miller, J.W.; Adamis, A.P. Constitutive expression of vegf, vegfr-1, and vegfr-2 in normal eyes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2115–2121. [Google Scholar]
- Stein, I.; Neeman, M.; Shweiki, D.; Itin, A.; Keshet, E. Stabilization of vascular endothelial growth factor mrna by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Mol. Cell. Biol. 1995, 15, 5363–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punglia, R.S.; Lu, M.; Hsu, J.; Kuroki, M.; Tolentino, M.J.; Keough, K.; Levy, A.P.; Levy, N.S.; Goldberg, M.A.; D’Amato, R.J.; et al. Regulation of vascular endothelial growth factor expression by insulin- like growth factor i. Diabetes 1997, 46, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, M.; Voest, E.E.; Amano, S.; Beerepoot, L.V.; Takashima, S.; Tolentino, M.; Kim, R.Y.; Rohan, R.M.; Colby, K.A.; Yeo, K.T.; et al. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J. Clin. Investig. 1996, 98, 1667–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Q.; Cao, X.; Bian, A.; Li, Y. C3a increases vegf and decreases pedf mrna levels in human retinal pigment epithelial cells. Biomed. Res. Int. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Cortright, D.N.; Meade, R.; Waters, S.M.; Chenard, B.L.; Krause, J.E. C5a, but not c3a, increases vegf secretion in arpe-19 human retinal pigment epithelial cells. Curr. Eye Res. 2009, 34, 57–61. [Google Scholar] [CrossRef]
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925. [Google Scholar] [CrossRef]
- Krause, T.A.; Alex, A.F.; Engel, D.R.; Kurts, C.; Eter, N. VEGF-production by ccr2-dependent macrophages contributes to laser-induced choroidal neovascularization. PLoS ONE 2014, 9, e94313. [Google Scholar] [CrossRef]
- Bhutto, I.A.; McLeod, D.S.; Jing, T.; Sunness, J.S.; Seddon, J.M.; Lutty, G.A. Increased choroidal mast cells and their degranulation in age-related macular degeneration. Br. J. Ophthalmol. 2016, 100, 720–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajardo, I.; Pejler, G. Tryptase is a gelatinase β human mast cell. J. Immunol Ref. 2003, 171, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, V.; Wielockx, B.; Munaut, C.; Galopin, C.; Jost, M.; Itoh, T.; Werb, Z.; Baker, A.; Libert, C.; Krell, H.-W.; et al. MMP-2 and mmp-9 synergize in promoting choroidal neovascularization. FASEB J. 2003, 17, 2290–2292. [Google Scholar] [CrossRef] [PubMed]
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 2010, 29, 500–519. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; McHarg, S.; Tilakaratna, V.; Brace, N.; Bishop, P.N. Bruch’s membrane compartmentalizes complement regulation in the eye with implications for therapeutic design in age-related macular degeneration. Front. Immunol. 2017, 8, 1778. [Google Scholar] [CrossRef] [Green Version]
- Liszewski, M.K.; Elvington, M.; Kulkarni, H.S.; Atkinson, J.P. Complement’s hidden arsenal: New insights and novel functions inside the cell. Mol. Immunol. 2017, 84, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.; Leffler, J.; Smolag, K.I.; Mytych, J.; Björk, A.; Chaves, L.D.; Alexander, J.J.; Quigg, R.J.; Blom, A.M. Factor h uptake regulates intracellular c3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016, 23, 903–911. [Google Scholar] [CrossRef]
- Luo, C.; Chen, M.; Xu, H. Complement gene expression and regulation in mouse retina and retinal pigment epithelium/choroid. Mol. Vis. 2011, 17, 1588–1597. [Google Scholar]
- Natoli, R.; Fernando, N.; Jiao, H.; Racic, T.; Madigan, M.; Barnett, N.L.; Chu-Tan, J.A.; Valter, K.; Provis, J.; Rutar, M. Retinal macrophages synthesize c3 and activate complement in amd and in models of focal retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2977–2990. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Muckersie, E.; Robertson, M.; Forrester, J.V.; Xu, H. Up-regulation of complement factor b in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp. Eye Res. 2008, 87, 543–550. [Google Scholar] [CrossRef]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor h by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, B.; Long, Q.; Coughlin, B.; Brooks Wilson, R.; Huang, Y.; Qiao, F.; Tang, P.H.; Kunchithapautham, K.; Gilkeson, G.S.; Tomlinson, S. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3056–3064. [Google Scholar] [CrossRef] [PubMed]
