The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases

 , ,
, , 
Abstract
:1. Introduction
2. Roles and Expression of MMPs and TIMPs under Normal Physiological Conditions
Normal Expression of MMPs and TIMPs in Adult Tissues
3. Regulation of MMPs and TIMPs
4. MMPs and TIMPs in Human Diseases
4.1. MMPs and TIMPs in Diabetes Mellitus
4.1.1. Gestational Diabetes Mellitus
4.1.2. Diabetic Nephropathy
4.2. MMPs and Wound Healing
4.3. MMPs and TIMPs in Renal Pathologies
Glomerular Diseases
4.4. MMPs and TIMPs in Neurodegenerative Diseases
4.4.1. Multiple Sclerosis
4.4.2. Parkinson’s Disease
4.4.3. Alzheimer’s Disease
4.4.4. Amyotrophic Lateral Sclerosis
4.5. MMPs and TIMPs in Cardiovascular Diseases
4.5.1. Hypertension
4.5.2. Atherosclerosis
4.5.3. Myocardial Infarction
4.5.4. Aortic Aneurysms
4.6. MMPs and TIMPs in Gynecological Disorders
4.6.1. Polycystic Ovarian Syndrome
4.6.2. Spontaneous Abortion
4.6.3. Preeclampsia
4.7. MMPs and TIMPs in Cancer
4.8. MMPs and TIMPs in Inflammatory Diseases
5. Targeting MMP and TIMP Function (Inhibitors)
6. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MMPs | Matrix metalloproteinases |
| TIMPs | Tissue inhibitors of metalloproteinases |
| ECM | Extracellular matrix |
| MT-MMPs | Membrane type matrix metalloproteases |
| GPI | Glycosylphosphatidylinositol |
| Ig | Immunoglobulin |
| TM | Transmembrane domain |
| ADAM | Disintegrin and metalloproteinase |
| WG | Weeks of gestation |
| WAT | White adipose tissue |
| BAT | Brown adipose tissue |
| DM | Diabetes mellitus |
| EVs | Extracellular vesicles |
| T2DM | Type 2 DM |
| GDM | Gestational diabetes mellitus |
| miRs | microRNAs |
| CVD | Cardiovascular disease |
| DFU | Diabetic foot ulcer |
| CKD | Kidney disease |
| GBM | Glomerular basement membrane |
| AKI | Acute kidney injury |
| CKD | Chronic kidney disease |
| CNS | Central nervous system |
| AD | Alzheimer’s disease |
| MS | Multiple sclerosis |
| BBB | Blood−brain barrier |
| PD | Parkinson’s disease |
| MHC | Major histocompatibility complex |
| MAPK | Mitogen-activated protein kinases |
| TNF-αALS | Tumor necrosis factor alphaAmyotrophic lateral sclerosis |
| CSF | Cerebrospinal fluid |
| APP | Amyloid precursor protein |
| CVD | Cardiovascular disease |
| TOD | Target organ damage |
| MI | Myocardial infarction |
| PCOS | Polycystic ovarian syndrome |
| SA | Spontaneous abortion |
| RPL | Recurrent pregnancy loss |
| PE | Preeclampsia |
| VEGF | Vascular endothelial growth factor |
| GCM1 | Factor glial cell missing-1 |
| BMDCs | Bone-marrow-derived cells |
| IL | Interleukin |
| HLA | Human leukocyte antigen |
| TGF-β | Transforming growth factor beta |
| ERK | Extracellular regulated kinase |
| FGF | Fibroblast growth factor |
| LOXL | Lysyl oxidase |
| RA | Rheumatoid arthritis |
| OA | Osteoarthritis |
| COPD | Chronic obstructive pulmonary disease |
| HF | Heart failure |
References
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and biological attributes of matrix metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, C.; Vaidya, S.; Wadhwan, V.; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28–35. [Google Scholar] [CrossRef]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallata, A.M.; Wyatt, R.A.; Levesque, J.M.; Dufour, A.; Overall, C.M.; Crawford, B.D. Intracellular Localization in Zebrafish Muscle and Conserved Sequence Features Suggest Roles for Gelatinase A Moonlighting in Sarcomere Maintenance. Biomedicines 2019, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.A.; Chow, A.K.; Kandasamy, A.D.; Fan, X.; West, L.J.; Crawford, B.D.; Simmen, T.; Schulz, R. Mechanisms of cytosolic targeting of matrix metalloproteinase-2. J. Cell. Physiol. 2012, 227, 3397–3404. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv. Pharm. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Greenlee, K.J.; Werb, Z.; Kheradmand, F. Matrix metalloproteinases in lung: Multiple, multifarious, and multifaceted. Physiol. Rev. 2007, 87, 69–98. [Google Scholar] [CrossRef] [PubMed]
- Domagała, Z.; Dąbrowski, P.; Kurlej, W.; Porwolik, M.; Woźniak, S.; Kacała, R.R.; Gworys, B. The sequence of lanugo pattern development on the trunk wall in human fetuses. Adv. Clin. Exp. Med. 2017, 26, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Miao, Y.; Wang, X.; Chen, C.; Lin, B.; Hu, Z. Expression of matrix metalloproteinases and tissue inhibitor of matrix metalloproteinases in the hair cycle. Exp. Med. 2016, 12, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharov, A.A.; Schroeder, M.; Sharova, T.Y.; Mardaryev, A.N.; Peters, E.M.; Tobin, D.J.; Botchkarev, V.A. Matrix metalloproteinase-9 is involved in the regulation of hair canal formation. J. Investig. Derm. 2011, 131, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.M.; Fox, H.L.; Hazen, S.A.; LaNoue, K.F.; Rannels, S.R.; Lynch, C.J. Role of the matrixin MMP-2 in multicellular organization of adipocytes cultured in basement membrane components. Am. J. Physiol. Cell Physiol. 1997, 272, C937–C949. [Google Scholar] [CrossRef]
- Alexander, C.M.; Selvarajan, S.; Mudgett, J.; Werb, Z. Stromelysin-1 regulates adipogenesis during mammary gland involution. J. Cell Biol. 2001, 152, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Ortega, N.; Behonick, D.J.; Werb, Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004, 14, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Xu, J.; Xue, M.; Jackson, C. Matrix metalloproteinases in bone development and pathology: Current knowledge and potential clinical utility. Met. Med. 2016, 3, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Johansson, N.; Saarialho-Kere, U.; Airola, K.; Herva, R.; Nissinen, L.; Westermarck, J.; Vuorio, E.; Heino, J.; Kähäri, V.M. Collagenase-3 (MMP-13) is expressed by hypertrophic chondrocytes, periosteal cells, and osteoblasts during human fetal bone development. Dev. Dyn. 1997, 208, 387–397. [Google Scholar] [CrossRef]
- Vadillo-Ortega, F.; Estrada-Gutierrez, G. Role of matrix metalloproteinases in preterm labor. Bjog. Int. J. Obstet. Gynaecol. 2005, 112 (Suppl. 1), 19–22. [Google Scholar] [CrossRef]
- López-Osma, F.A.; Ordóñez-Sánchez, S.A. Ruptura prematura de membranas fetales: De la fisiopatologia hacia los marcadores tempranos de la enfermedad. Rev. Colomb. Obstet. Y Ginecol. 2006, 57, 279–290. [Google Scholar] [CrossRef]
- Masumoto, K.; de Rooij, J.D.; Suita, S.; Rottier, R.; Tibboel, D.; de Krijger, R.R. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases during normal human pulmonary development. Histopathology 2005, 47, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouloumié, A.; Sengenès, C.; Portolan, G.; Galitzky, J.; Lafontan, M. Adipocyte produces matrix metalloproteinases 2 and 9: Involvement in adipose differentiation. Diabetes 2001, 50, 2080–2086. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.D.; Kim, H.R.; Chesler, L.; Tsao-Wu, G.; Bouck, N.; Polverini, P.J. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase. J. Cell. Physiol. 1994, 160, 194–202. [Google Scholar] [CrossRef]
- Lilla, J.; Stickens, D.; Werb, Z. Metalloproteases and adipogenesis: A weighty subject. Am. J. Pathol. 2002, 160, 1551–1554. [Google Scholar] [CrossRef] [Green Version]
- Dali-Youcef, N.; Hnia, K.; Blaise, S.; Messaddeq, N.; Blanc, S.; Postic, C.; Valet, P.; Tomasetto, C.; Rio, M.C. Matrix metalloproteinase 11 protects from diabesity and promotes metabolic switch. Sci. Rep. 2016, 6, 25140. [Google Scholar] [CrossRef]
- Andarawewa, K.L.; Motrescu, E.R.; Chenard, M.P.; Gansmuller, A.; Stoll, I.; Tomasetto, C.; Rio, M.C. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Res. 2005, 65, 10862–10871. [Google Scholar] [CrossRef] [Green Version]
- Rohani, M.G.; Parks, W.C. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015, 44–46, 113–121. [Google Scholar] [CrossRef]
- Revilla, G.; Darwin, E.; Rantam, F.A. Effect of allogeneic bone marrow-mesenchymal stem cells (BM-MSCs) to accelerate burn healing of rat on the expression of collagen type I and integrin alpha2beta1. Pak. J. Biol. Sci. 2016, 19, 345–351. [Google Scholar] [CrossRef]
- Morillas, P.; Quiles, J.; de Andrade, H.; Castillo, J.; Tarazon, E.; Rosello, E.; Portoles, M.; Rivera, M.; Bertomeu-Martinez, V. Circulating biomarkers of collagen metabolism in arterial hypertension: Relevance of target organ damage. J. Hypertens. 2013, 31, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chen, M.; Hughes, D.; Chumanevich, A.A.; Altilia, S.; Kaza, V.; Lim, C.U.; Kiaris, H.; Mythreye, K.; Pena, M.M.; et al. CDK8 selectively promotes the growth of colon cancer metastases in the liver by regulating gene expression of TIMP3 and matrix metalloproteinases. Cancer Res. 2018, 78, 6594–6606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.F.; Ding, L.; Tian, Y.; Han, N.; Li, Z.Q. Interaction of STAT3 and RelB modulates MMP-1 in colon cancer. Chem.-Biol. Interact. 2018, 293, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Fanjul-Fernandez, M.; Folgueras, A.R.; Fueyo, A.; Balbin, M.; Suarez, M.F.; Fernandez-Garcia, M.S.; Shapiro, S.D.; Freije, J.M.P.; Lopez-Otin, C. Matrix metalloproteinase Mmp-1a is dispensable for normal growth and fertility in mice and promotes lung cancer progression by modulating inflammatory responses. J. Biol. Chem. 2018, 293, 11970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjomsland, V.; Pomianowska, E.; Aasrum, M.; Sandnes, D.; Verbeke, C.S.; Gladhaug, I.P. Profile of MMP and TIMP expression in human pancreatic stellate cells: Regulation by IL-1alpha and TGFbeta and implications for migration of pancreatic cancer cells. Neoplasia 2016, 18, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catania, J.M.; Chen, G.; Parrish, A.R. Role of matrix metalloproteinases in renal pathophysiologies. Am. J. Physiol. Ren. Physiol. 2007, 292, F905–F911. [Google Scholar] [CrossRef]
- Kobusiak-Prokopowicz, M.; Krzysztofik, J.; Kaaz, K.; Jolda-Mydlowska, B.; Mysiak, A. MMP-2 and TIMP-2 in patients with heart failure and chronic kidney disease. Open Med. (Wars) 2018, 13, 237–246. [Google Scholar] [CrossRef]
- Derosa, G.; Avanzini, M.A.; Geroldi, D.; Fogari, R.; Lorini, R.; De Silvestri, A.; Tinelli, C.; Rondini, G.; d’Annunzio, G. Matrix metalloproteinase 2 may be a marker of microangiopathy in children and adolescents with type 1 diabetes mellitus. Diabetes Res. Clin. Pr. 2005, 70, 119–125. [Google Scholar] [CrossRef]
- Lorenzl, S.; Albers, D.S.; Narr, S.; Chirichigno, J.; Beal, M.F. Expression of MMP-2, MMP-9, and MMP-1 and their endogenous counterregulators TIMP-1 and TIMP-2 in postmortem brain tissue of Parkinson’s disease. Exp. Neurol. 2002, 178, 13–20. [Google Scholar] [CrossRef]
- Wetzl, V.; Tiede, S.L.; Faerber, L.; Weissmann, N.; Schermuly, R.T.; Ghofrani, H.A.; Gall, H. Plasma MMP2/TIMP4 ratio at follow-up assessment predicts disease progression of idiopathic pulmonary arterial hypertension. Lung 2017, 195, 489–496. [Google Scholar] [CrossRef]
- Godbole, G.; Suman, P.; Gupta, S.K.; Modi, D. Decidualized endometrial stromal cell derived factors promote trophoblast invasion. Fertil. Steril. 2011, 95, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, D.V.; Lippert, C.N.; Crisp, J.L.; Savariar, E.N.; Hasselmann, J.P.C.; Kuo, C.; Nguyen, Q.T.; Tsien, R.Y.; Whitney, M.A.; Ellies, L.G. Impact of MMP-2 and MMP-9 enzyme activity on wound healing, tumor growth and RACPP cleavage. PLoS ONE 2018, 13, e0198464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Wang, K.Y.; Gao, J.Q.; Li, R.J.; Guan, Q.B.; Song, L. Study on the expression of p53 and MMP-2 in patients with lung cancer after interventional therapy. Oncol. Lett. 2018, 16, 4291–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na’ara, S.; Amit, M.; Gil, Z. L1CAM induces perineural invasion of pancreas cancer cells by upregulation of metalloproteinase expression. Oncogene 2019, 38, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, B.; Xie, J.; Hou, D.; Zhang, H.; Huang, H. Melatonin inhibits epithelialtomesenchymal transition in gastric cancer cells via attenuation of IL1beta/NFkappaB/MMP2/MMP9 signaling. Int. J. Mol. Med. 2018, 42, 2221–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, A.I.; Chan, L.Y.; Lai, K.N.; Tang, S.C.; Chow, C.W.; Lam, M.F.; Leung, J.C. Distinct role of matrix metalloproteinase-3 in kidney injury molecule-1 shedding by kidney proximal tubular epithelial cells. Int. J. Biochem. Cell Biol. 2012, 44, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kim, S.S.; Cho, J.J.; Choi, D.H.; Hwang, O.; Shin, D.H.; Chun, H.S.; Beal, M.F.; Joh, T.H. Matrix metalloproteinase-3: A novel signaling proteinase from apoptotic neuronal cells that activates microglia. J. Neurosci. 2005, 25, 3701–3711. [Google Scholar] [CrossRef] [Green Version]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. Friends or foes: Matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediat. Inflamm. 2015, 2015, 620581. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Desantis, S.M.; Baicu, C.F.; Stroud, R.E.; Thompson, S.B.; McClure, C.D.; Mehurg, S.M.; Spinale, F.G. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ. Heart Fail. 2011, 4, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Kumari, N.; Singh, R.K.; Krishnani, N.; Shukla, P. Matrix metalloproteinase 7 expression in ampullary carcinoma. Indian J. Pathol. Microbiol. 2015, 58, 274–278. [Google Scholar] [CrossRef]
