Macrophage Proinflammatory Responses to Microorganisms and Transplanted Organs



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Types of Macrophages
Tumor-Associated Macrophages (TAMs)
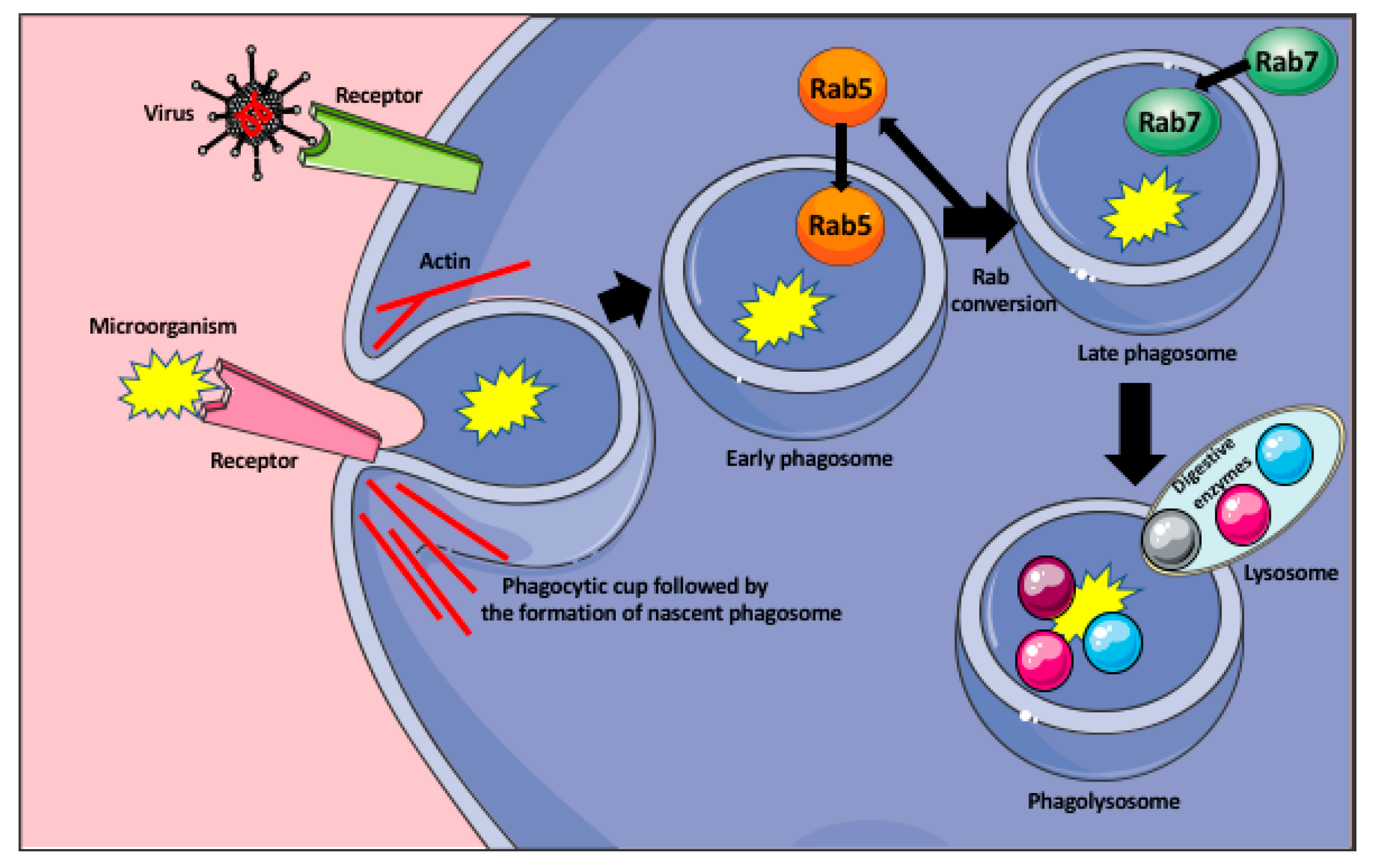
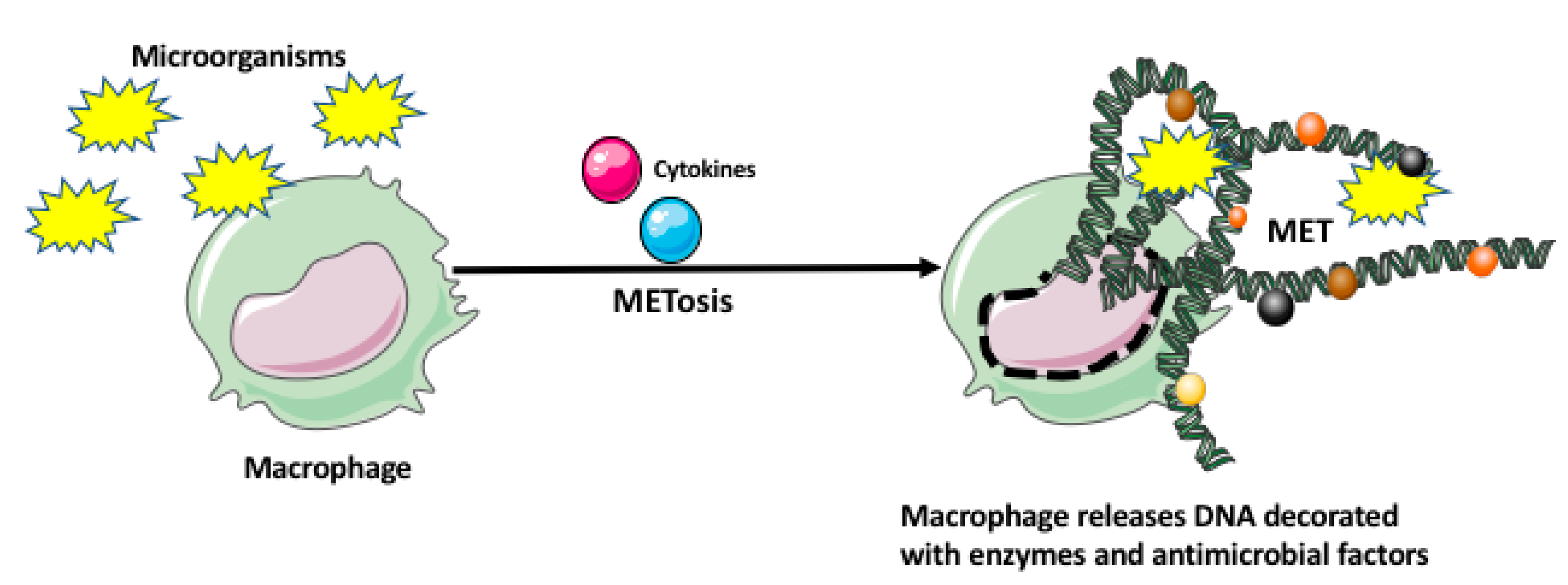
2. Macrophage Response to Microorganisms
2.1. How Bacteria Evade Macrophages
2.2. Macrophage Role in Virus Dissemination
3. The Specificity of Macrophage Response
3.1. Fungal Infection
3.2. Mycobacterium Tuberculosis Complex Infection
3.3. Mycobacterium Leprae Infection
3.4. Brain-Eating Amoeba Naegleria Fowleri Infection
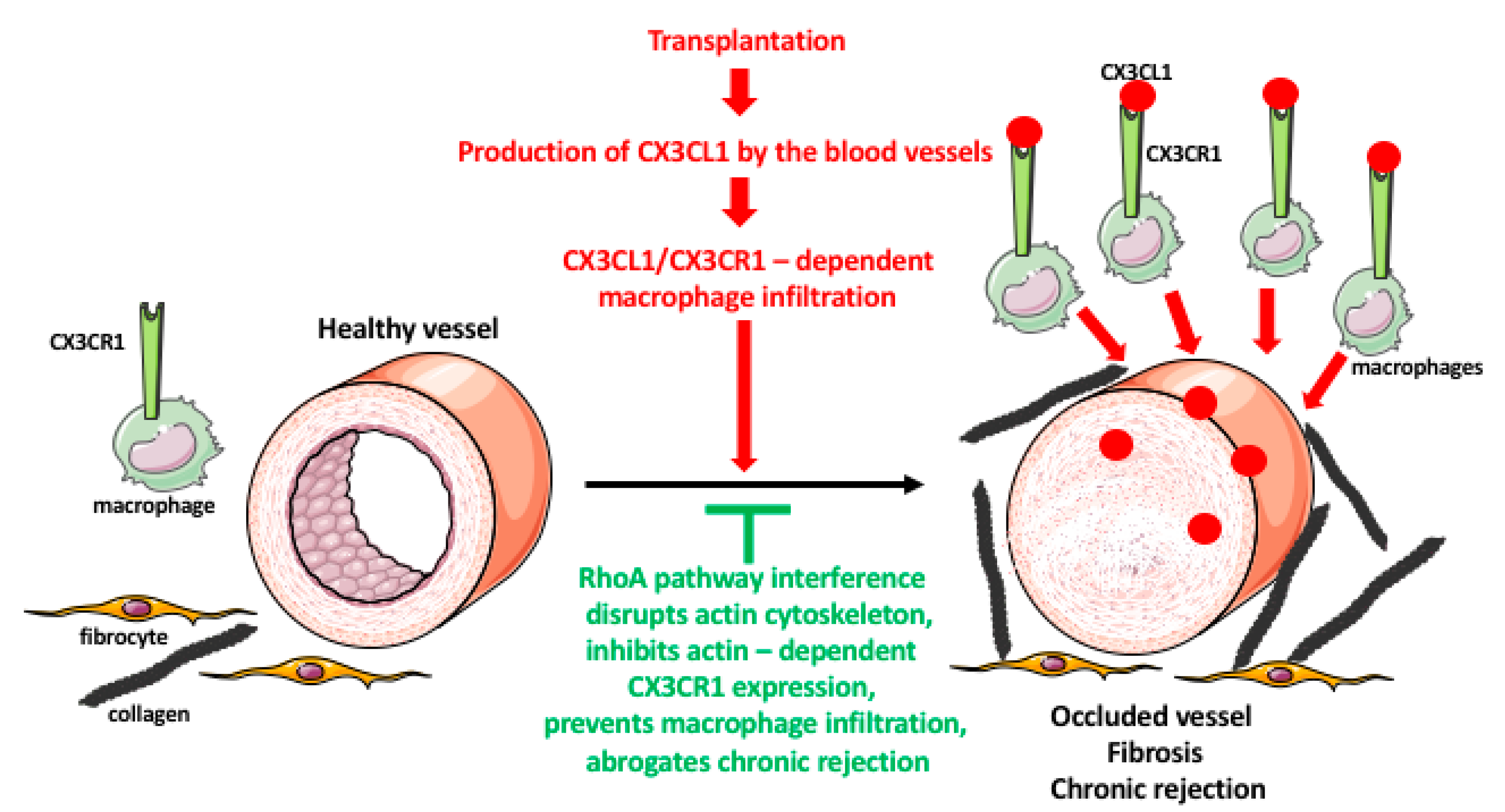
4. Macrophage Response to Organ Transplantation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liao, X.; Shen, Y.; Zhang, R.; Sugi, K.; Vasudevan, N.T.; Alaiti, M.A.; Sweet, D.R.; Zhou, L.; Qing, Y.; Gerson, S.L.; et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2018, 115, E4661–E4669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Panariti, A.; Martin, J.G. Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front. Immunol. 2018, 9, 1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apovian, C.M.; Apovian, C.M. Macrophage functions in lean and obese adipose tissue. Metabolism 2017, 72, 120–143. [Google Scholar] [CrossRef]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer Cells in the Liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar]
- Nagelkerke, S.Q.; Bruggeman, C.W.; Haan, J.M.D.; Mul, E.P.J.; Berg, T.K.V.D.; Van Bruggen, R.; Kuijpers, T.W. Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-γ receptors. Blood Adv. 2018, 2, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sandoval, H.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M.; Kloc, M. The phenotype of peritoneal mouse macrophages depends on the mitochondria and ATP/ADP homeostasis. Cell. Immunol. 2018, 324, 1–7. [Google Scholar] [CrossRef]
- Schliefsteiner, C.; Ibesich, S.; Wadsack, C. Placental Hofbauer Cell Polarization Resists Inflammatory Cues In Vitro. Int. J. Mol. Sci. 2020, 21, 736. [Google Scholar] [CrossRef] [Green Version]
- Kloc, M.; Li, X.C.; Ghobrial, R.M. Are Macrophages Responsible for Cancer Metastasis? J. Immunol. Biol. 2016, 1, 103. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 1–18. [Google Scholar] [CrossRef]
- Kloc, M.; Uosef, A.; Leśniak, M.; Kubiak, J.Z.; Ghobrial, R.M. Reciprocal interaction between mesenchymal stem cells and macrophages. Int. J. Dev. Biol. 2020, (in press). [Google Scholar] [CrossRef]