- Combadière, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodéro, M.; Pézard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [Green Version]
- Thurman, J.M.; Renner, B.; Kunchithapautham, K.; Ferreira, V.P.; Pangburn, M.K.; Ablonczy, Z.; Tomlinson, S.; Holers, V.M.; Rohrer, B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 2009, 284, 16939–16947. [Google Scholar] [CrossRef] [Green Version]
- Bhutto, I.A.; Baba, T.; Merges, C.; Juriasinghani, V.; McLeod, D.S.; Lutty, G.A. C-reactive protein and complement factor h in aged human eyes and eyes with age-related macular degeneration. Br. J. Ophthalmol. 2011, 95, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Moskovich, O.; Herzog, L.O.; Ehrlich, M.; Fishelson, Z. Caveolin-1 and dynamin-2 are essential for removal of the complement c5b-9 complex via endocytosis. J. Biol. Chem. 2012, 287, 19904–19915. [Google Scholar] [CrossRef] [Green Version]
- Georgiannakis, A.; Burgoyne, T.; Lueck, K.; Futter, C.; Greenwood, J.; Moss, S.E. Retinal pigment epithelial cells mitigate the effects of complement attack by endocytosis of c5b-9. J. Immunol. 2015, 195, 3382–3389. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Schnabolk, G.; Coughlin, B.; Joseph, K.; Kunchithapautham, K.; Bandyopadhyay, M.; O’Quinn, E.C.; Nowling, T.; Rohrer, B. Local production of the alternative pathway component factor b is sufficient to promote laser-induced choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1850–1863. [Google Scholar] [CrossRef] [Green Version]
- Khandhadia, S.; Hakobyan, S.; Heng, L.Z.; Gibson, J.; Adams, D.H.; Alexander, G.J.; Gibson, J.M.; Martin, K.R.; Menon, G.; Nash, K.; et al. Age-related macular degeneration and modification of systemic complement factor h production through liver transplantation. Ophthalmology 2013, 120, 1612–1618. [Google Scholar] [CrossRef]
- Booij, J.C.; Baas, D.C.; Beisekeeva, J.; Gorgels, T.G.M.F.; Bergen, A.A.B. The dynamic nature of bruch’s membrane. Prog. Retin. Eye Res. 2010, 29, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Spraul, C.W.; Lang, G.E.; Grossniklaus, H.E.; Lang, G.K. Histologic and morphometric analysis of the choroid, bruch’s membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv. Ophthalmol. 1999, 44, 10–32. [Google Scholar] [CrossRef]
- Chong, N.H.V.; Keonin, J.; Luthert, P.J.; Frennesson, C.I.; Weingeist, D.M.; Wolf, R.L.; Mullins, R.F.; Hageman, G.S. Decreased thickness and integrity of the macular elastic layer of bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am. J. Pathol. 2005, 166, 241–251. [Google Scholar] [CrossRef]
- Spraul, C.W.; Grossnihlatis, H.E. Characteristics of drusen and bruch’s membrane in postmortem eyes with age-related macular degeneration. Arch. Ophthalmol. 1997, 115, 267–273. [Google Scholar] [CrossRef]
- Rohrer, B.; Coughlin, B.; Kunchithapautham, K.; Long, Q.; Tomlinson, S.; Takahashi, K.; Holers, V.M. The alternative pathway is required, but not alone sufficient, for retinal pathology in mouse laser-induced choroidal neovascularization. Mol. Immunol. 2011, 48, e1–e8. [Google Scholar] [CrossRef] [Green Version]
