Rethinking Intellectual Disability from Neuro- to Astro-Pathology



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Astrocyte Function in the Healthy Brain
3. Astrocyte Involvement in Learning and Memory in the Healthy Brain
4. Astrocyte Dysfunction in Neurodevelopmental Disorders
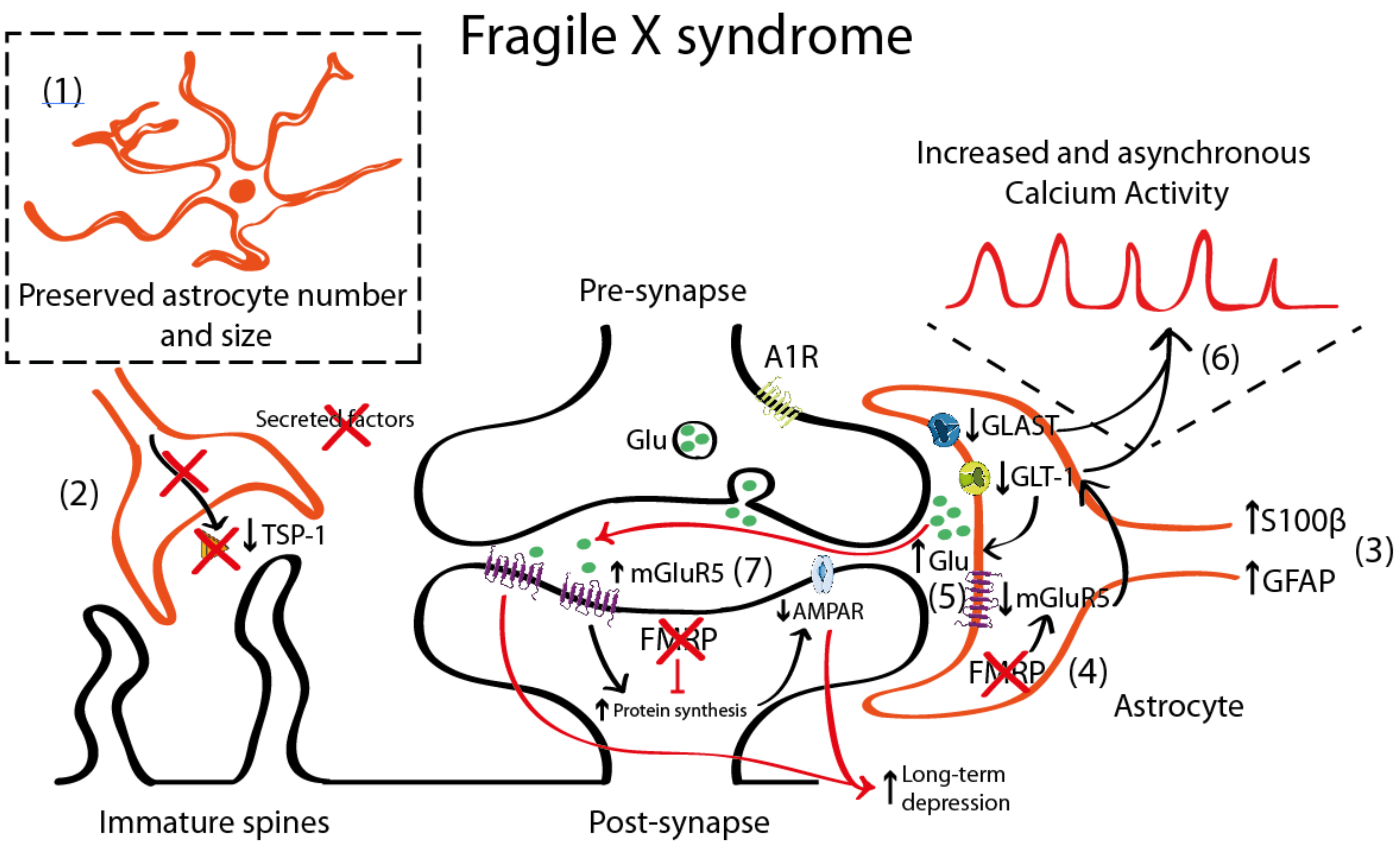
4.1. Astrocyte Pathology in Fragile X Syndrome (FXS)
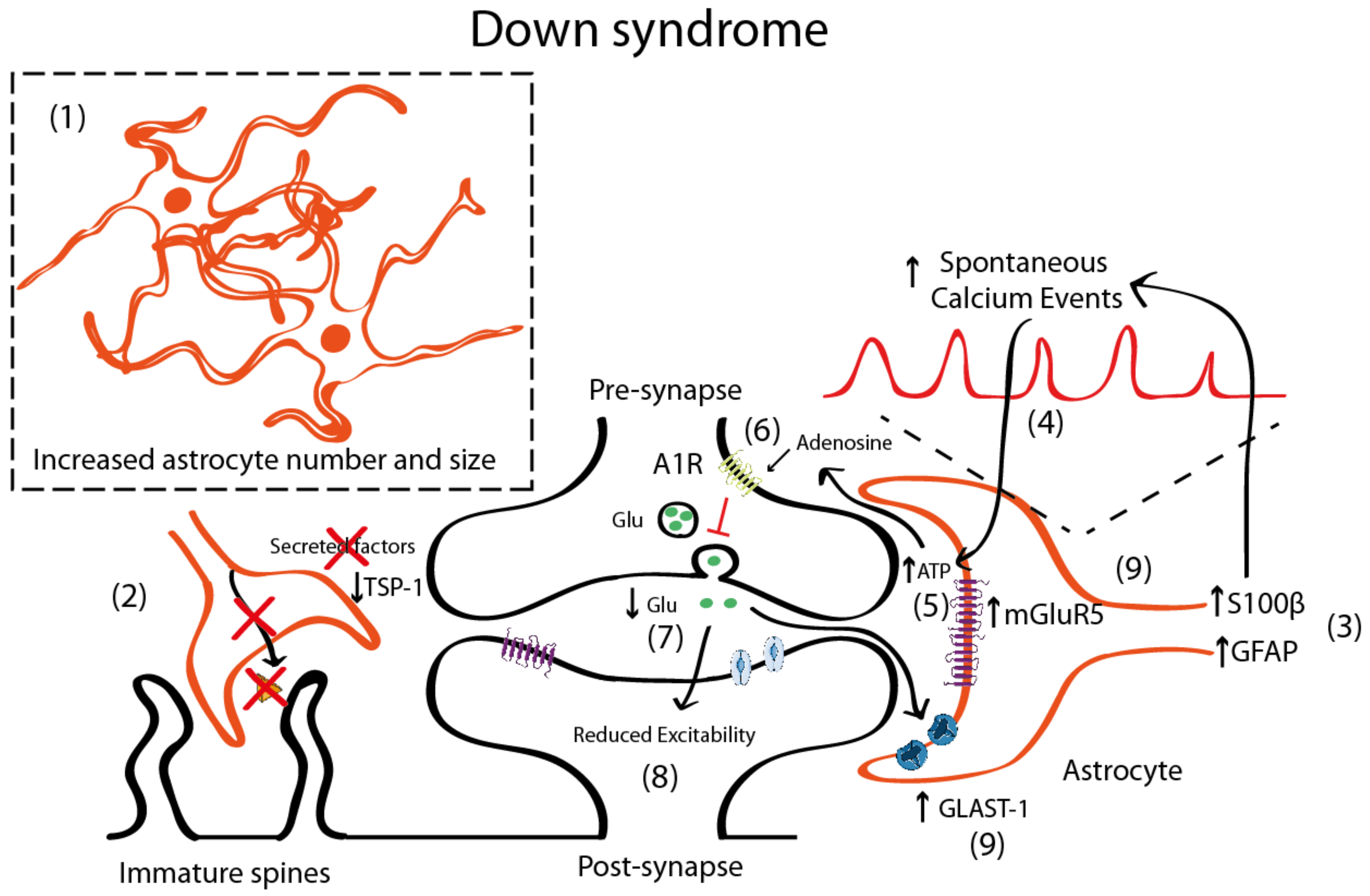
4.2. Astrocyte Pathology in Down Syndrome
5. Astrocytic Phenotypes in DS and FXS: Same Players for Different Phenotypes?
6. Astrocyte Involvement in Memory Pathology in Neurodevelopmental Disorders: A Look into the Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DS | Down syndrome |
| FXS | Fragile X syndrome |
| S100β | S100 calcium-binding protein β |
| HSA21 | Homo Sapiens Autosome 21 |
| GFAP | Glial Fibrillary Acidic Protein |
| GABA | Gamma aminobutyric acid |
| ATP | Adenosine triphosphate |
| PAP | Perisynaptic astrocytic process |
| LTP | Long-term potentiation |
| LTD | Long-term depression |
| TeNT | Tetanus Neurotoxin |
| hM3Dq | human Gq-coupled M3 muscarinic receptor |
| CA1 | Cornu Ammonis 1 |
| CA3 | Cornu Ammonis 3 |
| ACC | Anterior Cingulate Cortex |
| FMR1 | fragile X mental retardation 1 |
| FMRP | Fragile X Mental Retardation Protein |
| CA4 | Cornu Ammonis 4 |
| KO | Knock-out |
| mGluR5 | Metabotropic Glutamate Receptor 5 |
| GLT-1 | Glutamate Transporter 1 |
| GLAST-1 | Glutamate Aspartate Transporter 1 |
| TSP-1 | Thrombospondin-1 |
| iPSC | Induced pluripotent stem cells |
| YRK1A | Dual-specificity tyrosine phosphorylation-regulated kinase-1 |
| STAT | Signal Transducer and Activator of Transcription |
| NPC | Neural Progenitor Cell |
| A1R | Adenosine 1 Receptor |
| DPCPX | 8-Cyclopentyl-1,3-dipropyl xanthine |
References
- Dierssen, M. Top ten discoveries of the year: Neurodevelopmental disorders. Free Neuropathol. 2020, 1, 13. Available online: https://www.uni-muenster.de/Ejournals/index.php/fnp/article/view/2672 (accessed on 26 October 2020).
- Papazoglou, A.; Jacobson, L.A.; McCabe, M.; Kaufmann, W.; Zabel, T.A. To ID or Not to ID? Changes in Classification Rates of Intellectual Disability Using DSM-5. Intellect. Dev. Disabil. 2014, 52, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Ardiles, A.O.; Grabrucker, A.M.; Scholl, F.G.; Rudenko, G.; Borsello, T. Molecular and Cellular Mechanisms of Synaptopathies. Neural Plast. 2017, 2017, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Norris, R.; Gordon, S.; Nithianantharajah, J. Neurodevelopmental synaptopathies: Insights from behaviour in rodent models of synapse gene mutations. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 84, 424–439. [Google Scholar] [CrossRef]