- Hutchinson, J.A.; Riquelme, P.; Geissler, E.K.; Fändrich, F. Human Regulatory Macrophages. Risk Manag. Technol. 2010, 677, 181–192. [Google Scholar] [CrossRef]
- Maggini, J.; Mirkin, G.; Bognanni, I.; Holmberg, J.; Piazzón, I.M.; Nepomnaschy, I.; Costa, H.; Cañones, C.; Raiden, S.; Vermeulen, M.; et al. Mouse Bone Marrow-Derived Mesenchymal Stromal Cells Turn Activated Macrophages into a Regulatory-Like Profile. PLoS ONE 2010, 5, e9252. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2020, 77, 1919–1932. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a Novel Macrophage Phenotype That Develops in Response to Atherogenic Phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, J.R.; Neto, F.D.L.; Sotto, M.N.; Quaresma, J.A.S. Immunohistochemical characterization of the M4 macrophage population in leprosy skin lesions. BMC Infect. Dis. 2018, 18, 576. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Ural, B.B.; Yeung, S.T.; Damani-Yokota, P.; Devlin, J.C.; De Vries, M.; Vera-Licona, P.; Samji, T.; Sawai, C.M.; Jang, G.; Perez, O.A.; et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci. Immunol. 2020, 5, eaax8756. [Google Scholar] [CrossRef]
- Yeung, S.T.; Ural, B.B.; Damani-Yokota, P.; Devlin, J.C.; De Vries, M.; Samji, T.; Jang, G.; Loke, P.; Dittmann, M.; Reizis, B.; et al. Nerve associated lung resident interstitial macrophage subset exhibits distinct localization and polarization. J. Immunol. 2020, 204 (Suppl. 1), 149.3. [Google Scholar]
- Kloc, M.; Ghobrial, R.M.; Kubiak, J.Z. How nicotine can inhibit cytokine storm in the lungs and prevent or lessen the severity of COVID-19 infection? Immunol. Lett. 2020, 224, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.; Yang, S.-M. Macrophages in Tumor Microenvironments and the Progression of Tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C.F.; Gabay, J. Antimicrobial mechanisms of macrophages. In Mononuclear Phagocytes; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1992; pp. 259–267. [Google Scholar]
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens 2015, 4, 826–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doster, R.S.; Rogers, L.M.; Gaddy, J.A.; Aronoff, D.M. Macrophage Extracellular Traps: A Scoping Review. J. Innate Immun. 2018, 10, 3–13. [Google Scholar] [CrossRef]