- Bora, P.S.; Sohn, J.-H.; Cruz, J.M.C.; Jha, P.; Nishihori, H.; Wang, Y.; Kaliappan, S.; Kaplan, H.J.; Bora, N.S. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J. Immunol. 2005, 174, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Bora, N.S.; Kaliappan, S.; Jha, P.; Xu, Q.; Sivasankar, B.; Harris, C.L.; Morgan, B.P.; Bora, P.S. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J. Immunol. 2007, 178, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Bora, N.S.; Kaliappan, S.; Jha, P.; Xu, Q.; Sohn, J.-H.; Dhaulakhandi, D.B.; Kaplan, H.J.; Bora, P.S. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: Role of factor b and factor h. J. Immunol. 2006, 177, 1872–1878. [Google Scholar] [CrossRef]
- Brockmann, C.; Brockmann, T.; Dege, S.; Busch, C.; Kociok, N.; Vater, A.; Klussmann, S.; Strauß, O.; Joussen, A.M. Intravitreal inhibition of complement c5a reduces choroidal neovascularization in mice. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 1695–1704. [Google Scholar] [CrossRef]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components c3a and c5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [Green Version]
- Bora, N.S.; Jha, P.; Lyzogubov, V.V.; Kaliappan, S.; Liu, J.; Tytarenko, R.G.; Fraser, D.A.; Morgan, B.P.; Bora, P.S. Recombinant membrane-targeted form of cd59 inhibits the growth of choroidal neovascular complex in mice. J. Biol. Chem. 2010, 285, 33826–33833. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shang, Q.L.; Ma, J.X.; Liu, S.X.; Wang, C.X.; Ma, C. Complement factor b knockdown by short hairpin rna inhibits laser-induced choroidal neovascularization in rats. Int. J. Ophthalmol. 2020, 13, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Poor, S.H.; Qiu, Y.; Fassbender, E.S.; Shen, S.; Woolfenden, A.; Delpero, A.; Kim, Y.; Buchanan, N.; Gebuhr, T.C.; Hanks, S.M.; et al. Reliability of the mouse model of choroidal neovascularization induced by laser photocoagulation. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6525–6534. [Google Scholar] [CrossRef] [PubMed]
- Alawieh, A.; Elvington, A.; Zhu, H.; Yu, J.; Kindy, M.S.; Atkinson, C.; Tomlinson, S. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. J. Neuroinflammation 2015, 12, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.A.; D’Amore, P.A. A brief history of anti-vegf for the treatment of ocular angiogenesis. Am. J. Pathol. 2012, 181, 376–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, A.; Kay, B.K.; Lambris, J.D. Inhibition of human complement by a c3-binding peptide isolated from a phage-displayed random peptide library. J. Immunol. 1996, 157, 884–891. [Google Scholar] [PubMed]
- Skattum, L.; Van Deuren, M.; Van Der Poll, T.; Truedsson, L. Complement deficiency states and associated infections. Mol. Immunol. 2011, 48, 1643–1655. [Google Scholar] [CrossRef]
- Wu, J.; Sun, X. Complement system and age-related macular degeneration: Drugs and challenges. Drug Des. Devel. Ther. 2019, 13, 2413–2425. [Google Scholar] [CrossRef]
- A Dose Escalation Study of IBI302 in Patients With Wet Age-related Macular Degeneration. Available online: https://clinicaltrials.gov/ct2/show/NCT03814291 (accessed on 20 April 2020).
- Safety and Efficacy of Intravitreal LFG316 in Wet Age Related Macular Degeneration (AMD). Available online: https://clinicaltrials.gov/ct2/show/record/NCT01535950 (accessed on 4 April 2020).
- AAVCAGsCD59 for the Treatment of Wet AMD. Available online: https://clinicaltrials.gov/ct2/show/NCT03585556 (accessed on 5 April 2020).
- Safety and Tolerability of Intravenous LFG316 in Wet Age-related Macular Degeneration (AMD). Available online: https://clinicaltrials.gov/ct2/show/NCT01624636 (accessed on 4 April 2020).
- Safety and Efficacy of IONIS-FB-Lrx in up to 120 Patients 55 and Older With Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration (AMD). Available online: https://clinicaltrials.gov/ct2/show/NCT03446144 (accessed on 3 April 2020).
- A Study to Compare the Efficacy and Safety of Intravitreal APL-2 Therapy With Sham Injections in Patients With Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration. Available online: https://clinicaltrials.gov/ct2/show/NCT03525613 (accessed on 1 April 2020).