- Vilmi-Kerala, T.; Lauhio, A.; Tervahartiala, T.; Palomaki, O.; Uotila, J.; Sorsa, T.; Palomaki, A. Subclinical inflammation associated with prolonged TIMP-1 upregulation and arterial stiffness after gestational diabetes mellitus: A hospital-based cohort study. Cardiovasc. Diabetol. 2017, 16, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockelman, C.; Beilmann-Lehtonen, I.; Kaprio, T.; Koskensalo, S.; Tervahartiala, T.; Mustonen, H.; Stenman, U.H.; Sorsa, T.; Haglund, C. Serum MMP-8 and TIMP-1 predict prognosis in colorectal cancer. BMC Cancer 2018, 18, 679. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, A.; Hagstrom, J.; Mustonen, H.; Kokkola, A.; Tervahartiala, T.; Sorsa, T.; Bockelman, C.; Haglund, C. Serum MMP-8 and TIMP-1 as prognostic biomarkers in gastric cancer. Tumour Biol. J. Int. Soc. Oncodevelopmental. Biol. Med. 2018, 40, 1010428318799266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesi, C.; Dentali, F.; Nicolini, E.; Maresca, A.M.; Tayebjee, M.H.; Franz, M.; Guasti, L.; Venco, A.; Schiffrin, E.L.; Lip, G.Y.; et al. Plasma levels of matrix metalloproteinases and their inhibitors in hypertension: A systematic review and meta-analysis. J. Hypertens. 2012, 30, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Rajasinghe, L.D.; Pindiprolu, R.H.; Gupta, S.V. Delta-tocotrienol inhibits non-small-cell lung cancer cell invasion via the inhibition of NF-kappaB, uPA activator, and MMP-9. OncoTargets Ther. 2018, 11, 4301–4314. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Wang, S.; Shen, X.; Bai, Z.; Niu, Q.; Ma, D.; Xu, Y.; Liang, C. Polymorphism of MMP-9 gene is not associated with the risk of urinary cancers: Evidence from an updated meta-analysis. Pathol. Res. Pract. 2018, 214, 1966–1973. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, M.; Dorfman, R.G.; Pan, Y.; Tang, D.; Xu, L.; Zhao, Z.; Zhou, Q.; Zhou, L.; Wang, Y.; et al. SIRT2 promotes the migration and invasion of gastric cancer through RAS/ERK/JNK/MMP-9 pathway by increasing PEPCK1-related metabolism. Neoplasia 2018, 20, 745–756. [Google Scholar] [CrossRef]
- Liu, J.F.; Lee, C.W.; Tsai, M.H.; Tang, C.H.; Chen, P.C.; Lin, L.W.; Lin, C.Y.; Lu, C.H.; Lin, Y.F.; Yang, S.H.; et al. Thrombospondin 2 promotes tumor metastasis by inducing matrix metalloproteinase-13 production in lung cancer cells. Biochem. Pharmacol. 2018, 155, 537–546. [Google Scholar] [CrossRef]
- Ren, J.; Liu, J.; Sui, X. Correlation of COX-2 and MMP-13 expressions with gastric cancer and their effects on prognosis. J. B.U.ON. Off. J. Balk. Union Oncol. 2018, 23, 665–671. [Google Scholar]
- Bar-Or, A.; Gold, R.; Kappos, L.; Arnold, D.L.; Giovannoni, G.; Selmaj, K.; O’Gorman, J.; Stephan, M.; Dawson, K.T. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: Subgroup analyses of the DEFINE study. J. Neurol. 2013, 260, 2297–2305. [Google Scholar] [CrossRef]
- Dechaphunkul, A.; Phukaoloun, M.; Kanjanapradit, K.; Graham, K.; Ghosh, S.; Santos, C.; Mackey, J.R. Prognostic significance of tissue inhibitor of metalloproteinase-1 in breast cancer. Int. J. Breast Cancer 2012, 2012, 290854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groft, L.L.; Muzik, H.; Rewcastle, N.B.; Johnston, R.N.; Knauper, V.; Lafleur, M.A.; Forsyth, P.A.; Edwards, D.R. Differential expression and localization of TIMP-1 and TIMP-4 in human gliomas. Br. J. Cancer 2001, 85, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, T.; Gunawan, B.; Schulz, M.; Fuzesi, L.; Binder, C. mRNA expression of matrix metalloproteases and their inhibitors differs in subtypes of renal cell carcinomas. Eur. J. Cancer 2001, 37, 1839–1846. [Google Scholar] [CrossRef]
- Ambrosch, A.; Halevy, D.; Fwity, B.; Brin, T.; Lobmann, R. Effect of daptomycin on local interleukin-6, matrix metalloproteinase-9, and metallopeptidase inhibitor 1 in patients with MRSA-infected diabetic foot. Int. J. Low. Extrem. Wounds 2013, 12, 100–105. [Google Scholar] [CrossRef]
- Kelly, D.; Squire, I.B.; Khan, S.Q.; Dhillon, O.; Narayan, H.; Ng, K.H.; Quinn, P.; Davies, J.E.; Ng, L.L. Usefulness of plasma tissue inhibitors of metalloproteinases as markers of prognosis after acute myocardial infarction. Am. J. Cardiol. 2010, 106, 477–482. [Google Scholar] [CrossRef]
- Baka, S.; Zourla, K.; Kouskouni, E.; Makrakis, E.; Demeridou, S.; Tzanakaki, D.; Hassiakos, D.; Creatsas, G. Matrix metalloproteinases 2 and 9 and their tissue inhibitors in the follicular fluid of patients with polycystic ovaries undergoing in vitro fertilisation. In Vivo 2010, 24, 293–296. [Google Scholar]
- Cui, H.; Seubert, B.; Stahl, E.; Dietz, H.; Reuning, U.; Moreno-Leon, L.; Ilie, M.; Hofman, P.; Nagase, H.; Mari, B.; et al. Tissue inhibitor of metalloproteinases-1 induces a pro-tumourigenic increase of miR-210 in lung adenocarcinoma cells and their exosomes. Oncogene 2015, 34, 3640–3650. [Google Scholar] [CrossRef]
- Gomes, V.A.; Vieira, C.S.; Jacob-Ferreira, A.L.; Belo, V.A.; Soares, G.M.; Fernandes, J.B.; Ferriani, R.A.; Tanus-Santos, J.E. Imbalanced circulating matrix metalloproteinases in polycystic ovary syndrome. Mol. Cell. Biochem. 2011, 353, 251–257. [Google Scholar] [CrossRef]
- Fiorentino, L.; Cavalera, M.; Menini, S.; Marchetti, V.; Mavilio, M.; Fabrizi, M.; Conserva, F.; Casagrande, V.; Menghini, R.; Pontrelli, P.; et al. Loss of TIMP3 underlies diabetic nephropathy via FoxO1/STAT1 interplay. EMBO Mol. Med. 2013, 5, 441–455. [Google Scholar] [CrossRef]
- Haddock, G.; Cross, A.K.; Plumb, J.; Surr, J.; Buttle, D.J.; Bunning, R.A.; Woodroofe, M.N. Expression of ADAMTS-1, -4, -5 and TIMP-3 in normal and multiple sclerosis CNS white matter. Mult. Scler. 2006, 12, 386–396. [Google Scholar] [CrossRef]
- Takawale, A.; Zhang, P.; Azad, A.; Wang, W.; Wang, X.; Murray, A.G.; Kassiri, Z. Myocardial overexpression of TIMP3 after myocardial infarction exerts beneficial effects by promoting angiogenesis and suppressing early proteolysis. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H224–H236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksjoki, S.; Rahkonen, O.; Haarala, M.; Vuorio, E.; Anttila, L. Differences in connective tissue gene expression between normally functioning, polycystic and post-menopausal ovaries. Mol. Hum. Reprod. 2004, 10, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.W.; Tsai, L.H.; Chen, P.M.; Lee, M.C.; Wang, L.; Chen, C.Y.; Cheng, Y.W.; Lee, H. Loss of TIMP-3 promotes tumor invasion via elevated IL-6 production and predicts poor survival and relapse in HPV-infected non-small cell lung cancer. Am. J. Pathol. 2012, 181, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Gao, W.; Li, Q.; Li, P.F.; Yao, P.Y.; Chen, K. Tissue inhibitor of metalloproteinases-4 suppresses vascular smooth muscle cell migration and induces cell apoptosis. Life Sci. 2004, 75, 2483–2493. [Google Scholar] [CrossRef] [PubMed]
- Liss, M.; Sreedhar, N.; Keshgegian, A.; Sauter, G.; Chernick, M.R.; Prendergast, G.C.; Wallon, U.M. Tissue inhibitor of metalloproteinase-4 is elevated in early-stage breast cancers with accelerated progression and poor clinical course. Am. J. Pathol. 2009, 175, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Rorive, S.; Lopez, X.M.; Maris, C.; Trepant, A.L.; Sauvage, S.; Sadeghi, N.; Roland, I.; Decaestecker, C.; Salmon, I. TIMP-4 and CD63: New prognostic biomarkers in human astrocytomas. Mod. Pathol. 2010, 23, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Pilka, R.; Domanski, H.; Hansson, S.; Eriksson, P.; Casslen, B. Endometrial TIMP-4 mRNA is high at midcycle and in hyperplasia, but down-regulated in malignant tumours. Coordinated expression with MMP-26. Mol. Hum. Reprod. 2004, 10, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin proteases and their inhibitors: Foes or friends in nervous system physiology? J. Neurosci. 2010, 30, 15337–15357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamze, A.B.; Wei, S.; Bahudhanapati, H.; Kota, S.; Acharya, K.R.; Brew, K. Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 2007, 16, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matchett, E.F.; Wang, S.; Crawford, B.D. Paralogues of Mmp11 and Timp4 Interact during the Development of the Myotendinous Junction in the Zebrafish Embryo. J. Dev. Biol. 2019, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Barichello, T.; Generoso, J.S.; Michelon, C.M.; Simoes, L.R.; Elias, S.G.; Vuolo, F.; Comim, C.M.; Dal-Pizzol, F.; Quevedo, J. Inhibition of matrix metalloproteinases-2 and -9 prevents cognitive impairment induced by pneumococcal meningitis in Wistar rats. Exp. Biol. Med. (Maywood) 2014, 239, 225–231. [Google Scholar] [CrossRef]
- Amalinei, C.; Caruntu, I.D.; Giusca, S.E.; Balan, R.A. Matrix metalloproteinases involvement in pathologic conditions. Rom. J. Morphol. Embryol. 2010, 51, 215–228. [Google Scholar]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Koye, D.N.; Magliano, D.J.; Nelson, R.G.; Pavkov, M.E. The Global Epidemiology of Diabetes and Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 121–132. [Google Scholar] [CrossRef]
- Jameson, J.L.; Fauci, A.S.; Kasper, D.L.; Hauser, S.L.; Longo, D.L.; Loscalzo, J. Harrison. Principles of Internal Medicine, 20th ed.; McGraw-Hill: New York, NY, USA, 2018. [Google Scholar]
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and Gender Differences in Risk, Pathophysiology and Complications of Type 2 Diabetes Mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef] [Green Version]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Chaturvedi, N.; Fuller, J.H.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Theilade, S.; Rossing, P.; et al. Circulating matrix metalloproteinases are associated with arterial stiffness in patients with type 1 diabetes: Pooled analysis of three cohort studies. Cardiovasc. Diabetol. 2017, 16, 139. [Google Scholar] [CrossRef] [Green Version]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, S.; Matsushita, H.; Li, W.; Glassford, A.J.; Asagami, T.; Lee, K.H.; Harrison, D.G.; Tsao, P.S. Diabetes mellitus enhances vascular matrix metalloproteinase activity: Role of oxidative stress. Circ. Res. 2001, 88, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Zhu, H.; Chen, X.; Peng, Y.; Wang, J.; Liu, F.; Shi, S.; Fu, B.; Lu, Y.; Hong, Q.; et al. TIMP-1 transgenic mice recover from diabetes induced by multiple low-dose streptozotocin. Diabetes 2007, 56, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Song, B.; He, S. Interleukin 29 activates expression of tissue inhibitor of metalloproteinase 1 in macrophages via tolllike receptor 2. Mol. Med. Rep. 2018, 17, 8363–8368. [Google Scholar] [CrossRef]
- Kologrivova, I.V.; Suslova, T.E.; Koshel’skaya, O.A.; Vinnitskaya, I.V.; Trubacheva, O.A. System of matrix metalloproteinases and cytokine secretion in type 2 diabetes mellitus and impaired carbohydrate tolerance associated with arterial hypertension. Bull. Exp. Biol Med. 2014, 156, 635–638. [Google Scholar] [CrossRef]
- Cardellini, M.; Menghini, R.; Martelli, E.; Casagrande, V.; Marino, A.; Rizza, S.; Porzio, O.; Mauriello, A.; Solini, A.; Ippoliti, A.; et al. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SirT1. Diabetes 2009, 58, 2396–2401. [Google Scholar] [CrossRef] [Green Version]
- Waldron, A.L.; Schroder, P.A.; Bourgon, K.L.; Bolduc, J.K.; Miller, J.L.; Pellegrini, A.D.; Dubois, A.L.; Blaszkiewicz, M.; Townsend, K.L.; Rieger, S. Oxidative stress-dependent MMP-13 activity underlies glucose neurotoxicity. J. Diabetes Its Complicat. 2018, 32, 249–257. [Google Scholar] [CrossRef]
- Staff, N.P.; Fehrenbacher, J.C.; Caillaud, M.; Damaj, M.I.; Segal, R.A.; Rieger, S. Pathogenesis of paclitaxel-induced peripheral neuropathy: A current review of in vitro and in vivo findings using rodent and human model systems. Exp. Neurol. 2020, 324, 113121. [Google Scholar] [CrossRef]
- Garcia-Fernandez, N.; Jacobs-Cacha, C.; Mora-Gutierrez, J.M.; Vergara, A.; Orbe, J.; Soler, M.J. Matrix Metalloproteinases in Diabetic Kidney Disease. J. Clin. Med. 2020, 9, 472. [Google Scholar] [CrossRef] [Green Version]
- Chavey, C.; Mari, B.; Monthouel, M.N.; Bonnafous, S.; Anglard, P.; Van Obberghen, E.; Tartare-Deckert, S. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J. Biol. Chem. 2003, 278, 11888–11896. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Lee, S.J.; Lee, M.Y.; Park, M.W.; Kim, S.S.; Shin, N.; Lovett, D.H.; Bae, S.S.; Ahn, J.; Park, J.S.; et al. Enhanced cardiac expression of two isoforms of matrix metalloproteinase-2 in experimental diabetes mellitus. PLoS ONE 2019, 14, e0221798. [Google Scholar] [CrossRef] [PubMed]