- Dierssen, M.; Ramakers, G.J.A. Dendritic pathology in mental retardation: From molecular genetics to neurobiology. Genes Brain Behav. 2006, 5, 48–60. [Google Scholar] [CrossRef]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef]
- Dierssen, M.; Benavides-Piccione, R.; Martínez-Cué, C.; Estivill, X.; Flórez, J.; Elston, G.; DeFelipe, J. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: Effects of environmental enrichment. Cereb. Cortex 2003, 13, 758–764. [Google Scholar] [CrossRef] [Green Version]
- Belichenko, P.V.; Masliah, E.; Kleschevnikov, A.M.; Villar, A.J.; Epstein, C.J.; Salehi, A.; Mobley, W.C. Synaptic structural abnormalities in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2004, 480, 281–298. [Google Scholar] [CrossRef]
- Catuara-Solarz, S.; Espinosa-Carrasco, J.; Erb, I.; Langohr, K.; Gonzalez, J.R.; Notredame, C.; Dierssen, M. Combined Treatment with Environmental Enrichment and (-)-Epigallocatechin-3-Gallate Ameliorates Learning Deficits and Hippocampal Alterations in a Mouse Model of Down Syndrome. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef] [Green Version]
- Hinton, V.J.; Brown, W.T.; Wisniewski, K.; Rudelli, R.D. Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 1991, 41, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Miller, E.C.; Pozzo-Miller, L. Dendritic spine dysgenesis in Rett syndrome. Front. Neuroanat. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, S.; Putignano, E.; Boggio, E.M.; Giustetto, M.; Pizzorusso, T.; Ratto, G.M. The short-time structural plasticity of dendritic spines is altered in a model of Rett syndrome. Sci. Rep. 2011, 1, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, O.; Torres, M.; Helguera, P.; Coskun, P.; Busciglio, J. A Role for Thrombospondin-1 Deficits in Astrocyte-Mediated Spine and Synaptic Pathology in Down’s Syndrome. PLoS ONE 2010, 5, e14200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagni, F.; Salvalai, M.E.; Giacomini, A.; Emili, M.; Uguagliati, B.; Xia, E.; Grilli, M.; Bartesaghi, R.; Bartesaghi, R. Neonatal treatment with cyclosporine A restores neurogenesis and spinogenesis in the Ts65Dn model of Down syndrome. Neurobiol. Dis. 2019, 129, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Doering, L.C. Astrocytes Prevent Abnormal Neuronal Development in the Fragile X Mouse. J. Neurosci. 2010, 30, 4508–4514. [Google Scholar] [CrossRef]
- Fukuda, T.; Itoh, M.; Ichikawa, T.; Washiyama, K.; Goto, Y.-I. Delayed Maturation of Neuronal Architecture and Synaptogenesis in Cerebral Cortex ofMecp2-Deficient Mice. J. Neuropathol. Exp. Neurol. 2005, 64, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Kleschevnikov, A.M.; Belichenko, P.V.; Villar, A.J.; Epstein, C.J.; Malenka, R.C.; Mobley, W.C. Hippocampal Long-Term Potentiation Suppressed by Increased Inhibition in the Ts65Dn Mouse, a Genetic Model of Down Syndrome. J. Neurosci. 2004, 24, 8153–8160. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.-G.; Toyoda, H.; Ko, S.W.; Ding, H.-K.; Wu, L.-J.; Zhuo, M. Deficits in Trace Fear Memory and Long-Term Potentiation in a Mouse Model for Fragile X Syndrome. J. Neurosci. 2005, 25, 7385–7392. [Google Scholar] [CrossRef]