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteomics 2002, 1, 845–867. [Google Scholar] [CrossRef] [Green Version]
- Evaluation of AL-78898A in Exudative Age-Related Macular Degeneration. Available online: https://clinicaltrials.gov/ct2/show/NCT01157065 (accessed on 1 April 2020).
- Lenier, S. APL-2 slows growth of GA in phase II safety and efficacy trial. Available online: https://www.modernretina.com/view/bausch-health-spin-off-of-eye-health-business (accessed on 6 August 2020).
- Schramm, E.C.; Clark, S.J.; Triebwasser, M.P.; Raychaudhuri, S.; Seddon, J.M.; Atkinson, J.P. Genetic variants in the complement system predisposing to age-related macular degeneration: A review. Mol. Immunol. 2014, 61, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Sweigard, J.H.; Yanai, R.; Gaissert, P.; Saint-Geniez, M.; Kataoka, K.; Thanos, A.; Stahl, G.L.; Lambris, J.D.; Connor, K.M. The alternative complement pathway regulates pathological angiogenesis in the retina. FASEB J. 2014, 28, 3171–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cashman, S.M.; Desai, A.; Ramo, K.; Kumar-Singh, R. Expression of complement component 3 (c3) from an adenovirus leads to pathology in the murine retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Godino, R.; Bujakowska, K.M.; Pierce, E.A. Changes in extracellular matrix cause rpe cells to make basal deposits and activate the alternative complement pathway. Hum. Mol. Genet. 2018, 27, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnabolk, G.; Parsons, N.; Obert, E.; Annamalai, B.; Nasarre, C.; Tomlinson, S.; Lewin, A.S.; Rohrer, B. Delivery of cr2-fh using aav vector therapy as treatment strategy in the mouse model of choroidal neovascularization. Mol. Ther. Methods Clin. Dev. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fakhouri, F.; De Jorge, E.G.; Brune, F.; Azam, P.; Cook, H.T.; Pickering, M.C. Treatment with human complement factor h rapidly reverses renal complement deposition in factor h-deficient mice. Kidney Int. 2010, 78, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Cashman, S.M.; Gracias, J.; Adhi, M.; Kumar-Singh, R. Adenovirus-mediated delivery of factor h attenuates complement c3 induced pathology in the murine retina: A potential gene therapy for age-related macular degeneration. J. Gene Med. 2015, 17, 229–243. [Google Scholar] [CrossRef]
- Zhang, Y.; Nester, C.M.; Holanda, D.G.; Marsh, H.C.; Hammond, R.A.; Thomas, L.J.; Meyer, N.C.; Hunsicker, L.G.; Sethi, S.; Smith, R.J.H. Soluble cr1 therapy improves complement regulation in c3 glomerulopathy. J. Am. Soc. Nephrol. 2013, 24, 1820–1829. [Google Scholar] [CrossRef] [Green Version]
- Simmons, K.T.; Mazzilli, J.L.; Mueller-Ortiz, S.L.; Domozhirov, A.Y.; Garcia, C.A.; Zsigmond, E.M.; Wetsel, R.A. Complement receptor 1 (cr1/cd35)-expressing retinal pigment epithelial cells as a potential therapy for age-related macular degeneration. Mol. Immunol. 2020, 118, 91–98. [Google Scholar] [CrossRef]
- Rochowiak, A.; Niemir, Z.I. The structure and role of cr1 complement receptor in physiology. Pol. Merkur. Lek. 2010, 28, 79–83. [Google Scholar]
- Cashman, S.M.; Ramo, K.; Kumar-Singh, R. A non membrane-targeted human soluble cd59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS ONE 2011, 6, e19078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hom, G.L.; Singh, R.P. Complement Inhibitors for Treatment of Geographic Atrophy and Advanced Non-exudative AMD. Available online: https://www.retinalphysician.com/issues/2019/march-2019/complement-inhibitors-for-treatment-of-geographic (accessed on 16 November 2020).
- Corporation, O. Ophthotech Announces Results from Phase 2a Safety Trial of Zimura® in Combination with Lucentis® in Wet Age-Related Macular Degeneration. Available online: https://www.businesswire.com/news/home/20181112005203/en/Ophthotech-Announces-Results-Phase-2a-Safety-Trial (accessed on 16 November 2020).