- Opdenakker, G.; Abu El-Asrar, A. Metalloproteinases mediate diabetes-induced retinal neuropathy and vasculopathy. Cell. Mol. Life Sci. 2019, 76, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Jayashree, K.; Yasir, M.; Senthilkumar, G.P.; Ramesh Babu, K.; Mehalingam, V.; Mohanraj, P.S. Circulating matrix modulators (MMP-9 and TIMP-1) and their association with severity of diabetic retinopathy. Diabetes Metab. Syndr. 2018, 12, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Sakamuri, S.; Watts, R.; Takawale, A.; Wang, X.; Hernandez-Anzaldo, S.; Bahitham, W.; Fernandez-Patron, C.; Lehner, R.; Kassiri, Z. Absence of Tissue Inhibitor of Metalloproteinase-4 (TIMP4) ameliorates high fat diet-induced obesity in mice due to defective lipid absorption. Sci. Rep. 2017, 7, 6210. [Google Scholar] [CrossRef]
- D’Arpino, M.C.; Fuchs, A.G.; Sanchez, S.S.; Honore, S.M. Extracellular matrix remodeling and TGF-beta1/Smad signaling in diabetic colon mucosa. Cell Biol. Int. 2018, 42, 443–456. [Google Scholar] [CrossRef]
- Tokarz, A.; Szuscik, I.; Kusnierz-Cabala, B.; Kapusta, M.; Konkolewska, M.; Zurakowski, A.; Georgescu, A.; Stepien, E. Extracellular vesicles participate in the transport of cytokines and angiogenic factors in diabetic patients with ocular complications. Folia Med. Cracov. 2015, 55, 35–48. [Google Scholar]
- McKittrick, I.B.; Bogaert, Y.; Nadeau, K.; Snell-Bergeon, J.; Hull, A.; Jiang, T.; Wang, X.; Levi, M.; Moulton, K.S. Urinary matrix metalloproteinase activities: Biomarkers for plaque angiogenesis and nephropathy in diabetes. Am. J. Physiol. Ren. Physiol. 2011, 301, F1326–F1333. [Google Scholar] [CrossRef] [Green Version]
- Thrailkill, K.M.; Clay Bunn, R.; Fowlkes, J.L. Matrix metalloproteinases: Their potential role in the pathogenesis of diabetic nephropathy. Endocrine 2009, 35, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Abreu, B.J.; de Brito Vieira, W.H. Metalloproteinase Changes in Diabetes. Adv. Exp. Med. Biol. 2016, 920, 185–190. [Google Scholar] [CrossRef]
- Chang, M. Restructuring of the extracellular matrix in diabetic wounds and healing: A perspective. Pharm. Res. 2016, 107, 243–248. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Chen, X.; Yang, Y.; Wang, F.; Li, W.; Awuti, M.; Sun, Y.; Lian, C.; Li, Z.; et al. miR-365 promotes diabetic retinopathy through inhibiting Timp3 and increasing oxidative stress. Exp. Eye Res. 2018, 168, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Afkarian, M.; Sachs, M.C.; Kestenbaum, B.; Hirsch, I.B.; Tuttle, K.R.; Himmelfarb, J.; De Boer, I.H. Kidney disease and increased mortality risk in Type 2 diabetes. J. Am. Soc. Nephrol. 2013, 24, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassila, M.; Jandeleit-Dahm, K.; Seah, K.K.; Smith, C.M.; Calkin, A.C.; Allen, T.J.; Cooper, M.E. Imatinib attenuates diabetic nephropathy in apolipoprotein E-knockout mice. J. Am. Soc. Nephrol. 2005, 16, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Inada, A.; Nagai, K.; Arai, H.; Miyazaki, J.; Nomura, K.; Kanamori, H.; Toyokuni, S.; Yamada, Y.; Bonner-Weir, S.; Weir, G.C.; et al. Establishment of a diabetic mouse model with progressive diabetic nephropathy. Am. J. Pathol. 2005, 167, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Dimas, G.; Iliadis, F.; Grekas, D. Matrix metalloproteinases, atherosclerosis, proteinuria and kidney disease: Linkage-based approaches. Hippokratia 2013, 17, 292–297. [Google Scholar]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [Green Version]
- Aldemir, O.; Turgut, F.; Gokce, C. The association between methylation levels of targeted genes and albuminuria in patients with early diabetic kidney disease. Ren. Fail. 2017, 39, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gao, Y.; Ma, M.; Li, M.; Zou, D.; Yang, J.; Zhu, Z.; Zhao, X. Effect of miR-21 on renal fibrosis by regulating MMP-9 and TIMP1 in kk-ay diabetic nephropathy mice. Cell Biochem. Biophys. 2013, 67, 537–546. [Google Scholar] [CrossRef]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in Wound Healing: A Comprehensive Review. Adv. Wound Care (New Rochelle) 2014, 3, 445–464. [Google Scholar] [CrossRef] [Green Version]
- Olczyk, P.; Mencner, L.; Komosinska-Vassev, K. The role of the extracellular matrix components in cutaneous wound healing. Biomed. Res. Int. 2014, 2014, 747584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Fernandez, A.; Inada, M.; Balbin, M.; Fueyo, A.; Pitiot, A.S.; Astudillo, A.; Hirose, K.; Hirata, M.; Shapiro, S.D.; Noel, A.; et al. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8). FASEB J. 2007, 21, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.C.; Wu, C.S.; Kuo, H.Y.; Huang, S.M.; Chen, G.S. Hyperglycaemic conditions hamper keratinocyte locomotion via sequential inhibition of distinct pathways: New insights on poor wound closure in patients with diabetes. Br. J. Derm. 2009, 160, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Makela, M.; Larjava, H.; Pirila, E.; Maisi, P.; Salo, T.; Sorsa, T.; Uitto, V.J. Matrix metalloproteinase 2 (gelatinase A) is related to migration of keratinocytes. Exp. Cell Res. 1999, 251, 67–78. [Google Scholar] [CrossRef]
- Schenk, S.; Hintermann, E.; Bilban, M.; Koshikawa, N.; Hojilla, C.; Khokha, R.; Quaranta, V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J. Cell Biol. 2003, 161, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Salo, T.; Makela, M.; Kylmaniemi, M.; Autio-Harmainen, H.; Larjava, H. Expression of matrix metalloproteinase-2 and -9 during early human wound healing. Lab. Investig. 1994, 70, 176–182. [Google Scholar]
- Muller, M.; Trocme, C.; Morel, F.; Halimi, S.; Benhamou, P.Y. Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers: Response to Liu et al. Diabetes Care 2009, 32, e138. [Google Scholar] [CrossRef] [Green Version]
- Lobmann, R.; Ambrosch, A.; Schultz, G.; Waldmann, K.; Schiweck, S.; Lehnert, H. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia 2002, 45, 1011–1016. [Google Scholar] [CrossRef]
- Ferianec, V.; Fulop, M.; Jezovicova, M.; Radosinska, J.; Husseinova, M.; Feriancova, M.; Radosinska, D.; Barancik, M.; Muchova, J.; Hogger, P.; et al. The oak-wood extract Robuvit((R)) improves recovery and oxidative stress after hysterectomy: A randomized, double-blind, placebo-controlled pilot study. Nutrients 2020, 12, 913. [Google Scholar] [CrossRef] [Green Version]
- Tokito, A.; Jougasaki, M. Matrix metalloproteinases in non-neoplastic disorders. Int. J. Mol. Sci. 2016, 17, 1178. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.J.; Liu, Y. Matrix metalloproteinases in kidney homeostasis and diseases. Am. J. Physiol. Ren. Physiol. 2012, 302, F1351–F1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, T.A.; Kelly, K.J.; Mang, H.E.; Plotkin, Z.; Sandoval, R.M.; Dagher, P.C. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am. J. Physiol. Ren. Physiol. 2005, 288, F91–F97. [Google Scholar] [CrossRef] [PubMed]
- Horstrup, J.H.; Gehrmann, M.; Schneider, B.; Ploger, A.; Froese, P.; Schirop, T.; Kampf, D.; Frei, U.; Neumann, R.; Eckardt, K.U. Elevation of serum and urine levels of TIMP-1 and tenascin in patients with renal disease. Nephrol. Dial. Transpl. 2002, 17, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Sui, W.; Wang, B.; Zou, H.; Zou, G.; Luo, H. Expression of MMP-2 and TIMP-1 in renal tissue of patients with chronic active antibody-mediated renal graft rejection. Diagn. Pathol. 2012, 7, 141. [Google Scholar] [CrossRef] [Green Version]
- Ries, C. Cytokine functions of TIMP-1. Cell. Mol. Life Sci. 2014, 71, 659–672. [Google Scholar] [CrossRef]
- Musial, K.; Zwolinska, D. Pleiotropic functions of TIMP-1 in patients with chronic kidney disease. Cell. Mol. Life Sci. 2014, 71, 1547–1548. [Google Scholar] [CrossRef] [Green Version]
- Caliskan, Y.; Kiryluk, K. Novel biomarkers in glomerular disease. Adv. Chronic Kidney Dis. 2014, 21, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Pani, A.; Porta, C.; Cosmai, L.; Melis, P.; Floris, M.; Piras, D.; Gallieni, M.; Rosner, M.; Ponticelli, C. Glomerular diseases and cancer: Evaluation of underlying malignancy. J. Nephrol. 2016, 29, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Conley, S.M.; Abais, J.M.; Boini, K.M.; Li, P.-L. Inflammasome activation in chronic glomerular diseases. Curr. Drug Targets 2017, 18, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Ismail, O.Z. Kidney Injury Molecule-1 Signalling in Ischemic Acute Kidney Injury and Phagocytosis. Doctoral’s Thesis, The University of Western Ontario, London, ON, Canada, 2015. [Google Scholar]
- Lennon, R.; Hosawi, S. Glomerular cell crosstalk. PubMed 2016, 25, 187–193. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Zhao, H.; Chen, D.; Huang, Q.; Wang, S.; Zou, W.; Zhang, Y.; Li, X.; Huang, H. Increase in extracellular cross-linking by tissue transglutaminase and reduction in expression of MMP-9 contribute differentially to focal segmental glomerulosclerosis in rats. Mol. Cell. Biochem. 2006, 284, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Nee, L.E.; McMorrow, T.; Campbell, E.; Slattery, C.; Ryan, M.P. TNF-alpha and IL-1beta-mediated regulation of MMP-9 and TIMP-1 in renal proximal tubular cells. Kidney Int. 2004, 66, 1376–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.G.; Sun, W.X.; Gao, B.S.; Lian, X.; Zhou, H.L. Cell cycle arrest as a therapeutic target of acute kidney injury. Curr. Protein Pept. Sci. 2017, 18, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Small, C.D.; Crawford, B.D. Matrix metalloproteinases in neural development: A phylogenetically diverse perspective. Neural. Regen. Res. 2016, 11, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Lin, S.; Zhang, C.; Tao, W.; Dong, W.; Hao, Z.; Liu, M.; Wu, B. Activation of cerebral recovery by matrix metalloproteinase-9 after intracerebral hemorrhage. Neuroscience 2013, 230, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Swarnakar, S. Implication of matrix metalloproteinases in regulating neuronal disorder. Mol. Biol. Rep. 2015, 42, 1–11. [Google Scholar] [CrossRef]
- Liuzzi, G.M.; Trojano, M.; Fanelli, M.; Avolio, C.; Fasano, A.; Livrea, P.; Riccio, P. Intrathecal synthesis of matrix metalloproteinase-9 in patients with multiple sclerosis: Implication for pathogenesis. Mult. Scler. 2002, 8, 222–228. [Google Scholar] [CrossRef]
- Perez-Martinez, L.; Jaworski, D.M. Tissue inhibitor of metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. J. Neurosci. 2005, 25, 4917–4929. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, D.M.; Perez-Martinez, L. Tissue inhibitor of metalloproteinase-2 (TIMP-2) expression is regulated by multiple neural differentiation signals. J. Neurochem. 2006, 98, 234–247. [Google Scholar] [CrossRef]
- Nedivi, E.; Hevroni, D.; Naot, D.; Israeli, D.; Citri, Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 1993, 363, 718–722. [Google Scholar] [CrossRef]
- Jourquin, J.; Tremblay, E.; Bernard, A.; Charton, G.; Chaillan, F.A.; Marchetti, E.; Roman, F.S.; Soloway, P.D.; Dive, V.; Yiotakis, A.; et al. Tissue inhibitor of metalloproteinases-1 (TIMP-1) modulates neuronal death, axonal plasticity, and learning and memory. Eur. J. Neurosci. 2005, 22, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksenberg, J.R.; McCauley, J.L. Chapter 4—Genetics of multiple sclerosis. In Translational Neuroimmunology in Multiple Sclerosis; Academic Press: Cambridge, MA, USA, 2016; pp. 45–54. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Bittner, S.; Ruck, T.; Wiendl, H.; Grauer, O.M.; Meuth, S.G. Targeting B cells in relapsing–remitting multiple sclerosis: From pathophysiology to optimal clinical management. Ther. Adv. Neurol. Disord. 2017, 10, 51–66. [Google Scholar] [CrossRef]
- Lindberg, R.L.; De Groot, C.J.; Montagne, L.; Freitag, P.; van der Valk, P.; Kappos, L.; Leppert, D. The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain 2001, 124, 1743–1753. [Google Scholar] [CrossRef]
- Leppert, D.; Ford, J.; Stabler, G.; Grygar, C.; Lienert, C.; Huber, S.; Miller, K.M.; Hauser, S.L.; Kappos, L. Matrix metalloproteinase-9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain 1998, 121, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Chattopadhyay, S.; Kato, K.; Dolkas, J.; Kikuchi, S.; Myers, R.R.; Shubayev, V.I. MMPs initiate Schwann cell-mediated MBP degradation and mechanical nociception after nerve damage. Mol. Cell. Neurosci. 2008, 39, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Benesova, Y.; Vasku, A.; Novotna, H.; Litzman, J.; Stourac, P.; Beranek, M.; Kadanka, Z.; Bednarik, J. Matrix metalloproteinase-9 and matrix metalloproteinase-2 as biomarkers of various courses in multiple sclerosis. Mult. Scler. 2009, 15, 316–322. [Google Scholar] [CrossRef]
- Fainardi, E.; Castellazzi, M.; Tamborino, C.; Trentini, A.; Manfrinato, M.C.; Baldi, E.; Tola, M.R.; Dallocchio, F.; Granieri, E.; Bellini, T. Potential relevance of cerebrospinal fluid and serum levels and intrathecal synthesis of active matrix metalloproteinase-2 (MMP-2) as markers of disease remission in patients with multiple sclerosis. Mult. Scler. 2009, 15, 547–554. [Google Scholar] [CrossRef]
- Avolio, C.; Filippi, M.; Tortorella, C.; Rocca, M.A.; Ruggieri, M.; Agosta, F.; Tomassini, V.; Pozzilli, C.; Stecchi, S.; Giaquinto, P.; et al. Serum MMP-9/TIMP-1 and MMP-2/TIMP-2 ratios in multiple sclerosis: Relationships with different magnetic resonance imaging measures of disease activity during IFN-beta-1a treatment. Mult. Scler. 2005, 11, 441–446. [Google Scholar] [CrossRef]