- Martin, H.G.S.; Lassalle, O.; Brown, J.T.; Manzoni, O.J. Age-Dependent Long-Term Potentiation Deficits in the Prefrontal Cortex of theFmr1Knockout Mouse Model of Fragile X Syndrome. Cereb. Cortex 2015, 26, 2084–2092. [Google Scholar] [CrossRef] [Green Version]
- Weng, S.-M.; McLeod, F.; Bailey, M.E.S.; Cobb, S.R. Synaptic plasticity deficits in an experimental model of rett syndrome: Long-term potentiation saturation and its pharmacological reversal. Neuroscience 2011, 180, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Fred Attneave, M.B.; Hebb, D.O. The Organization of Behavior; A Neuropsychological Theory. Am. J. Psychol. 1950, 63, 633. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ramirez, S.; Pang, P.T.; Puryear, C.B.; Govindarajan, A.; Deisseroth, K.; Tonegawa, S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nat. Cell Biol. 2012, 484, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; De Lagran, M.M.; Farré, M.; Fitó, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, R.; De Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Jacquemont, S.; Berry-Kravis, E.; Hagerman, R.; Von Raison, F.; Gasparini, F.; Apostol, G.; Ufer, M.; Portes, V.D.; Gomez-Mancilla, B. The challenges of clinical trials in fragile X syndrome. Psychopharmacology 2014, 231, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Perea, G.; Araque, A. Astrocytes Potentiate Transmitter Release at Single Hippocampal Synapses. Science 2007, 317, 1083–1086. [Google Scholar] [CrossRef]
- Adamsky, A.; Kol, A.; Kreisel, T.; Doron, A.; Ozeri-Engelhard, N.; Melcer, T.; Refaeli, R.; Horn, H.; Regev, L.; Groysman, M.; et al. Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell 2018, 174, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Kol, A.; Adamsky, A.; Groysman, M.; Kreisel, T.; London, M.; Goshen, I. Astrocytes contribute to remote memory formation by modulating hippocampal–cortical communication during learning. Nat. Neurosci. 2020, 23, 1229–1239. [Google Scholar] [CrossRef]
- Lee, H.S.; Ghetti, A.; Pinto-Duarte, A.; Wang, X.; Dziewczapolski, G.; Galimi, F.; Huitron-Resendiz, S.; Piña-Crespo, J.C.; Roberts, A.J.; Verma, I.M.; et al. Astrocytes contribute to gamma oscillations and recognition memory. Proc. Natl. Acad. Sci. USA 2014, 111, E3343–E3352. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Lau, S.K.M.; Doering, L.C. Astrocyte-secreted thrombospondin-1 modulates synapse and spine defects in the fragile X mouse model. Mol. Brain 2016, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, J.L.; Yu, X.; Gilmore, A.; Bennett, H.; Tjia, M.; Perna, J.F.; Chen, C.