- Clinicaltrials.gov ZIMURA in Combination with LUCENTIS in Patients with Neovascular Age Related Macular Degeneration (NVAMD). Available online: https://clinicaltrials.gov/ct2/show/NCT03362190 (accessed on 4 April 2020).
- Ren, X.; Li, J.; Xu, X.; Wang, C.; Cheng, Y. IBI302, a promising candidate for amd treatment, targeting both the vegf and complement system with high binding affinity in vitro and effective targeting of the ocular tissue in healthy rhesus monkeys. Exp. Eye Res. 2016, 145, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Chung, K.J.; Orlova, V.V.; Choi, E.Y.; Kaul, S.; Kruhlak, M.J.; Alatsatianos, M.; DeAngelis, R.A.; Roche, P.A.; Magotti, P.; et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010, 116, 4395–4403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijnsma, K.L.; ter Heine, R.; Moes, D.J.A.R.; Langemeijer, S.; Schols, S.E.M.; Volokhina, E.B.; van den Heuvel, L.P.; Wetzels, J.F.M.; van de Kar, N.C.A.J.; Brüggemann, R.J. Pharmacology, pharmacokinetics and pharmacodynamics of eculizumab, and possibilities for an individualized approach to eculizumab. Clin. Pharmacokinet. 2019, 58, 859–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Aspects Med. 2012, 33, 487–509. [Google Scholar] [CrossRef] [Green Version]
- Volz, C.; Pauly, D. Antibody therapies and their challenges in the treatment of age-related macular degeneration. Eur. J. Pharm. Biopharm. 2015, 95, 158–172. [Google Scholar] [CrossRef]
- Corydon, T.J. Antiangiogenic eye gene therapy. Hum. Gene Ther. 2015, 26, 525–537. [Google Scholar] [CrossRef]
- Kumaran, N.; Michaelides, M.; Smith, A.J.; Ali, R.R.; Bainbridge, J.W.B. Retinal gene therapy. Br. Med. Bull. 2018, 126, 13–25. [Google Scholar] [CrossRef]
- Jackson, T.L.; Antcliff, R.J.; Hillenkamp, J.; Marshall, J. Human retinal molecular weight exclusion limit and estimate of species variation. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2141–2146. [Google Scholar] [CrossRef] [Green Version]
- El Sanharawi, M.; Kowalczuk, L.; Touchard, E.; Omri, S.; de Kozak, Y.; Behar-Cohen, F. Protein delivery for retinal diseases: From basic considerations to clinical applications. Prog. Retin. Eye Res. 2010, 29, 443–465. [Google Scholar] [CrossRef]
- Heiduschka, P.; Fietz, H.; Hofmeister, S.; Schultheiss, S.; Mack, A.F.; Peters, S.; Ziemssen, F.; Niggemann, B.; Julien, S.; Bartz-Schmidt, K.U.; et al. Penetration of bevacizumab through the retina after intravitreal injection in the monkey. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2814–2823. [Google Scholar] [CrossRef] [Green Version]