- Lee, M.A.; Palace, J.; Stabler, G.; Ford, J.; Gearing, A.; Miller, K. Serum gelatinase B, TIMP-1 and TIMP-2 levels in multiple sclerosis. A longitudinal clinical and MRI study. Brain 1999, 122, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, S.A.; Savinov, A.Y.; Cieplak, P.; Ratnikov, B.I.; Motamedchaboki, K.; Smith, J.W.; Strongin, A.Y. Matrix metalloproteinase proteolysis of the myelin basic protein isoforms is a source of immunogenic peptides in autoimmune multiple sclerosis. PLoS ONE 2009, 4, e4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichmann, H.; Brandt, M.D.; Klingelhoefer, L. The nonmotor features of Parkinson’s disease: Pathophysiology and management advances. Curr. Opin. Neurol. 2016, 29, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. Available online: https://www.nature.com/articles/nrdp201713#supplementary-information (accessed on 19 December 2020). [CrossRef] [PubMed]
- Magrinelli, F.; Picelli, A.; Tocco, P.; Federico, A.; Roncari, L.; Smania, N.; Zanette, G.; Tamburin, S. Pathophysiology of motor dysfunction in parkinson’s disease as the rationale for drug treatment and rehabilitation. Parkinson’s Disease 2016, 2016, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Crocker, S.J.; Milner, R.; Pham-Mitchell, N.; Campbell, I.L. Cell and agonist-specific regulation of genes for matrix metalloproteinases and their tissue inhibitors by primary glial cells. J. Neurochem. 2006, 98, 812–823. [Google Scholar] [CrossRef]
- Chung, Y.C.; Kim, Y.S.; Bok, E.; Yune, T.Y.; Maeng, S.; Jin, B.K. MMP-3 contributes to nigrostriatal dopaminergic neuronal loss, BBB damage, and neuroinflammation in an MPTP mouse model of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 370526. [Google Scholar] [CrossRef] [Green Version]
- Lorenzl, S.; Albers, D.S.; LeWitt, P.A.; Chirichigno, J.W.; Hilgenberg, S.L.; Cudkowicz, M.E.; Beal, M.F. Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J. Neurol. Sci. 2003, 207, 71–76. [Google Scholar] [CrossRef]
- Chen, Y.C.; Wu, Y.R.; Mesri, M.; Chen, C.M. Associations of matrix metalloproteinase-9 and tissue inhibitory factor-1 polymorphisms with parkinson disease in Taiwan. Medicine 2016, 95, e2672. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Qiao, W.; Bu, G. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ayllón, M.-S.; Lopez-Font, I.; Boix, C.P.; Fortea, J.; Sánchez-Valle, R.; Lleó, A.; Molinuevo, J.-L.; Zetterberg, H.; Blennow, K.; Sáez-Valero, J. C-terminal fragments of the amyloid precursor protein in cerebrospinal fluid as potential biomarkers for Alzheimer disease. Sci. Rep. 2017, 7, 2477. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, E.M.; Adcock, K.H.; Morgenstern, D.A.; Clayton, R.; von Stillfried, N.; Rhodes, K.; Ellis, C.; Fawcett, J.W.; Rogers, J.H. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res. Mol. Brain Res. 2002, 100, 103–117. [Google Scholar] [CrossRef]
- Deb, S.; Zhang, J.W.; Gottschall, P.E. Beta-amyloid induces the production of active, matrix-degrading proteases in cultured rat astrocytes. Brain Res. 2003, 970, 205–213. [Google Scholar] [CrossRef]
- Kalani, A.; Kamat, P.K.; Familtseva, A.; Chaturvedi, P.; Muradashvili, N.; Narayanan, N.; Tyagi, S.C.; Tyagi, N. Role of microRNA29b in blood-brain barrier dysfunction during hyperhomocysteinemia: An epigenetic mechanism. J. Cereb. Blood Flow Metab. 2014, 34, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Takagi, Y.; Aoki, T.; Hayase, M.; Marumo, T.; Gomi, M.; Nishimura, M.; Kataoka, H.; Hashimoto, N.; Nozaki, K. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1674–1685. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chintala, S.K. Influence of interleukin-1 beta induction and mitogen-activated protein kinase phosphorylation on optic nerve ligation-induced matrix metalloproteinase-9 activation in the retina. Exp. Eye Res. 2004, 78, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.; Gerhauser, I.; Seeliger, F.; Baumgartner, W.; Alldinger, S. Matrix metalloproteinases and their inhibitors in the developing mouse brain and spinal cord: A reverse transcription quantitative polymerase chain reaction study. Dev. Neurosci. 2005, 27, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Prada, C.; Lattarulo, C.; Fine, S.; Borrelli, L.A.; Betensky, R.; Greenberg, S.M.; Frosch, M.P.; Bacskai, B.J. Matrix metalloproteinase inhibition reduces oxidative stress associated with cerebral amyloid angiopathy in vivo in transgenic mice. J. Neurochem. 2009, 109, 1636–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Guillamon, M.; Delgado, P.; Ortega, L.; Pares, M.; Rosell, A.; Garcia-Bonilla, L.; Fernandez-Cadenas, I.; Borrell-Pages, M.; Boada, M.; Montaner, J. Neuronal TIMP-1 release accompanies astrocytic MMP-9 secretion and enhances astrocyte proliferation induced by beta-amyloid 25–35 fragment. J. Neurosci. Res. 2009, 87, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Stomrud, E.; Bjorkqvist, M.; Janciauskiene, S.; Minthon, L.; Hansson, O. Alterations of matrix metalloproteinases in the healthy elderly with increased risk of prodromal Alzheimer’s disease. Alzheimers Res. 2010, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Mroczko, B.; Groblewska, M.; Barcikowska, M. The role of matrix metalloproteinases and tissue inhibitors of metalloproteinases in the pathophysiology of neurodegeneration: A literature study. J. Alzheimer’s Dis. 2013, 37, 273–283. [Google Scholar] [CrossRef]
- Tuna, G.; Yener, G.G.; Oktay, G.; Islekel, G.H.; FG, K.I. Evaluation of matrix metalloproteinase-2 (MMP-2) and -9 (MMP-9) and their tissue inhibitors (TIMP-1 and TIMP-2) in plasma from patients with neurodegenerative dementia. J. Alzheimer’s Dis. 2018, 66, 1265–1273. [Google Scholar] [CrossRef]
- Qin, W.; Jia, X.; Wang, F.; Zuo, X.; Wu, L.; Zhou, A.; Li, D.; Min, B.; Wei, C.; Tang, Y.; et al. Elevated plasma angiogenesis factors in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 245–252. [Google Scholar] [CrossRef]
- Ethell, I.M.; Ethell, D.W. Matrix metalloproteinases in brain development and remodeling: Synaptic functions and targets. J. Neurosci. Res. 2007, 85, 2813–2823. [Google Scholar] [CrossRef]
- Peress, N.; Perillo, E.; Zucker, S. Localization of tissue inhibitor of matrix metalloproteinases in Alzheimer’s disease and normal brain. J. Neuropathol. Exp. Neurol. 1995, 54, 16–22. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2016, 13, 96. Available online: https://www.nature.com/articles/nrneurol.2016.182#supplementary-information (accessed on 19 December 2020). [CrossRef] [Green Version]
- Katz, J.S.; Woolley, S.C. Amyotrophic lateral sclerosis. In Physician’s Field Guide to Neuropsychology: Collaboration through Case Example; Sanders, K.M., Ed.; Springer: New York, NY, USA, 2019; pp. 255–265. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Lukaszewicz-Zajac, M.; Mroczko, B.; Slowik, A. Matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in amyotrophic lateral sclerosis (ALS). J. Neural Transm. 2014, 121, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Huber-Abel, F.; Teuchert, M.; Hendrich, C.; Dorst, J.; Schattauer, D.; Zettlmeissel, H.; Wlaschek, M.; Scharffetter-Kochanek, K.; Tumani, H.; et al. Linking neuron and skin: Matrix metalloproteinases in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 2009, 285, 62–66. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Woods, R.L., 3rd; Louis, M.K.; Zesiewicz, T.A.; Kuzmin-Nichols, N.; Sullivan, K.L.; Miller, A.M.; Hernandez-Ontiveros, D.G.; Sanberg, P.R. Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS ONE 2010, 5, e10614. [Google Scholar] [CrossRef]
- Niebroj-Dobosz, I.; Janik, P.; Sokolowska, B.; Kwiecinski, H. Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2010, 17, 226–231. [Google Scholar] [CrossRef]
- Bossolasco, P.; Cova, L.; Calzarossa, C.; Servida, F.; Mencacci, N.E.; Onida, F.; Polli, E.; Lambertenghi Deliliers, G.; Silani, V. Metalloproteinase alterations in the bone marrow of ALS patients. J. Mol. Med. 2010, 88, 553–564. [Google Scholar] [CrossRef]
- Lee, J.K.; Shin, J.H.; Suh, J.; Choi, I.S.; Ryu, K.S.; Gwag, B.J. Tissue inhibitor of metalloproteinases-3 (TIMP-3) expression is increased during serum deprivation-induced neuronal apoptosis in vitro and in the G93A mouse model of amyotrophic lateral sclerosis: A potential modulator of Fas-mediated apoptosis. Neurobiol. Dis. 2008, 30, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Hopps, E.; Caimi, G. Matrix metalloproteases as a pharmacological target in cardiovascular diseases. Eur. Rev. Med. Pharm. Sci. 2015, 19, 2583–2589. [Google Scholar]
- Gao, L.; Zheng, Y.J.; Gu, S.S.; Tan, J.L.; Paul, C.; Wang, Y.G.; Yang, H.T. Degradation of cardiac myosin light chain kinase by matrix metalloproteinase-2 contributes to myocardial contractile dysfunction during ischemia/reperfusion. J. Mol. Cell. Cardiol. 2014, 77, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Jobin, P.G.; Butler, G.S.; Overall, C.M. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Cauwe, B.; Opdenakker, G. Intracellular substrate cleavage: A novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 351–423. [Google Scholar] [CrossRef] [PubMed]
- Sariahmetoglu, M.; Crawford, B.D.; Leon, H.; Sawicka, J.; Li, L.; Ballermann, B.J.; Holmes, C.; Berthiaume, L.G.; Holt, A.; Sawicki, G.; et al. Regulation of matrix metalloproteinase-2 (MMP-2) activity by phosphorylation. FASEB J. 2007, 21, 2486–2495. [Google Scholar] [CrossRef] [Green Version]
- Jacob-Ferreira, A.L.; Kondo, M.Y.; Baral, P.K.; James, M.N.; Holt, A.; Fan, X.; Schulz, R. Phosphorylation status of 72 kDa MMP-2 determines its structure and activity in response to peroxynitrite. PLoS ONE 2013, 8, e71794. [Google Scholar] [CrossRef] [Green Version]
- Baghirova, S.; Hughes, B.G.; Poirier, M.; Kondo, M.Y.; Schulz, R. Nuclear matrix metalloproteinase-2 in the cardiomyocyte and the ischemic-reperfused heart. J. Mol. Cell. Cardiol. 2016, 94, 153–161. [Google Scholar] [CrossRef]
- De Coux, A.; Lindsey, M.L.; Villarreal, F.; Garcia, R.A.; Schulz, R. Myocardial matrix metalloproteinase-2: Inside out and upside down. J. Mol. Cell. Cardiol. 2014, 77, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Cho, W.J.; Hudson, B.; Kassiri, Z.; Granzier, H.; Schulz, R. Titin is a target of matrix metalloproteinase-2: Implications in myocardial ischemia/reperfusion injury. Circulation 2010, 122, 2039–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.Y.; Bergman, M.R.; Nguyen, A.P.; Turcato, S.; Swigart, P.M.; Rodrigo, M.C.; Simpson, P.C.; Karliner, J.S.; Lovett, D.H.; Baker, A.J. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc. Res. 2006, 69, 688–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierevogel, M.J.; Pasterkamp, G.; de Kleijn, D.P.; Strauss, B.H. Matrix metalloproteinases: A therapeutic target in cardiovascular disease. Curr. Pharm. Des. 2003, 9, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Radosinska, J.; Barancik, M.; Vrbjar, N. Heart failure and role of circulating MMP-2 and MMP-9. Panminerva Med. 2017, 59, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Giannakos, E.; Vardali, E.; Bartekova, M.; Fogarassyova, M.; Barancik, M.; Radosinska, J. Changes in activities of circulating MMP-2 and MMP-9 in patients suffering from heart failure in relation to gender, hypertension and treatment: A cross-sectional study. Physiol. Res. 2016, 65 (Suppl. 1), S149–S152. [Google Scholar] [CrossRef] [PubMed]
- Alp, E.; Yilmaz, A.; Tulmac, M.; Dikmen, A.U.; Cengel, A.; Yalcin, R.; Menevse, E.S. Analysis of MMP-7 and TIMP-2 gene polymorphisms in coronary artery disease and myocardial infarction: A Turkish case-control study. Kaohsiung J. Med. Sci. 2017, 33, 78–85. [Google Scholar] [CrossRef]
- Barker, H.E.; Chang, J.; Cox, T.R.; Lang, G.; Bird, D.; Nicolau, M.; Evans, H.R.; Gartland, A.; Erler, J.T. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011, 71, 1561–1572. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: The task force for the management of arterial hypertension of the european society of cardiology and the european society of hypertension: The task force for the management of arterial hypertension of the european society of cardiology and the european society of hypertension. J. Hypertens. 2018, 36, 1953–2041. [Google Scholar] [CrossRef] [Green Version]
- Rust, P.; Ekmekcioglu, C. Impact of salt intake on the pathogenesis and treatment of hypertension. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2016; Volume 956. [Google Scholar]
- Samadian, F.; Dalili, N.; Jamalian, A. Lifestyle modifications to prevent and control hypertension. Iran. J. Kidney Dis. 2016, 10, 237–263. [Google Scholar]
- Trojanek, J.B. Role of matrix metalloproteinases and tissue inhibitors of metalloproteinases in hypertension. Pathogenesis of hypertension and obesity. Postepy Biochem. 2015, 61, 356–363. [Google Scholar]