-C.; Li, X.; Lu, J.; Zuo, Y. Astrocytic Contributions to Synaptic and Learning Abnormalities in a Mouse Model of Fragile X Syndrome. Biol. Psychiatry 2017, 82, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.D.; Garcia, O.; Tang, C.; Busciglio, J. Dendritic spine pathology and thrombospondin-1 deficits in Down syndrome. Free. Radic. Biol. Med. 2017, 114, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Dossi, E.; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.; Ince, P.; Lace, G.; Forster, G.; Shaw, P.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S.B. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 2010, 31, 578–590. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [Green Version]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Suárez, E.; Caldwell, A.L.M.; Allen, N.J. Role of astrocyte-synapse interactions in CNS disorders. J. Physiol. 2017, 595, 1903–1916. [Google Scholar] [CrossRef] [Green Version]
- Bally, B.P.; Farmer, W.T.; Jones, E.V.; Jessa, S.; Kacerovsky, J.B.; Mayran, A.; Peng, H.; Lefebvre, J.L.; Drouin, J.; Hayer, A.; et al. Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum. Mol. Genet. 2020, 29, 785–802. [Google Scholar] [CrossRef]
- Mizuno, G.O.; Wang, Y.; Shi, G.; Wang, Y.; Sun, J.; Papadopoulos, S.; Broussard, G.J.; Unger, E.K.; Deng, W.; Weick, J.; et al. Aberrant Calcium Signaling in Astrocytes Inhibits Neuronal Excitability in a Human Down Syndrome Stem Cell Model. Cell Rep. 2018, 24, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mito, T.; Becker, L.E. Developmental Changes of S-100 Protein and Glial Fibrillary Acidic Protein in the Brain in Down Syndrome. Exp. Neurol. 1993, 120, 170–176. [Google Scholar] [CrossRef] [PubMed]
- J∅Rgensen, O.S.; Brooksbank, B.W.; Balazs, R. Neuronal plasticity and astrocytic reaction in Down syndrome and Alzheimer disease. J. Neurol. Sci. 1990, 98, 63–79. [Google Scholar] [CrossRef]
- Quinlan, R.A.; Brenner, M.; Goldman, J.E.; Messing, A. GFAP and its role in Alexander disease. Exp. Cell Res. 2007, 313, 2077–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Gerlai, R.; Wojtowicz, J.M.; Marks, A.; Roder, J. Overexpression of a calcium-binding protein, S100 beta, in astrocytes alters synaptic plasticity and impairs spatial learning in transgenic mice. Learn. Mem. 1995, 2, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte–neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef]
- Panatier, A.; Robitaille, R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience 2016, 323, 29–34. [Google Scholar] [CrossRef]
- Haydon, P.G. Glia: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Achour, S.; Pont-Lezica, L.; Béchade, C.; Pascual, O. Is astrocyte calcium signaling relevant for synaptic plasticity? Neuron Glia Biol. 2010, 6, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Mederos, S.; Perea, G. GABAergic-astrocyte signaling: A refinement of inhibitory brain networks. Glia 2019, 67, 1842–1851. [Google Scholar] [CrossRef] [Green Version]