- Boe, D.M.; Curtis, B.J.; Chen, M.M.; Ippolito, J.A.; Kovacs, E.J. Extracellular traps and macrophages: New roles for the versatile phagocyte. J. Leukoc. Biol. 2015, 97, 1023–1035. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef]
- Reddick, L.E.; Alto, N.M. Bacteria Fighting Back: How Pathogens Target and Subvert the Host Innate Immune System. Mol. Cell 2014, 54, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Vieira, O.V.; Bucci, C.; Harrison, R.E.; Trimble, W.S.; Lanzetti, L.; Gruenberg, J.; Schreiber, A.D.; Stahl, P.D.; Grinstein, S. Modulation of Rab5 and Rab7 Recruitment to Phagosomes by Phosphatidylinositol 3-Kinase. Mol. Cell. Biol. 2003, 23, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Hackam, D.J.; Rotstein, O.D.; Zhang, W.J.; Demaurex, N.; Woodside, M.; Tsai, O.; Grinstein, S. Regulation of phagosomal acidification. Differential targeting of Na+/H+ exchangers, Na+/K+-ATPases, and vacuolar-type H+-atpases. J. Biol. Chem. 1997, 272, 29810–29820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Haenseler, E.; Leist, T.; Cerny, A.; Hengartner, H.; Althage, A. T cell-mediated hepatitis in mice infected with lymphocytic choriomeningitis virus. Liver cell destruction by H-2 class I-restricted virus-specific cytotoxic T cells as a physiological correlate of the 51Cr-release assay? J. Exp. Med. 1986, 164, 1075–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.A.; Recher, M.; Honke, N.; Scheu, S.; Borkens, S.; Gailus, N.; Krings, C.; Meryk, A.; Kulawik, A.; Cervantes-Barragan, L.; et al. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 2010, 52, 25–32. [Google Scholar] [CrossRef]
- Brophy, M.B.; Nolan, E.M. Manganese and Microbial Pathogenesis: Sequestration by the Mammalian Immune System and Utilization by Microorganisms. ACS Chem. Biol. 2015, 10, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.-D.; Williamson, P.R. Masking the Pathogen: Evolutionary Strategies of Fungi and Their Bacterial Counterparts. J. Fungi 2015, 1, 397–421. [Google Scholar] [CrossRef] [Green Version]
- Ematsuura, M. Structural Modifications of Bacterial Lipopolysaccharide that Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front. Immunol. 2013, 4, 109. [Google Scholar] [CrossRef] [Green Version]