- Tan, J.; Hua, Q.; Xing, X.; Wen, J.; Liu, R.; Yang, Z. Impact of the metalloproteinase-9/tissue inhibitor of metalloproteinase-1 system on large arterial stiffness in patients with essential hypertension. Hypertens. Res. 2007, 30, 959–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, V.J.; Le, T.T.; Bryant, J.; Wong, J.I.; Su, B.; Lee, C.H.; Pua, C.J.; Sim, C.P.Y.; Ang, B.; Aw, T.C.; et al. Novel Index of Maladaptive Myocardial Remodeling in Hypertension. Circ. Cardiovasc. Imaging 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehoux, S.; Lemarie, C.A.; Esposito, B.; Lijnen, H.R.; Tedgui, A. Pressure-induced matrix metalloproteinase-9 contributes to early hypertensive remodeling. Circulation 2004, 109, 1041–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onal, I.K.; Altun, B.; Onal, E.D.; Kirkpantur, A.; Gul Oz, S.; Turgan, C. Serum levels of MMP-9 and TIMP-1 in primary hypertension and effect of antihypertensive treatment. Eur. J. Intern. Med. 2009, 20, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Tayebjee, M.H.; Nadar, S.K.; MacFadyen, R.J.; Lip, G.Y. Tissue inhibitor of metalloproteinase-1 and matrix metalloproteinase-9 levels in patients with hypertension Relationship to tissue Doppler indices of diastolic relaxation. Am. J. Hypertens. 2004, 17, 770–774. [Google Scholar] [CrossRef]
- Khezheva, F.M.; Mazur, N.A.; Masenko, V.P. Metalloproteinase activity of the blood in patients with arterial hypertension with paroxysmal form of atrial fibrillation. Kardiologiia 2007, 47, 10–14. [Google Scholar] [PubMed]
- Gai, X.; Zhang, Z.; Liang, Y.; Chen, Z.; Yang, X.; Hou, J.; Lan, X.; Zheng, W.; Hou, J.; Huang, M. MMP-2 and TIMP-2 gene polymorphisms and susceptibility to atrial fibrillation in Chinese Han patients with hypertensive heart disease. Clin. Chim. Acta 2010, 411, 719–724. [Google Scholar] [CrossRef]
- Basu, R.; Lee, J.; Morton, J.S.; Takawale, A.; Fan, D.; Kandalam, V.; Wang, X.; Davidge, S.T.; Kassiri, Z. TIMP3 is the primary TIMP to regulate agonist-induced vascular remodelling and hypertension. Cardiovasc. Res. 2013, 98, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Bei, Y.; Shen, S.; Zhang, J.; Lu, Y.; Xiao, J.; Li, X. MicroRNA-222 promotes the proliferation of pulmonary arterial smooth muscle cells by targeting P27 and TIMP3. Cell. Physiol. Biochem. 2017, 43, 282–292. [Google Scholar] [CrossRef]
- Torres, N.; Guevara-Cruz, M.; Velazquez-Villegas, L.A.; Tovar, A.R. Nutrition and atherosclerosis. Arch. Med. Res. 2015, 46, 408–426. [Google Scholar] [CrossRef]
- Lopes, J.; Adiguzel, E.; Gu, S.; Liu, S.L.; Hou, G.; Heximer, S.; Assoian, R.K.; Bendeck, M.P. Type VIII collagen mediates vessel wall remodeling after arterial injury and fibrous cap formation in atherosclerosis. Am. J. Pathol. 2013, 182, 2241–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, P.; Basili, S.; Martini, F.; Cardarello, C.M.; Ceci, F.; Di Franco, M.; Bertazzoni, G.; Gazzaniga, P.P.; Alessandri, C. Serum metalloproteinase 9 levels in patients with coronary artery disease: A novel marker of inflammation. J. Investig. Med. 2003, 51, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lelongt, B.; Bengatta, S.; Delauche, M.; Lund, L.R.; Werb, Z.; Ronco, P.M. Matrix metalloproteinase 9 protects mice from anti-glomerular basement membrane nephritis through its fibrinolytic activity. J. Exp. Med. 2001, 193, 793–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology 2013, 28, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskivirta, I.; Rahkonen, O.; Mayranpaa, M.; Pakkanen, S.; Husheem, M.; Sainio, A.; Hakovirta, H.; Laine, J.; Jokinen, E.; Vuorio, E.; et al. Tissue inhibitor of metalloproteinases 4 (TIMP4) is involved in inflammatory processes of human cardiovascular pathology. Histochem. Cell Biol. 2006, 126, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Rouis, M.; Adamy, C.; Duverger, N.; Lesnik, P.; Horellou, P.; Moreau, M.; Emmanuel, F.; Caillaud, J.M.; Laplaud, P.M.; Dachet, C.; et al. Adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-1 reduces atherosclerotic lesions in apolipoprotein E-deficient mice. Circulation 1999, 100, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Baker, A.H.; Oka, K.; Chan, L.; Newby, A.C.; Jackson, C.L.; George, S.J. Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: Involvement of macrophage migration and apoptosis. Circulation 2006, 113, 2435–2444. [Google Scholar] [CrossRef] [Green Version]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D.; ESC Scientific Document Group. Fourth universal definition of myocardial infarction (2018). Eur. Heart J. 2019, 40, 237–269. [Google Scholar] [CrossRef] [Green Version]
- Newby, A.C. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol. Rev. 2005, 85, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Halade, G.V.; Jin, Y.F.; Lindsey, M.L. Matrix metalloproteinase (MMP)-9: A proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol. Ther. 2013, 139, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.; Wedin, K.; Brown, M.D.; Keller, C.; Evans, A.J.; Smolen, J.; Burns, A.R.; Rossen, R.D.; Michael, L.; Entman, M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 2001, 103, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Escobar, G.P.; Dobrucki, L.W.; Goshorn, D.K.; Bouges, S.; Mingoia, J.T.; McClister, D.M., Jr.; Su, H.; Gannon, J.; MacGillivray, C.; et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H232–H239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, M.L.; Iyer, R.P.; Zamilpa, R.; Yabluchanskiy, A.; DeLeon-Pennell, K.Y.; Hall, M.E.; Kaplan, A.; Zouein, F.A.; Bratton, D.; Flynn, E.R.; et al. A novel collagen matricryptin reduces left ventricular dilation post-myocardial infarction by promoting scar formation and angiogenesis. J. Am. Coll. Cardiol. 2015, 66, 1364–1374. [Google Scholar] [CrossRef] [Green Version]
- Kandalam, V.; Basu, R.; Abraham, T.; Wang, X.; Soloway, P.D.; Jaworski, D.M.; Oudit, G.Y.; Kassiri, Z. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ. Res. 2010, 106, 796–808. [Google Scholar] [CrossRef] [Green Version]
- Hansson, J.; Lind, L.; Hulthe, J.; Sundstrom, J. Relations of serum MMP-9 and TIMP-1 levels to left ventricular measures and cardiovascular risk factors: A population-based study. Eur. J. Cardiovasc. Prev. Rehabil. 2009, 16, 297–303. [Google Scholar] [CrossRef]
- Kelly, D.; Khan, S.Q.; Thompson, M.; Cockerill, G.; Ng, L.L.; Samani, N.; Squire, I.B. Plasma tissue inhibitor of metalloproteinase-1 and matrix metalloproteinase-9: Novel indicators of left ventricular remodelling and prognosis after acute myocardial infarction. Eur. Heart J. 2008, 29, 2116–2124. [Google Scholar] [CrossRef] [Green Version]
- Weir, R.A.; Clements, S.; Steedman, T.; Dargie, H.J.; McMurray, J.J.; Squire, I.B.; Ng, L.L. Plasma TIMP-4 predicts left ventricular remodeling after acute myocardial infarction. J. Card. Fail. 2011, 17, 465–471. [Google Scholar] [CrossRef]
- Shi, J.; Bei, Y.; Kong, X.; Liu, X.; Lei, Z.; Xu, T.; Wang, H.; Xuan, Q.; Chen, P.; Xu, J.; et al. miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics 2017, 7, 664–676. [Google Scholar] [CrossRef]
- Balaz, P.; Bjorck, M. True aneurysm in autologous hemodialysis fistulae: Definitions, classification and indications for treatment. J. Vasc. Access 2015, 16, 446–453. [Google Scholar] [CrossRef]
- Joviliano, E.E.; Ribeiro, M.S.; Tenorio, E.J.R. MicroRNAs and current concepts on the pathogenesis of abdominal aortic aneurysm. Braz. J. Cardiovasc. Surg. 2017, 32, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegrum, S. Studies into the Effects of Hypoxia on Matrix Metalloproteinases within the Abdominal Aortic Aneurysm. Doctoral’s Thesis, University Of Bath, Bath, UK, 2017. [Google Scholar]
- Forsythe, R.O.; Newby, D.E.; Robson, J.M.J. Monitoring the biological activity of abdominal aortic aneurysms beyond Ultrasound. Heart 2016, 102, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef]
- Siefert, S.A.; Sarkar, R. Matrix metalloproteinases in vascular physiology and disease. Vascular 2012, 20, 210–216. [Google Scholar] [CrossRef]
- Zervoudaki, A.; Economou, E.; Stefanadis, C.; Pitsavos, C.; Tsioufis, K.; Aggeli, C.; Vasiliadou, K.; Toutouza, M.; Toutouzas, P. Plasma levels of active extracellular matrix metalloproteinases 2 and 9 in patients with essential hypertension before and after antihypertensive treatment. J. Hum. Hypertens. 2003, 17, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Bobryshev, Y.V.; Lord, R.S.; Parsson, H. Immunophenotypic analysis of the aortic aneurysm wall suggests that vascular dendritic cells are involved in immune responses. Cardiovasc. Surg. 1998, 6, 240–249. [Google Scholar] [CrossRef]
- Meng, X.; Yang, J.; Zhang, K.; An, G.; Kong, J.; Jiang, F.; Zhang, Y.; Zhang, C. Regulatory T cells prevent angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E knockout mice. Hypertension 2014, 64, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Li, Y.H.; Shi, G.Y.; Tsai, H.W.; Luo, C.Y.; Cheng, M.H.; Ma, C.Y.; Hsu, Y.Y.; Cheng, T.L.; Chang, B.I.; et al. Membrane-Bound Thrombomodulin Regulates Macrophage Inflammation in Abdominal Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2015, 35, 2412–2422. [Google Scholar] [CrossRef] [Green Version]
- Goodall, S.; Crowther, M.; Hemingway, D.M.; Bell, P.R.; Thompson, M.M. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation 2001, 104, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.A.; Barbour, J.R.; Lowry, A.S.; Bouges, S.; Beck, C.; McClister, D.M., Jr.; Mukherjee, R.; Ikonomidis, J.S. Spatiotemporal expression and localization of matrix metalloproteinas-9 in a murine model of thoracic aortic aneurysm. J. Vasc. Surg. 2006, 44, 1314–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, A.M.; Quintero, P.A.; Perkins, D.L.; Kobayashi, D.K.; Kelley, D.G.; Marconcini, L.A.; Mecham, R.P.; Senior, R.M.; Shapiro, S.D. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Investig. 2006, 116, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Dale, M.A.; Suh, M.K.; Zhao, S.; Meisinger, T.; Gu, L.; Swier, V.J.; Agrawal, D.K.; Greiner, T.C.; Carson, J.S.; Baxter, B.T.; et al. Background differences in baseline and stimulated MMP levels influence abdominal aortic aneurysm susceptibility. Atherosclerosis 2015, 243, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabkin, S.W. Differential expression of MMP-2, MMP-9 and TIMP proteins in thoracic aortic aneurysm - comparison with and without bicuspid aortic valve: A meta-analysis. Vasa 2014, 43, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Allaire, E.; Forough, R.; Clowes, M.; Starcher, B.; Clowes, A.W. Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. J. Clin. Investig. 1998, 102, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Gestrich, C.; Duerr, G.D.; Heinemann, J.C.; Meertz, A.; Probst, C.; Roell, W.; Schiller, W.; Zimmer, A.; Bindila, L.; Lutz, B.; et al. Activation of Endocannabinoid System Is Associated with Persistent Inflammation in Human Aortic Aneurysm. Biomed. Res. Int. 2015, 2015, 456582. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.R.; Schwalbe, E.C.; Jones, J.L.; Bell, P.R.; Thompson, M.M. Matrix metalloproteinase 8 (neutrophil collagenase) in the pathogenesis of abdominal aortic aneurysm. Br. J. Surg. 2005, 92, 828–833. [Google Scholar] [CrossRef]
- Cork, B.A.; Li, T.C.; Warren, M.A.; Laird, S.M. Interleukin-11 (IL-11) in human endometrium: Expression throughout the menstrual cycle and the effects of cytokines on endometrial IL-11 production in vitro. J. Reprod. Immunol. 2001, 50, 3–17. [Google Scholar] [CrossRef]
- Licht, P.; Fluhr, H.; Neuwinger, J.; Wallwiener, D.; Wildt, L. Is human chorionic gonadotropin directly involved in the regulation of human implantation? Mol. Cell. Endocrinol. 2007, 269, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Karthikeyan, V.J.; Lane, D.A.; Beevers, D.G.; Lip, G.Y.; Blann, A.D. Matrix metalloproteinases and their tissue inhibitors in hypertension-related pregnancy complications. J. Hum. Hypertens. 2013, 27, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Fauser, B.C.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.S.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S.; et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2012, 97, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Abraham Suzanne, O.J. Fundamentals of Obstetrics and Gynaecology, 10th ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- McCartney, C.R.; Marshall, J.C. Polycystic ovary syndrome. N. Engl. J. Med. 2016, 375, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orio, F.; Muscogiuri, G.; Nese, C.; Palomba, S.; Savastano, S.; Tafuri, D.; Colarieti, G.; La Sala, G.; Colao, A.; Yildiz, B.O. Obesity, type 2 diabetes mellitus and cardiovascular disease risk: An uptodate in the management of polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 207, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Hulboy, D.L.; Rudolph, L.A.; Matrisian, L.M. Matrix metalloproteinases as mediators of reproductive function. Mol. Hum. Reprod. 1997, 3, 27–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dambala, K.; Vavilis, D.; Bili, E.; Goulis, D.G.; Tarlatzis, B.C. Serum visfatin, vascular endothelial growth factor and matrix metalloproteinase-9 in women with polycystic ovary syndrome. Gynecol. Endocrinol. 2017, 33, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Shalev, E.; Goldman, S.; Ben-Shlomo, I. The balance between MMP-9 and MMP-2 and their tissue inhibitor (TIMP)-1 in luteinized granulosa cells: Comparison between women with PCOS and normal ovulatory women. Mol. Hum. Reprod. 2001, 7, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Cai, L.Y.; Lv, H.M.; Xia, L.; Zhang, Y.J.; Zhang, H.X.; Guan, Y.M. Raised serum levels of matrix metalloproteinase-9 in women with polycystic ovary syndrome and its association with insulin-like growth factor binding protein-1. Gynecol. Endocrinol. 2008, 24, 285–288. [Google Scholar] [CrossRef]