- Cavaccini, A.; Durkee, C.; Kofuji, P.; Tonini, R.; Araque, A. Astrocyte Signaling Gates Long-Term Depression at Corticostriatal Synapses of the Direct Pathway. J. Neurosci. 2020, 40, 5757–5768. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca2+ signalling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Ventura, R.; Harris, K.M. Three-Dimensional Relationships between Hippocampal Synapses and Astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef]
- Theodosis, D.T. Oxytocin-Secreting Neurons: A Physiological Model of Morphological Neuronal and Glial Plasticity in the Adult Hypothalamus. Front. Neuroendocr. 2002, 23, 101–135. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Okabe, S. Direct Astrocytic Contacts Regulate Local Maturation of Dendritic Spines. J. Neurosci. 2007, 27, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; Hülsmann, S.; Kirchhoff, F. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur. J. Neurosci. 2004, 20, 2235–2239. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [Green Version]
- Van Horn, M.R.; Ruthazer, E.S. Glial regulation of synapse maturation and stabilization in the developing nervous system. Curr. Opin. Neurobiol. 2019, 54, 113–119. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, H.; Knöpfel, T.; Endo, S.; Itohara, S. Glial protein S100B modulates long-term neuronal synaptic plasticity. Proc. Natl. Acad. Sci. USA 2002, 99, 4037–4042. [Google Scholar] [CrossRef] [Green Version]
- Morquette, P.; Verdier, D.; Kadala, A.; Féthière, J.; Philippe, A.G.; Robitaille, R.; Kolta, A. An astrocyte-dependent mechanism for neuronal rhythmogenesis. Nat. Neurosci. 2015, 18, 844–854. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Beier, H.; Semkova, I.; Schaper, C.; Krieglstein, J. S-100β protects cultured neurons against glutamate- and staurosporine-induced damage and is involved in the antiapoptotic action of the 5 HT1A-receptor agonist, Bay x 3702. Brain Res. 2000, 858, 121–128. [Google Scholar] [CrossRef]
- Mori, T.; Tan, J.; Arendash, G.W.; Koyama, N.; Nojima, Y.; Town, T. Overexpression of Human S100B Exacerbates Brain Damage and Periinfarct Gliosis after Permanent Focal Ischemia. Stroke 2008, 39, 2114–2121. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, A.; Avilés-Reyes, R.; Angelo, M.F.; Reines, A.G.; Ramos, A.J. S100B alters neuronal survival and dendrite extension via RAGE?mediated NF??B signaling. J. Neurochem. 2011, 117, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Benchenane, K.; Tiesinga, P.H.; Battaglia, F.P. Oscillations in the prefrontal cortex: A gateway to memory and attention. Curr. Opin. Neurobiol. 