- Montminy, S.W.; Khan, N.N.; McGrath, S.C.; Walkowicz, M.J.; Sharp, F.; Conlon, J.E.; Fukase, K.; Kusumoto, S.; Sweet, C.R.; Miyake, K.; et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 2006, 7, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Rebeil, R.; Ernst, R.K.; Gowen, B.B.; Miller, S.I.; Hinnebusch, B.J. Variation in lipid A structure in the pathogenic yersiniae. Mol. Microbiol. 2004, 52, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Rosowski, E.E. Illuminating Macrophage Contributions to Host-Pathogen Interactions In Vivo: The Power of Zebrafish. Infect. Immun. 2020, 88, e00906-19. [Google Scholar] [CrossRef]
- Levitte, S.; Adams, K.N.; Berg, R.D.; Cosma, C.L.; Urdahl, K.B.; Whitworth, L. Mycobacterial Acid Tolerance Enables Phagolysosomal Survival and Establishment of Tuberculous Infection In Vivo. Cell Host Microbe 2016, 20, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigden, R.C.; Carrasco, C.P.; Summerfield, A.; McCullough, K. Macrophage phagocytosis of foot-and-mouth disease virus may create infectious carriers. Immunology 2002, 106, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Klepper, A.; Branch, A.D. Macrophages and the Viral Dissemination Super Highway. EC Microbiol. 2016, 2, 328–336. [Google Scholar]
- Nikitina, E.; Larionova, I.; Choynzonov, E.L.; Kzhyshkowska, J. Monocytes and Macrophages as Viral Targets and Reservoirs. Int. J. Mol. Sci. 2018, 19, 2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloc, M.; Ghobrial, R.M.; Lewicki, S.; Kubiak, J.Z. Macrophages in diabetes mellitus (DM) and COVID-19: Do they trigger DM? J. Diabetes Metab. Disord. 2020, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, G.; Meng, G. Pathogenic Fungal Infection in the Lung. Front. Immunol. 2019, 10, 1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigliotti, F.; Limper, A.H.; Wright, T. Pneumocystis. Cold Spring Harb. Perspect. Med. 2014, 4, a019828. [Google Scholar] [CrossRef] [Green Version]
- Miceli, M.H. Central Nervous System Infections Due to Aspergillus and Other Hyaline Molds. J. Fungi 2019, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Fei, M.; Yarlagadda, M.; Qi, Z.; Akira, S.; Saijo, S.; Iwakura, Y.; Van Rooijen, N.; Gibson, G.A.; Croix, C.M.S.; et al. Rapid Host Defense against Aspergillus fumigatus Involves Alveolar Macrophages with a Predominance of Alternatively Activated Phenotype. PLoS ONE 2011, 6, e15943. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.N.; Shellito, J.E. Current understanding ofPneumocystisimmunology. Future Microbiol. 