- Akcali, A.; Bostanci, N.; Ozcaka, O.; Ozturk-Ceyhan, B.; Gumus, P.; Tervahartiala, T.; Husu, H.; Buduneli, N.; Sorsa, T.; Belibasakis, G.N. Elevated matrix metalloproteinase-8 in saliva and serum in polycystic ovary syndrome and association with gingival inflammation. Innate Immun. 2015, 21, 619–625. [Google Scholar] [CrossRef]
- Lewandowski, K.C.; Komorowski, J.; O’Callaghan, C.J.; Tan, B.K.; Chen, J.; Prelevic, G.M.; Randeva, H.S. Increased circulating levels of matrix metalloproteinase-2 and -9 in women with the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 1173–1177. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Shi, L.B.; Zhang, S.Y. Ovarian Fibrosis: A Phenomenon of Concern. Chin. Med. J. 2017, 130, 365–371. [Google Scholar] [CrossRef]
- Ben-Shlomo, I.; Goldman, S.; Shalev, E. Regulation of matrix metalloproteinase-9 (MMP-9), tissue inhibitor of MMP, and progesterone secretion in luteinized granulosa cells from normally ovulating women with polycystic ovary disease. Fertil. Steril. 2003, 79 (Suppl. 1), 694–701. [Google Scholar] [CrossRef]
- Barisic, A.; Devic Pavlic, S.; Ostojic, S.; Pereza, N. Matrix metalloproteinase and tissue inhibitors of metalloproteinases gene polymorphisms in disorders that influence fertility and pregnancy complications: A systematic review and meta-analysis. Gene 2018, 647, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Rouse, C.E.; Eckert, L.O.; Babarinsa, I.; Fay, E.; Gupta, M.; Harrison, M.S.; Kawai, A.T.; Kharbanda, E.O.; Kucuku, M.; Meller, L.; et al. Spontaneous abortion and ectopic pregnancy: Case definition & guidelines for data collection, analysis, and presentation of maternal immunization safety data. Vaccine 2017, 35, 6563–6574. [Google Scholar] [CrossRef] [PubMed]
- Meaney, S.; Corcoran, P.; Spillane, N.; O’Donoghue, K. Experience of miscarriage: An interpretative phenomenological analysis. BMJ Open 2017, 7, e011382. [Google Scholar] [CrossRef] [Green Version]
- Franasiak, J.M.; Scott, R.T. Contribution of immunology to implantation failure of euploid embryos. Fertil. Steril. 2017, 107, 1279–1283. [Google Scholar] [CrossRef] [Green Version]
- Pereza, N.; Ostojić, S.; Kapović, M.; Peterlin, B. Systematic review and meta-analysis of genetic association studies in idiopathic recurrent spontaneous abortion. Fertil. Steril. 2017, 107, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Schatz, F.; Guzeloglu-Kayisli, O.; Arlier, S.; Kayisli, U.A.; Lockwood, C.J. The role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding. Hum. Reprod. Update 2016, 22, 497–515. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.S.; Crocker, S.J. An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol. 2012, 180, 12–16. [Google Scholar] [CrossRef]
- Whiteside, E.J.; Jackson, M.M.; Herington, A.C.; Edwards, D.R.; Harvey, M.B. Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-3 are key regulators of extracellular matrix degradation by mouse embryos. Biol. Reprod. 2001, 64, 1331–1337. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Qi, Y. Detection of MMP-9 and TIMP-3 mRNA expression in the villi of patients undergoing early spontaneous abortion: A report of 30 cases. Exp. Med. 2015, 9, 1939–1943. [Google Scholar] [CrossRef] [Green Version]
- Onogi, A.; Naruse, K.; Sado, T.; Tsunemi, T.; Shigetomi, H.; Noguchi, T.; Yamada, Y.; Akasaki, M.; Oi, H.; Kobayashi, H. Hypoxia inhibits invasion of extravillous trophoblast cells through reduction of matrix metalloproteinase (MMP)-2 activation in the early first trimester of human pregnancy. Placenta 2011, 32, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Nissi, R.; Talvensaari-Mattila, A.; Kotila, V.; Niinimaki, M.; Jarvela, I.; Turpeenniemi-Hujanen, T. Circulating matrix metalloproteinase MMP-9 and MMP-2/TIMP-2 complex are associated with spontaneous early pregnancy failure. Reprod. Biol. Endocrinol. 2013, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anumba, D.O.; El Gelany, S.; Elliott, S.L.; Li, T.C. Circulating levels of matrix proteases and their inhibitors in pregnant women with and without a history of recurrent pregnancy loss. Reprod. Biol. Endocrinol. 2010, 8, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzypczak, J.; Wirstlein, P.; Mikolajczyk, M.; Ludwikowski, G.; Zak, T. TGF superfamily and MMP2, MMP9, TIMP1 genes expression in the endometrium of women with impaired reproduction. Folia Histochem. Cytobiol. 2007, 45 (Suppl. 1), S143–S148. [Google Scholar] [PubMed]
- Li, M.Q.; Hou, X.F.; Shao, J.; Tang, C.L.; Li, D.J. The DSCs-expressed CD82 controls the invasiveness of trophoblast cells via integrinbeta1/MAPK/MAPK3/1 signaling pathway in human first-trimester pregnancy. Biol. Reprod. 2010, 82, 968–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ACOG Committee on Obstetric Practice. ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obs. Gynecol. 2002, 99, 159–167. [Google Scholar]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staun-Ram, E.; Goldman, S.; Gabarin, D.; Shalev, E. Expression and importance of matrix metalloproteinase 2 and 9 (MMP-2 and -9) in human trophoblast invasion. Reprod. Biol. Endocrinol. 2004, 2, 59. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.Y.; Pang, Z.J.; Yu, Y.H. Regulation of trophoblast invasion: The role of matrix metalloproteinases. Rev. Obs. Gynecol. 2012, 5, e137–e143. [Google Scholar]
- Plaks, V.; Rinkenberger, J.; Dai, J.; Flannery, M.; Sund, M.; Kanasaki, K.; Ni, W.; Kalluri, R.; Werb, Z. Matrix metalloproteinase-9 deficiency phenocopies features of preeclampsia and intrauterine growth restriction. Proc. Natl. Acad. Sci. USA 2013, 110, 11109–11114. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Khalil, R.A. Matrix metalloproteinases in normal pregnancy and preeclampsia. Prog. Mol. Biol. Transl. Sci. 2017, 148, 87–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.E.; Merchant, S.J.; Macleod, M.; Mires, G.J.; Baker, P.N.; Davidge, S.T. MMP-2 levels are elevated in the plasma of women who subsequently develop preeclampsia. Hypertens. Pregnancy 2005, 24, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Gutierrez, G.; Cappello, R.E.; Mishra, N.; Romero, R.; Strauss, J.F., 3rd; Walsh, S.W. Increased expression of matrix metalloproteinase-1 in systemic vessels of preeclamptic women: A critical mediator of vascular dysfunction. Am. J. Pathol. 2011, 178, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fierro, M.L.; Perez-Favila, A.; Garza-Veloz, I.; Espinoza-Juarez, M.A.; Avila-Carrasco, L.; Delgado-Enciso, I.; Ortiz-Castro, Y.; Cardenas-Vargas, E.; Cid-Baez, M.A.; Ramirez-Santoyo, R.M.; et al. Matrix metalloproteinase multiplex screening identifies increased MMP-2 urine concentrations in women predicted to develop preeclampsia. Biomarkers 2018, 23, 18–24. [Google Scholar] [CrossRef]
- Staun-Ram, E.; Shalev, E. Human trophoblast function during the implantation process. Reprod. Biol. Endocrinol. 2005, 3, 56. [Google Scholar] [CrossRef] [Green Version]
- Luizon, M.R.; Palei, A.C.; Sandrim, V.C.; Amaral, L.M.; Machado, J.S.; Lacchini, R.; Cavalli, R.C.; Duarte, G.; Tanus-Santos, J.E. Tissue inhibitor of matrix metalloproteinase-1 polymorphism, plasma TIMP-1 levels, and antihypertensive therapy responsiveness in hypertensive disorders of pregnancy. Pharm. J. 2014, 14, 535–541. [Google Scholar] [CrossRef]
- Montagnana, M.; Lippi, G.; Albiero, A.; Scevarolli, S.; Salvagno, G.L.; Franchi, M.; Guidi, G.C. Evaluation of metalloproteinases 2 and 9 and their inhibitors in physiologic and pre-eclamptic pregnancy. J. Clin. Lab. Anal. 2009, 23, 88–92. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, Q.D.; Qiao, C. Expression of trophoblast invasion related genes mRNA and protein in human placenta in preeclampsia. Zhonghua Fu Chan Ke Za Zhi 2006, 41, 509–513. [Google Scholar]
- Deng, C.L.; Ling, S.T.; Liu, X.Q.; Zhao, Y.J.; Lv, Y.F. Decreased expression of matrix metalloproteinase-1 in the maternal umbilical serum, trophoblasts and decidua leads to preeclampsia. Exp. Med. 2015, 9, 992–998. [Google Scholar] [CrossRef]
- Lavee, M.; Goldman, S.; Daniel-Spiegel, E.; Shalev, E. Matrix metalloproteinase-2 is elevated in midtrimester amniotic fluid prior to the development of preeclampsia. Reprod. Biol. Endocrinol. 2009, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Gu, B.; Gu, Y.; Groome, L.J.; Wang, Y. Down-regulation of TIMP3 leads to increase in TACE expression and TNFalpha production by placental trophoblast cells. Am. J. Reprod. Immunol. 2014, 71, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, D.; Zhu, J.; Liu, Q.; Li, J.; Song, M.; Wang, K.; Zhou, Q.; Jia, Y.; Li, T. Dysregulation of HDAC9 represses trophoblast cell migration and invasion through TIMP3 activation in preeclampsia. Am. J. Hypertens. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhang, X.; Li, Q.; Xu, J.; Zhou, X.; Wang, T.; Xing, Q.; Liu, Y.; Wang, L.; He, L.; et al. Promoter hypomethylation of TIMP3 is associated with pre-eclampsia in a Chinese population. Mol. Hum. Reprod. 2013, 19, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandrim, V.C.; Diniz, S.; Eleuterio, N.M.; Gomes, K.B.; Dusse, L.M.S.; Cavalli, R.C. Higher levels of circulating TIMP-4 in preeclampsia is strongly associated with clinical parameters and microRNA. Clin. Exp. Hypertens. 2018, 40, 609–612. [Google Scholar] [CrossRef]
- Sandrim, V.C.; Luizon, M.R.; Machado, J.S.R.; Cavalli, R.C. C>T (rs17035945) polymorphism of TIMP-4 protects against preeclampsia. J. Obs. Gynaecol. 2019, 39, 135–137. [Google Scholar] [CrossRef]
- Drewlo, S.; Czikk, M.; Baczyk, D.; Lye, S.; Kingdom, J. Glial cell missing-1 mediates over-expression of tissue inhibitor of metalloproteinase-4 in severe pre-eclamptic placental villi. Hum. Reprod. 2011, 26, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.H.; Edwards, D.R.; Murphy, G. Metalloproteinase inhibitors: Biological actions and therapeutic opportunities. J. Cell Sci. 2002, 115, 3719–3727. [Google Scholar] [CrossRef] [Green Version]
- Dufour, A.; Overall, C.M. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharm. Sci. 2013, 34, 233–242. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, S.; Guo, J.; Zhou, L.; You, L.; Zhang, T.; Zhao, Y. Insights into the distinct roles of MMP-11 in tumor biology and future therapeutics (Review). Int. J. Oncol. 2016, 48, 1783–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlaki, M.; Zucker, S. Matrix metalloproteinase inhibitors (MMPIs): The beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. 2003, 22, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.S.; Cohen, A.M.; Zhang, Z.F.; Stetler-Stevenson, W.; Guillem, J.G. Elevated tissue inhibitor of metalloproteinase 1 RNA in colorectal cancer stroma correlates with lymph node and distant metastases. Clin. Cancer Res. 1995, 1, 899–906. [Google Scholar] [PubMed]
- Troeberg, L.; Lazenbatt, C.; Anower, E.K.M.F.; Freeman, C.; Federov, O.; Habuchi, H.; Habuchi, O.; Kimata, K.; Nagase, H. Sulfated glycosaminoglycans control the extracellular trafficking and the activity of the metalloprotease inhibitor TIMP-3. Chem. Biol. 2014, 21, 1300–1309. [Google Scholar] [CrossRef] [Green Version]
- Bachman, K.E.; Herman, J.G.; Corn, P.G.; Merlo, A.; Costello, J.F.; Cavenee, W.K.; Baylin, S.B.; Graff, J.R. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999, 59, 798–802. [Google Scholar]
- Baker, A.H.; George, S.J.; Zaltsman, A.B.; Murphy, G.; Newby, A.C. Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br. J. Cancer 1999, 79, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Ahonen, M.; Baker, A.H.; Kahari, V.M. Adenovirus-mediated gene delivery of tissue inhibitor of metalloproteinases-3 inhibits invasion and induces apoptosis in melanoma cells. Cancer Res. 1998, 58, 2310–2315. [Google Scholar]
- Anand-Apte, B.; Bao, L.; Smith, R.; Iwata, K.; Olsen, B.R.; Zetter, B.; Apte, S.S. A review of tissue inhibitor of metalloproteinases-3 (TIMP-3) and experimental analysis of its effect on primary tumor growth. Biochem. Cell Biol. 1996, 74, 853–862. [Google Scholar] [CrossRef]