- Avery, R.L.; Pieramici, D.J.; Rabena, M.D.; Castellarin, A.A.; Nasir, M.A.; Giust, M.J. Intravitreal bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmology 2006, 113, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, J.; Fei, D.; Beyer, J.C.; Ryan, A.; Rangell, L.; Shiu, V.; Damico, L.A. Pharmacokinetics and retinal distribution of ranibizumab, a humanized antibody fragment directed against vegf-a, following intravitreal administration in rabbits. Retina 2007, 27, 1260–1266. [Google Scholar] [CrossRef]
- Habot-Wilner, Z.; Noronha, G.; Wykoff, C.C. Suprachoroidally injected pharmacological agents for the treatment of chorio-retinal diseases: A targeted approach. Acta Ophthalmol. 2019, 97, 460–472. [Google Scholar] [CrossRef]
- Ding, K.; Shen, J.; Hafiz, Z.; Hackett, S.F.; e Silva, R.L.; Khan, M.; Lorenc, V.E.; Chen, D.; Chadha, R.; Zhang, M.; et al. AAV8-vectored suprachoroidal gene transfer produces widespread ocular transgene expression. J. Clin. Investig. 2019, 129, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- Yiu, G.; Chung, S.H.; Mollhoff, I.N.; Nguyen, U.T.; Thomasy, S.M.; Yoo, J.; Taraborelli, D.; Noronha, G. Suprachoroidal and subretinal injections of aav using transscleral microneedles for retinal gene delivery in nonhuman primates. Mol. Ther. Methods Clin. Dev. 2020, 16, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Spaide, R.F. Perspectives: Rationale for combination therapies for choroidal neovascularization. Am. J. Ophthalmol. 2006, 141, 149–156. [Google Scholar] [CrossRef]
- Askou, A.L.; Jakobsen, T.S.; Corydon, T.J. Retinal gene therapy: An eye-opener of the 21st century. Gene Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Askou, A.L.; Alsing, S.; Benckendorff, J.N.E.; Holmgaard, A.; Mikkelsen, J.G.; Aagaard, L.; Bek, T.; Corydon, T.J. Suppression of choroidal neovascularization by aav-based dual-acting antiangiogenic gene therapy. Mol. Ther. Nucleic Acids 2019, 16, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askou, A.L.; Corydon, T.J. Development of multigenic lentiviral vectors for cell-specific expression of antiangiogenic miRNAs and protein factors. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1715, pp. 47–60. [Google Scholar]
- Askou, A.L.; Aagaard, L.; Kostic, C.; Arsenijevic, Y.; Hollensen, A.K.; Bek, T.; Jensen, T.G.; Mikkelsen, J.G.; Corydon, T.J. Multigenic lentiviral vectors for combined and tissue-specific expression of mirna- and protein-based antiangiogenic factors. Mol. Ther. Methods Clin. Dev. 2015, 2, 14064. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
