Conformation of the Intermediates in the Reaction Catalyzed by Protoporphyrinogen Oxidase: An In Silico Analysis


Abstract
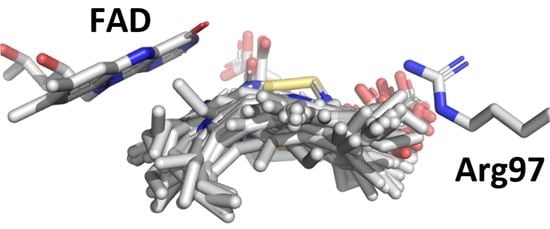
:
1. Introduction
2. Results and Discussion
2.1. Determining the Initial Conformation of Protogen
2.2. Docking of Protogen, Tautomeric Reaction Intermediates and Proto to PPO
2.3. Characterization of the Disordered Loop Crossing the Opening of PPO Binding Domain
3. Conclusions
4. Materials and Methods
4.1. Determining the Initial Conformation of Protogen and Construction of All the Reaction Intermediates
4.2. Docking of Protogen, Tautomeric Reaction Intermediates and Proto to PPO
4.3. Modeling of Disordered Loop Crossing the Opening of PPO Binding Domain
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| copro | Coproporphyrinogen III |
| PPO | Protoporphyrinogen oxidase |
| proto | Protoporphyrin IX |
| protogen | Protoporphyrinogen IX |
References
- Battersby, A.R. Tetrapyrroles: The pigments of life. Nat. Prod. Rep. 2000, 17, 507–526. [Google Scholar] [CrossRef]
- Thunell, S. Porphyrins, porphyrin metabolism and porphyrias. I. Update. Scand. J. Clin. Lab. Investig. 2000, 60, 509–540. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, N.; Tanaka, R.; Grimm, B.; Masuda, T.; Moulin, M.; Smith, A.G.; Tanaka, A.; Terry, M.J. The cell biology of tetrapyrroles: A life and death struggle. Trends Plant Sci. 2010, 15, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.E.; Barker, A.; Tranel, P.J. Origins and structure of chloroplastic and mitochondrial plant protoporphyrinogen oxidases: Implications for the evolution of herbicide resistance. Pest Manag. Sci. 2018, 74, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Rudnick, S.; Cengia, B.; Bonkovsky, H.L. Acute hepatic porphyrias: Review and recent progress. Hepatol. Commun. 2019, 3, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.; Jordan, P.M.; Akhtar, M. Mechanism and stereochemistry of the porphobilinogen deaminase and protoporphyrinogen IX oxidase reactions: Stereospecific manipulation of hydrogen atoms at the four methylene bridges during the biosynthesis of haem. J. Chem. Soc. Perkin Trans. 1984, 1, 2625–2633. [Google Scholar] [CrossRef]
- Koch, M.; Breithaupt, C.; Kiefersauer, R.; Freigang, J.; Huber, R.; Messerschmidt, A. Crystal structure of protoporphyrinogen IX oxidase: A key enzyme in haem and chlorophyll biosynthesis. EMBO J. 2004, 23, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.D.; Whitby, F.G.; Kushner, J.P.; Hill, C.P. Structural basis for tetrapyrrole coordination by uroporphyrinogen decarboxylase. EMBO J. 2003, 22, 6225–6233. [Google Scholar] [CrossRef] [Green Version]
- Hao, G.-F.; Tan, Y.; Yang, S.-G.; Wang, Z.-F.; Zhan, C.-G.; Xi, Z.; Yang, G.-F. Computational and experimental insights into the mechanism of substrate recognition and feedback inhibition of protoporphyrinogen oxidase. PLoS ONE 2013, 8, e69198. [Google Scholar] [CrossRef] [Green Version]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the mitochondrial heme metabolism complex. PLoS ONE 2015, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, G.C.; Andrew, T.L.; Karr, S.W.; Dailey, H.A. Organization of the terminal two enzymes of the heme biosynthetic pathway. Orientation of protoporphyrinogen oxidase and evidence for a membrane complex. J. Biol. Chem. 1988, 263, 3835–3839. [Google Scholar] [PubMed]
- Forli, S.; Botta, M. Lennard-Jones potential and dummy atom settings to overcome the AUTODOCK limitation in treating flexible ring systems. J. Chem. Inf. Model. 2007, 47, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caughey, W.S.; Ibers, J.A. Crystal and molecular structure of the free base porphyrin, protoporphyrin IX dimethyl ester. J. Am. Chem. Soc. 1977, 99, 6639–6645. [Google Scholar] [CrossRef]
- Ismail, A.; Leroux, V.; Smadja, M.; Gonzalez, L.; Lombard, M.; Pierrel, F.; Mellot-Draznieks, C.; Fontecave, M. Coenzyme Q biosynthesis: Evidence for a substrate access channel in the FAD-dependent monooxygenase Coq6. PLoS Comput. Biol. 2016, 12, e1004690. [Google Scholar] [CrossRef] [Green Version]
- Lennon, B.W.; Williams, C.H., Jr.; Ludwig, M.L. Crystal structure of reduced thioredoxin reductase from Escherichia coli: Structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 1999, 8, 2366–2379. [Google Scholar] [CrossRef] [Green Version]