- Sharma, S.; Free, A.; Mei, Y.; Peiper, S.C.; Wang, Z.; Cowell, J.K. Distinct molecular signatures in pediatric infratentorial glioblastomas defined by aCGH. Exp. Mol. Pathol. 2010, 89, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Xing, X.; Tanzer, M.; Rocken, C.; Weichert, W.; Ivanauskas, A.; Pross, M.; Peitz, U.; Malfertheiner, P.; Schmid, R.M.; et al. Frequent loss of TIMP-3 expression in progression of esophageal and gastric adenocarcinomas. Neoplasia 2008, 10, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.; De Young, N.J.; Tian, Z.Q.; Caruso, M.; Ruszkiewicz, A.R.; Liu, J.F.; Jamieson, G.G.; Drew, P.A. Methylation of TIMP3 in esophageal squamous cell carcinoma. World J. Gastroenterol. 2008, 14, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Zheng, W.; Luo, J.; Bai, Y.; Lu, Z. Multiple gene methylation of nonsmall cell lung cancers evaluated with 3-dimensional microarray. Cancer 2008, 112, 1325–1336. [Google Scholar] [CrossRef]
- Wild, A.; Ramaswamy, A.; Langer, P.; Celik, I.; Fendrich, V.; Chaloupka, B.; Simon, B.; Bartsch, D.K. Frequent methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene in pancreatic endocrine tumors. J. Clin. Endocrinol. Metab. 2003, 88, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Span, P.N.; Lindberg, R.L.; Manders, P.; Tjan-Heijnen, V.C.; Heuvel, J.J.; Beex, L.V.; Sweep, C.G. Tissue inhibitors of metalloproteinase expression in human breast cancer: TIMP-3 is associated with adjuvant endocrine therapy success. J. Pathol. 2004, 202, 395–402. [Google Scholar] [CrossRef]
- Guan, Z.; Zhang, J.; Song, S.; Dai, D. Promoter methylation and expression of TIMP3 gene in gastric cancer. Diagn. Pathol. 2013, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Voland, P.; Besig, S.; Rad, R.; Braun, T.; Baur, D.M.; Perren, A.; Langer, R.; Hofler, H.; Prinz, C. Correlation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinase expression in ileal carcinoids, lymph nodes and liver metastasis with prognosis and survival. Neuroendocrinology 2009, 89, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Riddick, A.C.; Shukla, C.J.; Pennington, C.J.; Bass, R.; Nuttall, R.K.; Hogan, A.; Sethia, K.K.; Ellis, V.; Collins, A.T.; Maitland, N.J.; et al. Identification of degradome components associated with prostate cancer progression by expression analysis of human prostatic tissues. Br. J. Cancer 2005, 92, 2171–2180. [Google Scholar] [CrossRef] [Green Version]
- Schrohl, A.S.; Look, M.P.; Meijer-van Gelder, M.E.; Foekens, J.A.; Brunner, N. Tumor tissue levels of Tissue Inhibitor of Metalloproteinases-1 (TIMP-1) and outcome following adjuvant chemotherapy in premenopausal lymph node-positive breast cancer patients: A retrospective study. BMC Cancer 2009, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Lipton, A.; Leitzel, K.; Chaudri-Ross, H.A.; Evans, D.B.; Ali, S.M.; Demers, L.; Hamer, P.; Brown-Shimer, S.; Pierce, K.; Gaur, V.; et al. Serum TIMP-1 and response to the aromatase inhibitor letrozole versus tamoxifen in metastatic breast cancer. J. Clin. Oncol. 2008, 26, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Zurakowski, D.; Wischhusen, J.; Frauenhoffer, C.; Hooshmand, S.; Kulke, M.; Moses, M.A. Urinary TIMP-1 and MMP-2 levels detect the presence of pancreatic malignancies. Br. J. Cancer 2014, 111, 1772–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, M.; Szylberg, L.; Kazmierczak, W.; Marszalek, A. Tumor progression driven by pathways activating matrix metalloproteinases and their inhibitors. J. Oral Pathol. Med. 2015, 44, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Kowalska, M.; Fuksiewicz, M.; Kotowicz, B.; Mierzejewska, E.; Kosela-Paterczyk, H.; Szamotulska, K.; Rutkowski, P. Serum markers in early-stage and locally advanced melanoma. Tumour Biol. J. Int. Soc. Oncodevelopmental. Biol. Med. 2015, 36, 8277–8285. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.C.; Chen, M.K.; Chen, L.Y.; Ho, E.S.; Ying, T.H.; Wang, P.H.; Yang, S.F. Genetic polymorphism of the tissue inhibitor of metalloproteinase-1 is associated with an increased risk of endometrial cancer. Clin. Chim. Acta 2009, 409, 127–131. [Google Scholar] [CrossRef]
- Pulukuri, S.M.; Patibandla, S.; Patel, J.; Estes, N.; Rao, J.S. Epigenetic inactivation of the tissue inhibitor of metalloproteinase-2 (TIMP-2) gene in human prostate tumors. Oncogene 2007, 26, 5229–5237. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, A.; Brinson, R.; Wei, B.; Stetler-Stevenson, W.G. Tissue Inhibitor of Metalloprotease-2 (TIMP-2): Bioprocess Development, Physicochemical, Biochemical, and Biological Characterization of Highly Expressed Recombinant Protein. Biochemistry 2017, 56, 6423–6433. [Google Scholar] [CrossRef]
- Nakopoulou, L.; Katsarou, S.; Giannopoulou, I.; Alexandrou, P.; Tsirmpa, I.; Panayotopoulou, E.; Mavrommatis, J.; Keramopoulos, A. Correlation of tissue inhibitor of metalloproteinase-2 with proliferative activity and patients’ survival in breast cancer. Mod. Pathol. 2002, 15, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yu, H.; Liu, S.Y.; Xiao, X.S.; Dong, W.H.; Chen, Y.N.; Xu, W.; Zhu, T. Prognostic value of tissue inhibitor of metalloproteinase-2 expression in patients with non-small cell lung cancer: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0124230. [Google Scholar] [CrossRef] [Green Version]
- Ring, P.; Johansson, K.; Hoyhtya, M.; Rubin, K.; Lindmark, G. Expression of tissue inhibitor of metalloproteinases TIMP-2 in human colorectal cancer--a predictor of tumour stage. Br. J. Cancer 1997, 76, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Kubben, F.J.; Sier, C.F.; Meijer, M.J.; van den Berg, M.; van der Reijden, J.J.; Griffioen, G.; van de Velde, C.J.; Lamers, C.B.; Verspaget, H.W. Clinical impact of MMP and TIMP gene polymorphisms in gastric cancer. Br. J. Cancer 2006, 95, 744–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsom, R.; Pignatelli, M.; Stetler-Stevenson, W.G.; Liotta, L.A.; Wright, P.A.; Jeffery, R.E.; Longcroft, J.M.; Rogers, L.; Stamp, G.W. Stromal expression of 72 kda type IV collagenase (MMP-2) and TIMP-2 mRNAs in colorectal neoplasia. Am. J. Pathol. 1992, 141, 389–396. [Google Scholar] [PubMed]
- Grignon, D.J.; Sakr, W.; Toth, M.; Ravery, V.; Angulo, J.; Shamsa, F.; Pontes, J.E.; Crissman, J.C.; Fridman, R. High levels of tissue inhibitor of metalloproteinase-2 (TIMP-2) expression are associated with poor outcome in invasive bladder cancer. Cancer Res. 1996, 56, 1654–1659. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Bertaux, B.; Hornebeck, W.; Eisen, A.Z.; Dubertret, L. Growth stimulation of human keratinocytes by tissue inhibitor of metalloproteinases. J. Investig. Derm. 1991, 97, 679–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yamashita, K.; Iwata, K.; Hayakawa, T. Both tissue inhibitors of metalloproteinases-1 (TIMP-1) and TIMP-2 activate Ras but through different pathways. Biochem. Biophys. Res. Commun. 2002, 296, 201–205. [Google Scholar] [CrossRef]
- Yamashita, K.; Suzuki, M.; Iwata, H.; Koike, T.; Hamaguchi, M.; Shinagawa, A.; Noguchi, T.; Hayakawa, T. Tyrosine phosphorylation is crucial for growth signaling by tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2). FEBS Lett. 1996, 396, 103–107. [Google Scholar] [CrossRef] [Green Version]
- D’Alessio, S.; Ferrari, G.; Cinnante, K.; Scheerer, W.; Galloway, A.C.; Roses, D.F.; Rozanov, D.V.; Remacle, A.G.; Oh, E.S.; Shiryaev, S.A.; et al. Tissue inhibitor of metalloproteinases-2 binding to membrane-type 1 matrix metalloproteinase induces MAPK activation and cell growth by a non-proteolytic mechanism. J. Biol. Chem. 2008, 283, 87–99. [Google Scholar] [CrossRef]
- Hoegy, S.E.; Oh, H.R.; Corcoran, M.L.; Stetler-Stevenson, W.G. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J. Biol. Chem. 2001, 276, 3203–3214. [Google Scholar] [CrossRef] [Green Version]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Goehlsdorf, D.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.G.; Steinle, A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008, 68, 6368–6376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.K.; Liu, X.W.; Chirco, R.; Fridman, R.; Kim, H.R. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006, 25, 3934–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetler-Stevenson, W.G. Tissue inhibitors of metalloproteinases in cell signaling: Metalloproteinase-independent biological activities. Sci. Signal. 2008, 1, re6. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Nishio, K.; Yamashita, K.; Hayakawa, T.; Hoshino, T. Cell cycle-dependent localization of tissue inhibitor of metalloproteinases-1 immunoreactivity in cultured human gingival fibroblasts. Nagoya J. Med. Sci. 1995, 58, 133–142. [Google Scholar]
- Smith, M.R.; Kung, H.; Durum, S.K.; Colburn, N.H.; Sun, Y. TIMP-3 induces cell death by stabilizing TNF-alpha receptors on the surface of human colon carcinoma cells. Cytokine 1997, 9, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, M.; Poukkula, M.; Baker, A.H.; Kashiwagi, M.; Nagase, H.; Eriksson, J.E.; Kahari, V.M. Tissue inhibitor of metalloproteinases-3 induces apoptosis in melanoma cells by stabilization of death receptors. Oncogene 2003, 22, 2121–2134. [Google Scholar] [CrossRef] [Green Version]
- Alexius-Lindgren, M.; Andersson, E.; Lindstedt, I.; Engstrom, W. The RECK gene and biological malignancy--its significance in angiogenesis and inhibition of matrix metalloproteinases. Anticancer Res. 2014, 34, 3867–3873. [Google Scholar]
- Eiro, N.; Gonzalez, L.; Martinez-Ordonez, A.; Fernandez-Garcia, B.; Gonzalez, L.O.; Cid, S.; Dominguez, F.; Perez-Fernandez, R.; Vizoso, F.J. Cancer-associated fibroblasts affect breast cancer cell gene expression, invasion and angiogenesis. Cell. Oncol. 2018, 41, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Koolwijk, P.; Sidenius, N.; Peters, E.; Sier, C.F.; Hanemaaijer, R.; Blasi, F.; van Hinsbergh, V.W. Proteolysis of the urokinase-type plasminogen activator receptor by metalloproteinase-12: Implication for angiogenesis in fibrin matrices. Blood 2001, 97, 3123–3131. [Google Scholar] [CrossRef] [Green Version]
- Handsley, M.M.; Edwards, D.R. Metalloproteinases and their inhibitors in tumor angiogenesis. Int. J. Cancer 2005, 115, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Wurtz, S.O.; Schrohl, A.S.; Sorensen, N.M.; Lademann, U.; Christensen, I.J.; Mouridsen, H.; Brunner, N. Tissue inhibitor of metalloproteinases-1 in breast cancer. Endocr.-Relat. Cancer 2005, 12, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, A.L.; Stetler-Stevenson, W.G.; Costouros, N.G.; Knezevic, V.; Baibakov, G.; Alexander, H.R., Jr.; Lorang, D.; Hewitt, S.M.; Seo, D.W.; Miller, M.S.; et al. Modulation of tumor-host interactions, angiogenesis, and tumor growth by tissue inhibitor of metalloproteinase 2 via a novel mechanism. Cancer Res. 2004, 64, 4481–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Takahashi, R.; Kondo, S.; Mizoguchi, A.; Adachi, E.; Sasahara, R.M.; Nishimura, S.; Imamura, Y.; Kitayama, H.; Alexander, D.B.; et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001, 107, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, C.A.; Butterfield, C.; Jackson, G.; Moses, M.A. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): Loop 6 is a novel angiogenesis inhibitor. J. Biol. Chem. 2003, 278, 40989–40995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.H.; Ebrahem, Q.; Moore, N.; Murphy, G.; Claesson-Welsh, L.; Bond, M.; Baker, A.; Anand-Apte, B. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): Inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 2003, 9, 407–415. [Google Scholar] [CrossRef]
- Kang, K.H.; Park, S.Y.; Rho, S.B.; Lee, J.H. Tissue inhibitor of metalloproteinases-3 interacts with angiotensin II type 2 receptor and additively inhibits angiogenesis. Cardiovasc. Res. 2008, 79, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Saunders, W.B.; Bohnsack, B.L.; Faske, J.B.; Anthis, N.J.; Bayless, K.J.; Hirschi, K.K.; Davis, G.E. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J. Cell Biol. 2006, 175, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Y.; Chen, B.; Ding, Y.Q. Metastasis-associated factors facilitating the progression of colorectal cancer. Asian Pac. J. Cancer Prev. 2012, 13, 2437–2444. [Google Scholar] [CrossRef] [Green Version]
- Radisky, E.S.; Raeeszadeh-Sarmazdeh, M.; Radisky, D.C. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell. Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef] [Green Version]
- Bosch, R.; Philips, N.; Suarez-Perez, J.A.; Juarranz, A.; Devmurari, A.; Chalensouk-Khaosaat, J.; Gonzalez, S. Mechanisms of photoaging and cutaneous photocarcinogenesis, and photoprotective strategies with phytochemicals. Antioxidants 2015, 4, 248–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland. Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, R.C.; Liu, X.W.; Najy, A.J.; Jung, Y.S.; Won, J.; Chai, K.X.; Fridman, R.; Kim, H.R. TIMP-1 via TWIST1 induces EMT phenotypes in human breast epithelial cells. Mol. Cancer Res. 2014, 12, 1324–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, A.T.; Voura, E.B.; Soloway, P.D.; Watson, K.L.; Khokha, R. MMP inhibitors augment fibroblast adhesion through stabilization of focal adhesion contacts and up-regulation of cadherin function. J. Biol. Chem. 2001, 276, 40215–40224. [Google Scholar] [CrossRef] [Green Version]