2011, 21, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Escuela, D.O.B.; Carlsson, J.; Ambrogini, P.; Narváez, M.; Wydra, K.; Tarakanov, A.O.; Li, X.; Millón, C.; Ferraro, L.; Cuppini, R.; et al. Understanding the Role of GPCR Heteroreceptor Complexes in Modulating the Brain Networks in Health and Disease. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Durkee, C.A.; Covelo, A.; Lines, J.; Kofuji, P.; Aguilar, J.; Araque, A. G i/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 2019, 67, 1076–1093. [Google Scholar] [CrossRef] [PubMed]
- Doron, A.; Goshen, I. Investigating the transition from recent to remote memory using advanced tools. Brain Res. Bull. 2018, 141, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Frankland, P.W.; Bontempi, B. The organization of recent and remote memories. Nat. Rev. Neurosci. 2005, 6, 119–130. [Google Scholar] [CrossRef]
- Moscovitch, M.; Cabeza, R.; Winocur, G.; Nadel, L. Episodic Memory and Beyond: The Hippocampus and Neocortex in Transformation. Annu. Rev. Psychol. 2016, 67, 105–134. [Google Scholar] [CrossRef] [Green Version]
- Araujo, B.H.S.; Kaid, C.; De Souza, J.S.; Da Silva, S.G.; Goulart, E.; Caires, L.C.J.; Musso, C.M.; Torres, L.B.; Ferrasa, A.; Herai, R.; et al. Down Syndrome iPSC-Derived Astrocytes Impair Neuronal Synaptogenesis and the mTOR Pathway In Vitro. Mol. Neurobiol. 2017, 55, 5962–5975. [Google Scholar] [CrossRef]
- Murphy, G.M.; Ellis, W.G.; Lee, Y.-L.; Stultz, K.E.; Shrivastava, R.; Tinklenberg, J.R.; Eng, L.F. Chapter 40: Astrocytic Gliosis in the Amygdala in Down’s Syndrome and Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 1992; Volume 94, pp. 475–483. [Google Scholar]
- Guttenplan, K.A.; Stafford, B.K.; El-Danaf, R.N.; Adler, D.I.; Münch, A.E.; Weigel, M.K.; Huberman, A.D.; Liddelow, S.A. Neurotoxic Reactive Astrocytes Drive Neuronal Death after Retinal Injury. Cell Rep. 2020, 31, 107776. [Google Scholar] [CrossRef]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Sofroniew, M. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekny, M.; Pekna, M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, W.; Sheng, J.G.; McKenzie, J.E.; Royston, M.C.; Gentleman, S.M.; Brumback, R.A.; Cork, L.C.; Del Bigio, M.R.; Roberts, G.W.; Mrak, R.E. Life-long Overexpression of S100β in Down’s Syndrome: Implications for Alzheimer Pathogenesis. Neurobiol. Aging 1999, 19, 401–405. [Google Scholar] [CrossRef]
- Banerjee, A.; Ifrim, M.F.; Valdez, A.N.; Raj, N.; Bassell, G.J. Aberrant RNA translation in fragile X syndrome: From FMRP mechanisms to emerging therapeutic strategies. Brain Res. 2018, 1693, 24–36. [Google Scholar] [CrossRef]
- Dockendorff, T.C.; Labrador, M. The Fragile X Protein and Genome Function. Mol. Neurobiol. 2018, 56, 711–721. [Google Scholar] [CrossRef]