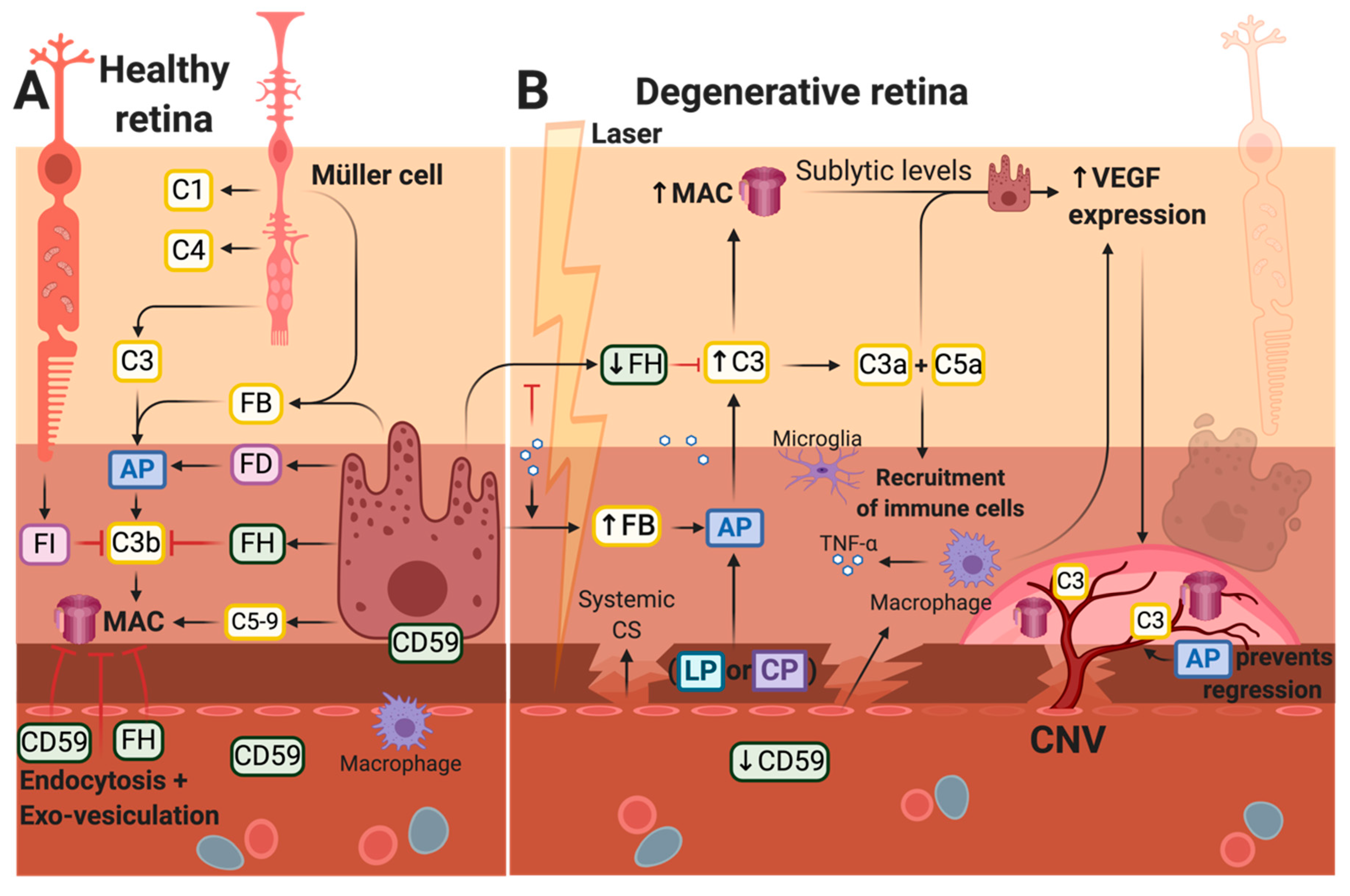{kind=link}
| Target | Drug | Injection Site | Phase (Name), Trial Number | Status/Outcome |
|---|---|---|---|---|
| C3 | POT-4 (Potentia/Alcon) - Compstatin analog - Peptide | IVT | 1 (AsAP) NCT00473928 | Completed, Some clinical efficacy and no safety concerns |
| IVT | 2 (RACE) NCT01157065 | Completed, Results from the phase I trial (AsAP) were not replicated | ||
| APL-2 (Apellis) - POT-4 derivative - Pegylated peptide | IVT | 1 (AsAP II) NCT02461771 | Completed, Unpublished | |
| IVT | 1b/2 NCT03465709 | Terminated (sufficient data were collected), Unpublished | ||
| CD59 | Combination of: AAVCAGsCD59 (Hemera) - Virus (AAV2) encoding soluble human CD59 and an anti-VEGF treatment - Bevacizumab (Avastin) - Ranibizumab (Lucentis) - Aflibercept (Eylea) | IVT | 1 NCT03585556 | Ongoing, Unpublished |
| C5 | LFG316 (Novartis Pharma AG) - Monoclonal human IgG1 ab | IVT | 2 NCT01535950 | Completed, Unpublished |
| IV | 2 NCT01624636 | Terminated, Unpublished | ||
| Combination of: ARC1905 (Zimura, IVERIC) - RNA aptamer and Ranibizumab (Lucentis) - Humanized monoclonal Fab-fragment | IVT | 1 NCT00709527 | Completed, Well-tolerated and no evidence of acute toxicity | |
| IVT | 2a NCT03362190 | Completed, Generally well-tolerated. Studies were halted to focus on other studies (e.g., Zimura for GA) | ||
| C3/C4 | IBI302 - Bispecific decoy receptor fusion protein - Binds and inhibits VEGF, C3b + C4b simultaneously | IVT | 1 NCT03814291 | Ongoing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, E.G.; Jakobsen, T.S.; Thiel, S.; Askou, A.L.; Corydon, T.J. Associations between the Complement System and Choroidal Neovascularization in Wet Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 9752. https://doi.org/10.3390/ijms21249752
Jensen EG, Jakobsen TS, Thiel S, Askou AL, Corydon TJ. Associations between the Complement System and Choroidal Neovascularization in Wet Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2020; 21(24):9752. https://doi.org/10.3390/ijms21249752
Chicago/Turabian StyleJensen, Emilie Grarup, Thomas Stax Jakobsen, Steffen Thiel, Anne Louise Askou, and Thomas J. Corydon. 2020. "Associations between the Complement System and Choroidal Neovascularization in Wet Age-Related Macular Degeneration" International Journal of Molecular Sciences 21, no. 24: 9752. https://doi.org/10.3390/ijms21249752