- Hojilla, C.V.; Kim, I.; Kassiri, Z.; Fata, J.E.; Fang, H.; Khokha, R. Metalloproteinase axes increase beta-catenin signaling in primary mouse mammary epithelial cells lacking TIMP3. J. Cell Sci. 2007, 120, 1050–1060. [Google Scholar] [CrossRef] [Green Version]
- Malanchi, I.; Huelsken, J. Cancer stem cells: Never Wnt away from the niche. Curr. Opin. Oncol. 2009, 21, 41–46. [Google Scholar] [CrossRef]
- Hancox, R.A.; Allen, M.D.; Holliday, D.L.; Edwards, D.R.; Pennington, C.J.; Guttery, D.S.; Shaw, J.A.; Walker, R.A.; Pringle, J.H.; Jones, J.L. Tumour-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res. 2009, 11, R24. [Google Scholar] [CrossRef] [Green Version]
- Nissinen, L.; Kahari, V.M. Matrix metalloproteinases in inflammation. Biochim. Biophys. Acta 2014, 1840, 2571–2580. [Google Scholar] [CrossRef]
- Folgueras, A.R.; Pendas, A.M.; Sanchez, L.M.; Lopez-Otin, C. Matrix metalloproteinases in cancer: From new functions to improved inhibition strategies. Int. J. Dev. Biol. 2004, 48, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Nagao, Y.; Hisaoka, M.; Matsuyama, A.; Kanemitsu, S.; Hamada, T.; Fukuyama, T.; Nakano, R.; Uchiyama, A.; Kawamoto, M.; Yamaguchi, K.; et al. Association of microRNA-21 expression with its targets, PDCD4 and TIMP3, in pancreatic ductal adenocarcinoma. Mod. Pathol. 2012, 25, 112–121. [Google Scholar] [CrossRef]
- Overall, C.M. Molecular determinants of metalloproteinase substrate specificity: Matrix metalloproteinase substrate binding domains, modules, and exosites. Mol. Biotechnol. 2002, 22, 51–86. [Google Scholar] [CrossRef]
- He, J.; Qin, M.; Chen, Y.; Hu, Z.; Xie, F.; Ye, L.; Hui, T. Epigenetic regulation of matrix metalloproteinases in inflammatory diseases: A narrative review. Cell Biosci. 2020, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Menou, A.; Duitman, J.; Crestani, B. The impaired proteases and anti-proteases balance in idiopathic pulmonary fibrosis. Matrix Biol. 2018, 68–69, 382–403. [Google Scholar] [CrossRef] [PubMed]
- Chow, Y.Y.; Chin, K.Y. The role of inflammation in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2020, 2020, 8293921. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.W.; Wan, R.Z.; Liu, Z.P. Recent research advances in selective matrix metalloproteinase-13 inhibitors as anti-osteoarthritis agents. ChemMedChem 2017, 12, 1157–1168. [Google Scholar] [CrossRef]
- Mitchell, P.G.; Magna, H.A.; Reeves, L.M.; Lopresti-Morrow, L.L.; Yocum, S.A.; Rosner, P.J.; Geoghegan, K.F.; Hambor, J.E. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J. Clin. Investig. 1996, 97, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Mehana, E.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Qu, R.; Chen, X.; Wang, W.; Qiu, C.; Ban, M.; Guo, L.; Vasilev, K.; Chen, J.; Li, W.; Zhao, Y. Ghrelin protects against osteoarthritis through interplay with Akt and NF-kappaB signaling pathways. FASEB J. 2018, 32, 1044–1058. [Google Scholar] [CrossRef] [Green Version]
- Nees, T.A.; Rosshirt, N.; Reiner, T.; Schiltenwolf, M.; Moradi, B. Inflammation and osteoarthritis-related pain. Schmerz 2019, 33, 4–12. [Google Scholar] [CrossRef]
- Hasan, U.H.; Uttra, A.M.; Qasim, S.; Ikram, J.; Saleem, M.; Niazi, Z.R. Phytochemicals targeting matrix metalloproteinases regulating tissue degradation in inflammation and rheumatoid arthritis. Phytomed. Int. J. Phytother. Phytopharm. 2020, 66, 153134. [Google Scholar] [CrossRef]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix metalloproteinases: Role in arthritis. Front. Biosci. A J. Virtual. Libr. 2006, 11, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokarewa, M.; Dahlberg, L.; Tarkowski, A. Expression and functional properties of antibodies to tissue inhibitors of metalloproteinases (TIMPs) in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1014–R1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casagrande, V.; Menghini, R.; Menini, S.; Marino, A.; Marchetti, V.; Cavalera, M.; Fabrizi, M.; Hribal, M.L.; Pugliese, G.; Gentileschi, P.; et al. Overexpression of tissue inhibitor of metalloproteinase 3 in macrophages reduces atherosclerosis in low-density lipoprotein receptor knockout mice. Arter. Thromb. Vasc. Biol. 2012, 32, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezentsev, A.; Nikolaev, A.; Bruskin, S. Matrix metalloproteinases and their role in psoriasis. Gene 2014, 540, 1–10. [Google Scholar] [CrossRef]
- Hattori, N.; Mochizuki, S.; Kishi, K.; Nakajima, T.; Takaishi, H.; D’Armiento, J.; Okada, Y. MMP-13 plays a role in keratinocyte migration, angiogenesis, and contraction in mouse skin wound healing. Am. J. Pathol. 2009, 175, 533–546. [Google Scholar] [CrossRef] [Green Version]
- Zibert, J.R.; Lovendorf, M.B.; Litman, T.; Olsen, J.; Kaczkowski, B.; Skov, L. MicroRNAs and potential target interactions in psoriasis. J. Dermatol. Sci. 2010, 58, 177–185. [Google Scholar] [CrossRef]
- Flisiak, I.; Porebski, P.; Chodynicka, B. Effect of psoriasis activity on metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 in plasma and lesional scales. Acta Derm.-Venereol. 2006, 86, 17–21. [Google Scholar] [CrossRef]
- Uysal, P.; Uzun, H. Relationship between circulating Serpina3g, matrix metalloproteinase-9, and tissue inhibitor of metalloproteinase-1 and -2 with chronic obstructive pulmonary disease severity. Biomolecules 2019, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C.J. Inhibition of MMPs and ADAM/ADAMTS. Biochem. Pharmacol. 2019, 165, 33–40. [Google Scholar] [CrossRef]
- Linton, M.F.; Moslehi, J.J.; Babaev, V.R. Akt signaling in macrophage polarization, survival, and atherosclerosis. Int. J. Mol. Sci. 2019, 20, 2703. [Google Scholar] [CrossRef] [Green Version]
- Noack, M.; Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, X. The role of PI3K/AKT/FOXO signaling in psoriasis. Arch. Dermatol. Res. 2019, 311, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem. J. 1997, 322, 809–814. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
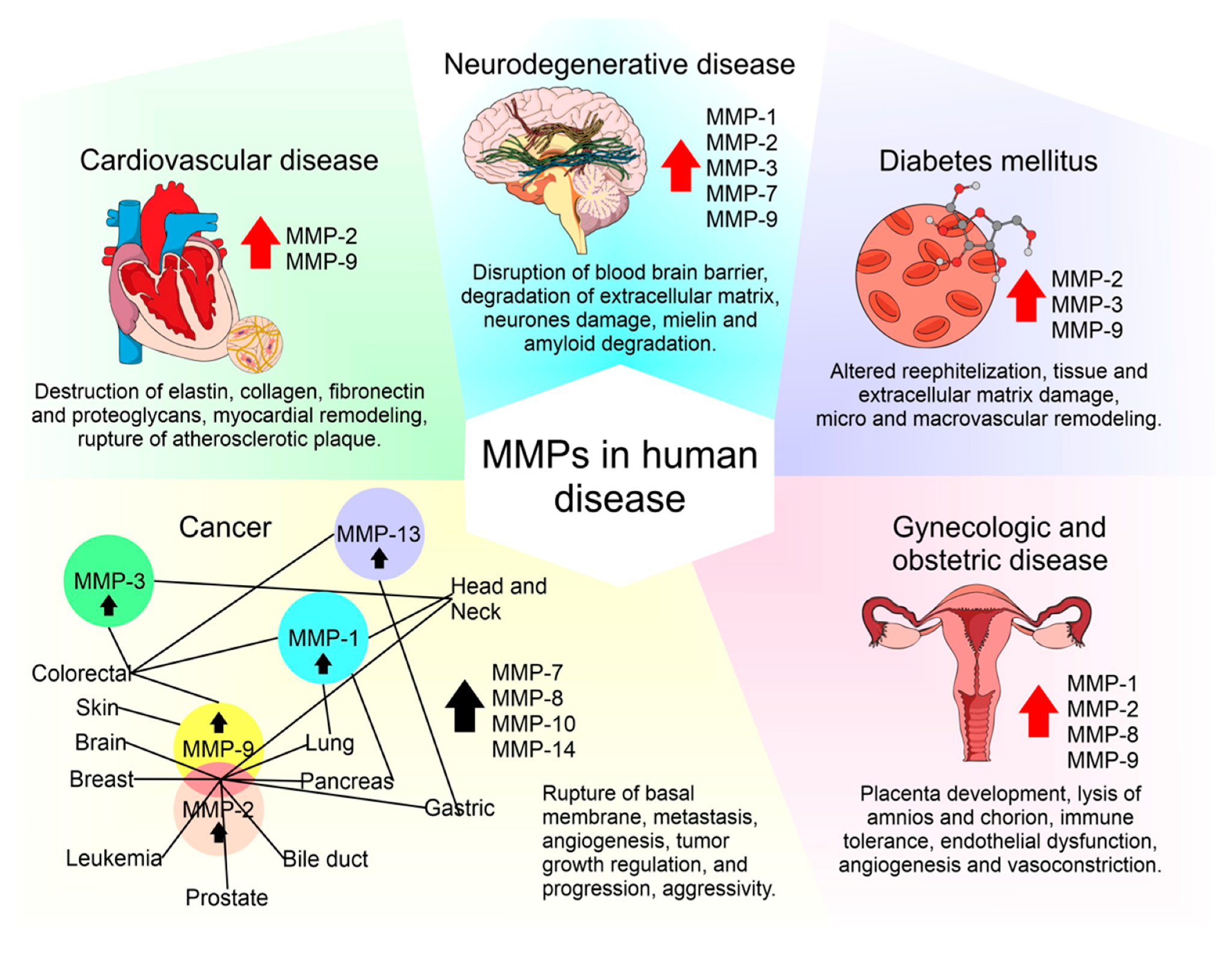{kind=link}
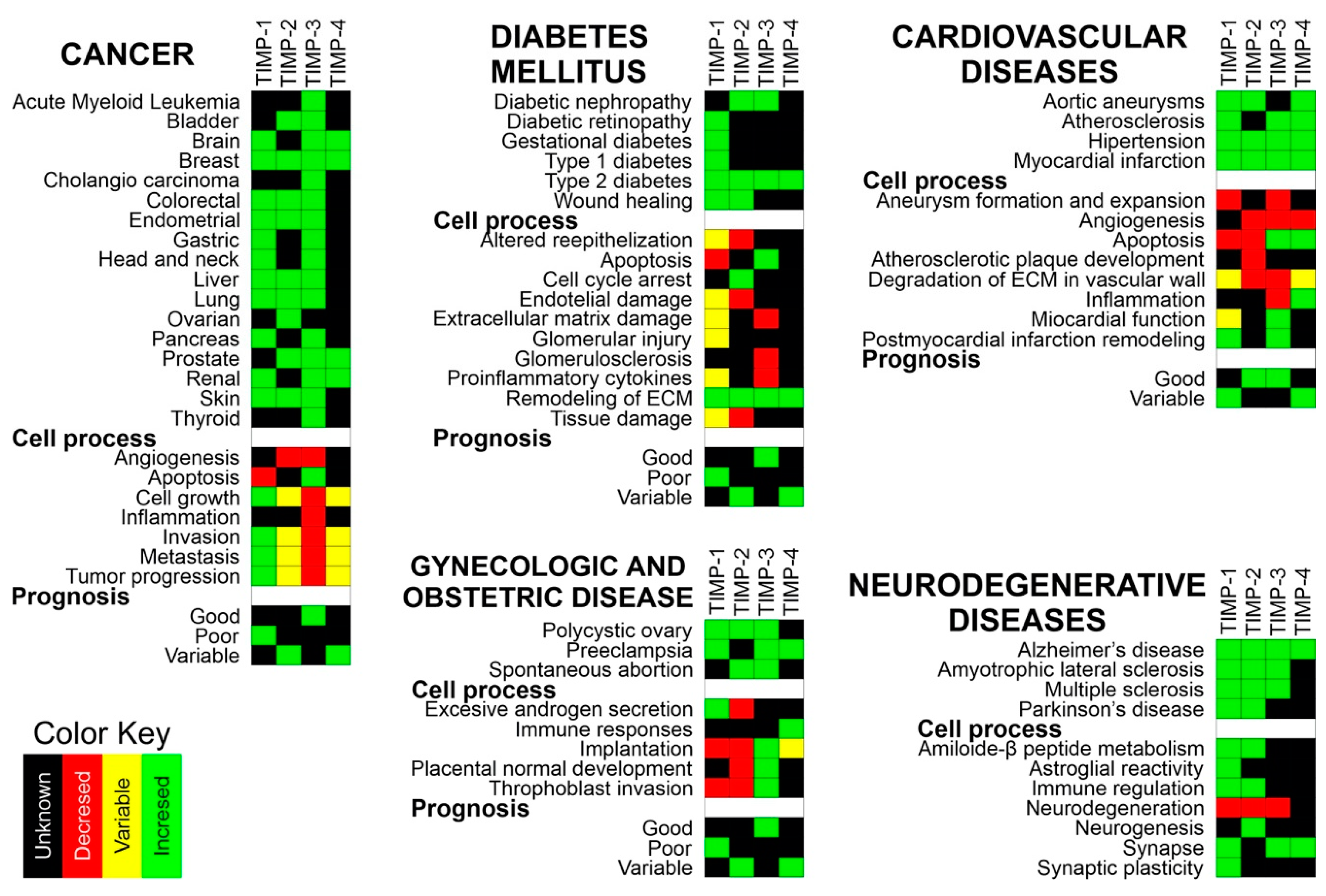{kind=link}
{kind=link}
| Feature | Subgroup | Description | MMPs |
|---|---|---|---|
| Structure | Minimal domain MMPs | Secreted | MMP-7, MMP-26 |
| Simple Hemopexin domain-containing MMPs | Secreted | MMP-1, -3, -8, 10, -12, -13, -18, -19, -22, -20, 27 | |
| Gelating binding MMPs | Secreted | MMP-2, MMP-9 | |
| Furin-activated MMPs | Secreted | MMP-11, MMP-28 | |
| Vitronectin-like insert MMPs | Secreted | MMP-21 | |
| Transmembrane MMPs (with cytoplasmic domain) | Membrane-type MMPs | TM: MMP-14, -15, -16, -24. | |
| glycosylphosphatidylinositol (GPI)-anchored MMPs | Membrane-type MMPs | MMP-17, MMP-25 | |
| Type II transmembrane MMPs (cysteine array and immunoglobulin-like domains) | Membrane-type MMPs | MMP-23 | |
| Substrate | Collagenases (1–4) | Substrates: Col II, II, III, VIII, X, gelatins, aggrecan, entactin | MMP-1, -8,-13, -18 |
| Gelatinases (A and B) | Substrates: gelatins, collagens I, IV, V, VII, X, XI, fibronectin elastin, laminin, vitronectin, proMMPs-9 and 13 | MMP-2, MMP-9 | |
| Stromelysins (1–3) | Substrates: proteoglycans, laminin, gelatins, fibronectin, entactin, collagens III, IV, V, IX, X, XI, proMMPs 1, 8, 9 and 13, vitronectin, α 1-proteinase inhibitor | MMP-3, -10, -11 | |
| Matrilysin | Substrates: proteoglycans, laminin, fibronectin, gelatins, entactin, collagen IV, elastin, tenascin | MMP-7, MMP-26 | |
| Membrane-type MMPs (MT-MMP-1, -3, -4) | Substrates: col I, II, III, gelatins, fibronectin, laminin, proteoglycans, proMMP-2 and 13 | MMP-14,-15, -16, -17, -24, -25 |
| Action Site | MMPs or TIMPs | Function | Secretory Cell | Reference |
|---|---|---|---|---|
| Lung | MMP-1, MMP-9, TIMP-1-3 | Branching Morphogenesis | Fetal epithelium | [11] |
| MMP-1, MMP-2, TIMP-2, TIMP-3 | Pulmonary vascular endothelium and media. | |||
| MMP-1, MMP-9 | Angiogenesis | |||
| MMP-14 | Alveolarization | |||
| Hair (Lanugo) | MMP-2, MMP-9 | Degrade gelatin, collagen IV, V, VII and X, fibronectin, and elastin. | [12,13,14] | |
| TIMP-2 | Inhibits MMP-2 | |||
| TIMP-1 | Inhibits MMP-9 | |||
| Adipocyte | ↓MMP-2, MMP-9 | Adipocyte Clustering | Adipose Tissue | [15,16] |
| ↑MMP-3 | Slow down adipogenic process | |||
| ↑MMP-2, ↑MMP-9 ↓TIMP-1 | Adipocyte differentiation | |||
| ↑TIMP-1 | Involutional adipogenesis | |||
| Bone | ↑MMP-2 | Bone mineralization | [17,18] | |
| MMP-9 | Implantation and bone resorption, bending strength and toughness of bone | Trophoblasts and osteoclasts | ||
| MMP-14 | Membrane-bound protein | Peri-skeletal and skeletal tissue | ||
| MMP-16 | Promoting bone growth and development | Osteoblasts and osteocytes | ||
| ↑TIMP-1 | Regulatory role in bone formation and remodeling. Stimulate the bone-resorbing activity of osteoclasts | Chondrocytes, osteoblasts, osteocytes, and osteoclasts. | ||
| ↑TIMP-2 | Stimulate the bone-resorbing activity of osteoclasts | Hypertrophic chondrocytes and osteoblasts | ||
| Cartilage | MMP-13 | Degradation of type II collagen | Hypertrophic cartilage during endochondral ossification | [19] |
| Membranes | MMP-8 | Membrane rupture (birth) | Polymorphonuclear cells | [20] |
| MMP-13 | Amnion | |||
| ↑MMP-2 | Amnion | |||
| MMP-9 | Amnion, trophoblast and decidual cells | |||
| MMP-3 | ||||
| MMP-2 | Protection | Amnion | [21] |
| MMP | Normal Expression (Tissues) | Expression in Human Diseases (Overexpression) | Reference |
|---|---|---|---|
| MMP-1 | Kidneys, liver, colon, placenta, small intestines, stomach, bladder, and pancreas | Skin, heart, colon, lungs, pancreas, and umbilical cord | [29,30,31,32,33,34,35] |
| MMP-2 | Lymph nodes, heart, adrenals, kidneys, liver, small intestines, colon, esophagus, lungs, ovaries, placenta, breasts, uterus, prostate, skin, bladder, testes, and CNS | Skin, kidneys, brain, heart, endometrium, bone marrow, ovaries, breasts, central nervous system (CNS), lungs, pancreas, circulatory system, and stomach | [29,36,37,38,39,40,41,42,43,44,45] |
| MMP-3 | Tibial nerve, skeletal muscle, small intestine, colon adipocyte, salivary glands, stomach, bladder, and breasts | Kidney, brain, endometrium, colon, and pancreas | [35,36,41,46,47] |
| MMP-7 | Kidneys, lung, bladder, pancreas, prostate, salivary gland, breasts, uterus, ovaries, and testis. | Blood vessels, heart, and pancreas | [48,49,50] |
| MMP-8 | Bone marrow, lungs, and spleen | Skin, colon, and stomach | [51,52,53] |
| MMP-9 | Bone marrow, colon, duodenum, lymph nodes, lungs, adrenals, ovaries, breasts, small intestine, placenta spleen, stomach, CNS, bladder, and kidneys | Skin, kidneys, brain, circulatory system, heart, endometrium, ovaries, colon, breasts, lungs, prostate, cardiovascular system, pancreas, and stomach | [29,32,36,41,42,44,45,48,51,54,55,56,57] |
| MMP-13 | Lung, skin, prostate, small intestine, breast, testis, and bladder | Kidneys, colon, lung, and stomach | [32,36,58,59] |
| MMP-14 | Adrenals, colon, uterus, esophagus, bladder, heart, brain, liver, kidneys, spleen, lungs, lymph node, prostate, skin, small intestine, stomach, breast, ovaries, and testes | Kidneys and brain | [36,60] |
| TIMP-1 | Lymph nodes, brain, heart, arteries, colon, kidneys, liver, lungs, bladder, breasts, skin, ovaries, uterus, prostate, and testis | Breasts, brain, lungs, liver, kidneys, placenta, colon, circulatory system, skin, ovaries, and heart (overexpression) | [61,62,63,64,65,66,67] |
| TIMP-2 | Lymph nodes, brain, heart, arteries, colon, kidneys, liver, breast, ovaries, prostate, and testes | Kidneys, brain, heart, and ovaries, bone marrow and amniotic fluid (over- and underexpression) | [37,39,65,68] |
| TIMP-3 | Brain, heart, colon, kidneys, lungs, liver, breast, ovaries, prostate and testes, and eyes | Kidneys, liver, brain, heart, ovaries, cardiovascular system lungs, placenta, and breasts (underexpression) | [69,70,71,72,73] |
| TIMP-4 | Brain, heart, breast, uterus, ovary, kidney, pancreas, colon, testes, adipose tissue and prostate | Heart, brain and uterus, cardiovascular system and circulatory system (over- and underexpression) | [63,74,75,76,77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. https://doi.org/10.3390/ijms21249739
Cabral-Pacheco GA, Garza-Veloz I, Castruita-De la Rosa C, Ramirez-Acuña JM, Perez-Romero BA, Guerrero-Rodriguez JF, Martinez-Avila N, Martinez-Fierro ML. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. International Journal of Molecular Sciences. 2020; 21(24):9739. https://doi.org/10.3390/ijms21249739
Chicago/Turabian StyleCabral-Pacheco, Griselda A, Idalia Garza-Veloz, Claudia Castruita-De la Rosa, Jesús M Ramirez-Acuña, Braulio A Perez-Romero, Jesús F Guerrero-Rodriguez, Nadia Martinez-Avila, and Margarita L Martinez-Fierro. 2020. "The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases" International Journal of Molecular Sciences 21, no. 24: 9739. https://doi.org/10.3390/ijms21249739