- Reiss, A.L.; Aylward, E.; Freund, L.S.; Joshi, P.K.; Bryan, R.N. Neuroanatomy of fragile X syndrome: The posterior fossa. Ann. Neurol. 1991, 29, 26–32. [Google Scholar] [CrossRef]
- Sabaratnam, M. Pathological and neuropathological findings in two males with fragile-X syndrome. J. Intellect. Disabil. Res. 2000, 44, 81–85. [Google Scholar] [CrossRef]
- The Dutch-Belgian Fragile X Consorthrum. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 1994, 78. [CrossRef]
- Higashimori, H.; Morel, L.; Huth, J.; Lindemann, L.; Dulla, C.; Taylor, A.; Freeman, M.; Yang, Y. Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum. Mol. Genet. 2013, 22, 2041–2054. [Google Scholar] [CrossRef] [Green Version]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veloz, M.F.V.; Buijsen, R.A.; Willemsen, R.; Cupido, A.; Bosman, L.W.; Koekkoek, S.K.E.; Potters, J.W.; Oostra, B.A.; De Zeeuw, C.I. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 2012, 11, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Pop, A.S.; Levenga, J.; De Esch, C.E.F.; Buijsen, R.A.; Nieuwenhuizen, I.M.; Li, T.; Isaacs, A.; Gasparini, F.; Oostra, B.A.; Willemsen, R. (Rob) Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology 2012, 231, 1227–1235. [Google Scholar] [CrossRef]
- Aloisi, E.; Le Corf, K.; Dupuis, J.; Zhang, P.; Ginger, M.; Labrousse, V.; Spatuzza, M.; Haberl, M.G.; Costa, L.; Shigemoto, R.; et al. Altered surface mGluR5 dynamics provoke synaptic NMDAR dysfunction and cognitive defects in Fmr1 knockout mice. Nat. Commun. 2017, 8, 1103. [Google Scholar] [CrossRef] [Green Version]
- Carroll, R.C.; Lissin, D.V.; Von Zastrow, M.; Nicoll, R.A.; Malenka, R.C. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat. Neurosci. 1999, 2, 454–460. [Google Scholar] [CrossRef]
- Snyder, E.M.; Philpot, B.D.; Huber, K.M.; Dong, X.; Fallon, J.R.; Bear, M.F. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 2001, 4, 1079–1085. [Google Scholar] [CrossRef]
- Zakharenko, S.S.; Zablow, L.; Siegelbaum, S.A. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 2002, 35, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [Green Version]
- Yuskaitis, C.J.; Beurel, E.; Jope, R.S. Evidence of reactive astrocytes but not peripheral immune system activation in a mouse model of Fragile X syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 1006–1012. [Google Scholar] [CrossRef]
- Pacey, L.K.K.; Guan, S.; Tharmalingam, S.; Thomsen, C.; Hampson, D.R. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. 