2010, 5, 43–65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhu, J.; Imrich, A.; Cushion, M.; Kinane, T.B.; Koziel, H. Pneumocystis activates human alveolar macrophage NF-κB signaling through mannose receptors. Infect. Immun. 2004, 72, 3147–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebron, F.; Vassallo, R.; Puri, V.; Limper, A.H. Pneumocystis carinii cell wall β-glucans initiate macrophage inflammatory responses through NF-κB activation. J. Biol. Chem. 2003, 278, 25001–25008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, C.; Marrero, L.; Swain, S.D.; Harmsen, A.G.; Zheng, M.; Brown, G.D.; Gordon, S.; Shellito, J.E.; Kolls, J.K. Alveolar Macrophage–mediated Killing of Pneumocystis carinii f. sp. muris Involves Molecular Recognition by the Dectin-1 β-Glucan Receptor. J. Exp. Med. 2003, 198, 1677–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, S.; Fujikado, N.; Furuta, T.; Chung, S.-H.; Kotaki, H.; Seki, K.; Sudo, K.; Akira, S.; Adachi, Y.; Ohno, N.; et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 2006, 8, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Forrellad, M.A.; Klepp, L.I.; Gioffré, A.; García, J.S.Y.; Morbidoni, H.R.; Santangelo, M.D.L.P.; Cataldi, A.A.; Bigi, F. Virulence factors of theMycobacterium tuberculosiscomplex. Virulence 2013, 4, 3–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.-X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, R.O.; Schmitz, V.; Silva, B.J.D.A.; Dias, A.A.; De Souza, B.J.; Barbosa, M.G.D.M.; Esquenazi, D.D.A.; Pessolani, M.C.V.; Sarno, E.N. Innate Immune Responses in Leprosy. Front. Immunol. 2018, 9, 518. [Google Scholar] [CrossRef]
- Ridley, D.S.; Jopling, W.H. Classification of leprosy according to immunity. A five-group system. Int. J. Lepr. Other Mycobact. Dis. 1966, 34, 255–273. [Google Scholar]
- Silva, B.J.D.A.; Barbosa, M.G.D.M.; Andrade, P.R.; Ferreira, H.; Nery, J.A.D.C.; Côrte-Real, S.; Da Silva, G.M.S.; Rosa, P.S.; Fabri, M.; Sarno, E.N.; et al. Autophagy Is an Innate Mechanism Associated with Leprosy Polarization. PLoS Pathog. 2017, 13, e1006103. [Google Scholar] [CrossRef]