- Eswaramoorthy, S.; Bonanno, J.B.; Burley, S.K.; Swaminathan, S. Mechanism of action of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. USA 2006, 103, 9832–9837. [Google Scholar] [CrossRef] [Green Version]
- Stirling, A.J.; Gilbert, S.E.; Conner, M.; Mallette, E.; Kimber, M.S.; Seah, S.Y.K. A key glycine in bacterial steroid-degrading acyl-CoA dehydrogenases allows flavin-ring repositioning and modulates substrate side chain specificity. Biochemistry 2020, 59, 4081–4092. [Google Scholar] [CrossRef]
- Lans, I.; Medina, M.; Rosta, E.; Hummer, G.; Garcia-Viloca, M.; Lluch, J.M.; González-Lafont, À. Theoretical study of the mechanism of the hydride transfer between ferredoxin–NADP+ reductase and NADP+: The role of Tyr303. J. Am. Chem. Soc. 2012, 134, 20544–20553. [Google Scholar] [CrossRef]
- Lans, I.; Peregrina, J.R.; Medina, M.; Garcia-Viloca, M.; González-Lafont, À.; Lluch, J.M. Mechanism of the hydride transfer between Anabaena Tyr303Ser FNRrd/FNRox and NADP+/H. A combined pre-steady-state kinetic/ensemble-averaged transition-state theory with multidimensional tunneling study. J. Phys. Chem. B 2010, 114, 3368–3379. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S. Comparison of hydride, hydrogen atom, and proton-coupled electron transfer reactions. ChemPhysChem 2002, 3, 33–42. [Google Scholar] [CrossRef]
- Peters, K.S. A theory-experiment conundrum for proton transfer. Acc. Chem. Res. 2009, 42, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Wencewicz, T.A. Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 2013, 30, 175–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archipowa, N.; Kutta, R.J.; Heyes, D.J.; Scrutton, N.S. Stepwise hydride transfer in a biological system: Insights into the reaction mechanism of the light-dependent protochlorophyllide oxidoreductase. Angew. Chem. 2018, 130, 2712–2716. [Google Scholar] [CrossRef] [Green Version]
- Piano, V.; Palfey, B.A.; Mattevi, A. Flavins as covalent catalysts: New mechanisms emerge. Trends Biochem. Sci. 2017, 42, 457–469. [Google Scholar] [CrossRef]
- Ghisla, S.; Massey, V. Mechanisms of flavoprotein-catalyzed reactions. In EJB Reviews 1989; Springer: Berlin/Heidelberg, Germany, 1989; pp. 29–45. [Google Scholar]
- Valdar, W.S. Scoring residue conservation. Proteins 2002, 48, 227–241. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Qin, X.; Tan, Y.; Wang, L.; Wang, Z.; Wang, B.; Wen, X.; Yang, G.; Xi, Z.; Shen, Y. Structural insight into human variegate porphyria disease. FASEB J. 2011, 25, 653–664. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Lohning, A.E.; Levonis, S.M.; Williams-Noonan, B.; Schweiker, S.S. A practical guide to molecular docking and homology modelling for medicinal chemists. Curr. Top. Med. Chem. 2017, 17, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.; Hönigschmid, P.; Schafferhans, A.; Roos, M.; Bernhofer, M. PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res 2014, 42, W337–W343. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Docked Molecules 1 | Distance between N5 of FAD and C10 of Tetrapyrrole (Å) | Distance between Propionate Group and Arg97 (Å) | |
|---|---|---|---|
| Protogen | 23 | 4.4 ± 0.5 | 3.3 ± 0.8 |
| 1a | 22 | 4.3 ± 0.7 | 3.5 ± 0.9 |
| 1c | 12 | 4.3 ± 0.5 | 3.3 ± 0.5 |
| 2a | 17 | 4.5 ± 0.6 | 3.7 ± 1.3 |
| 2d | 20 | 4.3 ± 0.5 | 4.1 ± 1.7 |
| Proto | 26 | 4.6 ± 0.2 | 4.1 ± 0.8 |
| Distance between C10 Methylene H and N5 of FAD (Å) 1 | Distance between Methylene H and Pyrrole N (Å) 1 | |||
|---|---|---|---|---|
| α-H | β-H | α-H | β-H | |
| Protogen | 3.7 ± 0.8 | 5.1 ± 0.7 | - | - |
| 1a | - | - | 3.0 ± 0.1 | 3.3 ± 0.1 |
| 1c | 3.6 ± 0.6 | 5.0 ± 0.8 | - | - |
| 2a | - | - | 2.9 ± 0.2 | 3.3 ± 0.1 |
| 2d | 3.6 ± 0.6 | 5.0 ± 0.7 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barker, A.L.; Barnes, H.; Dayan, F.E. Conformation of the Intermediates in the Reaction Catalyzed by Protoporphyrinogen Oxidase: An In Silico Analysis. Int. J. Mol. Sci. 2020, 21, 9495. https://doi.org/10.3390/ijms21249495
Barker AL, Barnes H, Dayan FE. Conformation of the Intermediates in the Reaction Catalyzed by Protoporphyrinogen Oxidase: An In Silico Analysis. International Journal of Molecular Sciences. 2020; 21(24):9495. https://doi.org/10.3390/ijms21249495
Chicago/Turabian StyleBarker, Abigail L., Hamlin Barnes, and Franck E. Dayan. 2020. "Conformation of the Intermediates in the Reaction Catalyzed by Protoporphyrinogen Oxidase: An In Silico Analysis" International Journal of Molecular Sciences 21, no. 24: 9495. https://doi.org/10.3390/ijms21249495