- Teles, R.M.B.; Graeber, T.G.; Krutzik, S.R.; Montoya, D.; Schenk, M.; Lee, D.J.; Komisopoulou, E.; Kelly-Scumpia, K.; Chun, R.; Iyer, S.S.; et al. Type I Interferon Suppresses Type II Interferon-Triggered Human Anti-Mycobacterial Responses. Science 2013, 339, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Grace, E.; Asbill, S.; Virga, K. Naegleria fowleri: Pathogenesis, Diagnosis, and Treatment Options. Antimicrob. Agents Chemother. 2015, 59, 6677–6681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moseman, E.A. Battling brain-eating amoeba: Enigmas surrounding immunity to Naegleria fowleri. PLoS Pathog. 2020, 16, e1008406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Sohn, H.-J.; Yoo, J.-K.; Kang, H.; Seong, G.-S.; Chwae, Y.-J.; Kim, K.; Park, S.; Shin, H.-J. NLRP3 Inflammasome Activation in THP-1 Target Cells Triggered by Pathogenic Naegleria fowleri. Infect. Immun. 2016, 84, 2422–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Ghobrial, R.M. Chronic allograft rejection: A significant hurdle to transplant success. Burn. Trauma 2014, 2, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Kloc, M.; Li, X.C. Macrophages as Effectors of Acute and Chronic Allograft Injury. Curr. Transplant. Rep. 2016, 3, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Julius, B.K.; Jost, C.H.A.; Sütsch, G.; Brunner, H.P.; Kuenzli, A.; Vogt, P.R.; Turina, M.; Hess, O.M.; Kiowski, W. Incidence, progression and functional significance of cardiac allograft vasculopathy after heart transplantation1. Transplantation 2000, 69, 847–853. [Google Scholar] [CrossRef]
- Mitchell, R.N. Graft Vascular Disease: Immune Response Meets the Vessel Wall. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 19–47. [Google Scholar] [CrossRef]
- Kloc, M.; Li, X.C.; Ghobrial, R.M. RhoA cytoskeletal pathway to transplantation. J. Immunol. Clin. Res. 2014, 2, 1012–2014. [Google Scholar]
- Liu, Y.; Tejpal, N.; You, J.; Li, X.C.; Ghobrial, R.M.; Kloc, M. ROCK inhibition impedes macrophage polarity and functions. Cell. Immunol. 2016, 300, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M.; Chen, W.; Kloc, M. Dissonant response of M0/M2 and M1 bone-marrow-derived macrophages to RhoA pathway interference. Cell Tissue Res. 2016, 366, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Minze, L.J.; Mumma, L.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Mouse macrophage polarity and ROCK1 activity depend on RhoA and non-apoptotic Caspase 3. Exp. Cell Res. 2016, 341, 225–236. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, Y.; Li, X.C.; Kubiak, J.Z.; Ghobrial, R.M.; Kloc, M. Rho-specific Guanine nucleotide exchange factors (Rho-GEFs) inhibition affects macrophage phenotype and disrupts Golgi complex. Int. J. Biochem. Cell Biol. 2017, 93, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, W.; Wu, C.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Kloc, M.; Ghobrial, R.M. Macrophage/monocyte-specific deletion of Ras homolog gene family member A (RhoA) downregulates fractalkine receptor and inhibits chronic rejection of mouse cardiac allografts. J. Hear. Lung Transplant. 2017, 36, 340–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Chen, S.; Chen, W.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Screening RhoA/ROCK inhibitors for the ability to prevent chronic rejection of mouse cardiac allografts. Transpl. Immunol. 2018, 50, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Ghobrial, R.M. The multiple sclerosis (MS) drugs as a potential treatment of ARDS in COVID-19 patients. Mult. Scler. Relat. Disord. 2020, 45, 102437. [Google Scholar] [CrossRef]
- Uosef, A.; Vaughn, N.; Chu, X.; Elshawwaf, M.; Abdelshafy, A.A.A.; Elsaid, K.M.K.; Ghobrial, R.M.; Kloc, M. Siponimod (Mayzent) Downregulates RhoA and Cell Surface Expression of the S1P1 and CX3CR1 Receptors in Mouse RAW 264.7 Macrophages. Arch. Immunol. Ther. Exp. 2020, 68, 19. [Google Scholar] [CrossRef]
- Chen, W.; Ghobrial, R.M.; Li, X.C.; Kloc, M. Inhibition of RhoA and mTORC2/Rictor by Fingolimod (FTY720) induces p21-activated kinase 1, PAK-1 and amplifies podosomes in mouse peritoneal macrophages. Immunobiol. 2018, 223, 634–647. [Google Scholar] [CrossRef]
- Chen, W.; Chen, W.; Chen, S.; Uosef, A.; Ghobrial, R.M.; Kloc, M. Fingolimod (FTY720) prevents chronic rejection of rodent cardiac allografts through inhibition of the RhoA pathway. Transpl. Immunol. 2020, 101347. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloc, M.; Uosef, A.; Kubiak, J.Z.; Ghobrial, R.M. Macrophage Proinflammatory Responses to Microorganisms and Transplanted Organs. Int. J. Mol. Sci. 2020, 21, 9669. https://doi.org/10.3390/ijms21249669
Kloc M, Uosef A, Kubiak JZ, Ghobrial RM. Macrophage Proinflammatory Responses to Microorganisms and Transplanted Organs. International Journal of Molecular Sciences. 2020; 21(24):9669. https://doi.org/10.3390/ijms21249669
Chicago/Turabian StyleKloc, Malgorzata, Ahmed Uosef, Jacek Z. Kubiak, and Rafik M. Ghobrial. 2020. "Macrophage Proinflammatory Responses to Microorganisms and Transplanted Organs" International Journal of Molecular Sciences 21, no. 24: 9669. https://doi.org/10.3390/ijms21249669