2015, 5, e00400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Hulsizer, S.; Cui, Y.; Pretto, D.L.; Kim, K.H.; Hagerman, P.J.; Tassone, F.; Pessah, I.N. Enhanced Asynchronous Ca2+Oscillations Associated with Impaired Glutamate Transport in Cortical Astrocytes ExpressingFmr1Gene Premutation Expansion. J. Biol. Chem. 2013, 288, 13831–13841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asch, A.S.; Leung, L.L.; Shapiro, J.; Nachman, R.L. Human brain glial cells synthesize thrombospondin. Proc. Natl. Acad. Sci. USA 1986, 83, 2904–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Ge, J.; Summers, J.B.; Li, F.; Liu, X.; Ma, P.; Kaminski, J.; Zhuang, J. TSP-1 Secreted by Bone Marrow Stromal Cells Contributes to Retinal Ganglion Cell Neurite Outgrowth and Survival. PLoS ONE 2008, 3, e2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.C.; Tucker, R.P. The thrombospondin type 1 repeat (TSR) superfamily: Diverse proteins with related roles in neuronal development. Dev. Dyn. 2000. [Google Scholar] [CrossRef]
- Lu, Z.; Kipnis, J. Thrombospondin 1—A key astrocyte-derived neurogenic factor. FASEB J. 2010, 24, 1925–1934. [Google Scholar] [CrossRef] [Green Version]
- Pinter, J.D.; Eliez, S.; Schmitt, J.E.; Capone, G.T.; Reiss, A.L. Neuroanatomy of Down’s Syndrome: A High-Resolution MRI Study. Am. J. Psychiatry 2001, 158, 1659–1665. [Google Scholar] [CrossRef]
- Raz, N.; Torres, I.J.; Briggs, S.D.; Spencer, W.D.; Thornton, A.E.; Loken, W.J.; Gunning, F.M.; McQuain, J.D.; Driesen, N.R.; Acker, J.D. Selective neuroanatornic abnormalities in Down’s syndrome and their cognitive correlates: Evidence from MRI morphometry. Neurology 1995, 45, 356–366. [Google Scholar] [CrossRef]
- De Lagran, M.M.; Benavides-Piccione, R.; Ballesteros-Yanez, I.; Calvo, M.; Morales, M.; Fillat, C.; DeFelipe, J.; Ramakers, G.J.A.; Dierssen, M. Dyrk1A Influences Neuronal Morphogenesis Through Regulation of Cytoskeletal Dynamics in Mammalian Cortical Neurons. Cereb. Cortex 2012, 22, 2867–2877. [Google Scholar] [CrossRef] [Green Version]
- Lott, I.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L.; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockrow, J.P.; Fortress, A.M.; Granholm, A.-C.E. Age-Related Neurodegeneration and Memory Loss in Down Syndrome. Curr. Gerontol. Geriatr. Res. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Goodison, K.L.; Parhad, I.M.; White, C.L.; Sima, A.A.F.; Clark, A.W. Neuronal and Glial Gene Expression in Neocortex of Downʼs Syndrome and Alzheimerʼs Disease. J. Neuropathol. Exp. Neurol. 1993, 52, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kanaumi, T.; Milenkovic, I.; Adle-Biassette, H.; Aronica, E.; Kovacs, G.G. Non-neuronal cell responses differ between normal and Down syndrome developing brains. Int. J. Dev. Neurosci. 2013, 31, 796–803. [Google Scholar] [CrossRef]
- Guidi, S.; Bonasoni, P.; Ceccarelli, C.; Santini, D.; Gualtieri, F.; Ciani, E.; Bartesaghi, R. RESEARCH ARTICLE: Neurogenesis Impairment and Increased Cell Death Reduce Total Neuron Number in the Hippocampal Region of Fetuses with Down Syndrome. Brain Pathol. 2007, 18, 180–197. [Google Scholar] [CrossRef]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Sailani, M.R.; Dahoun, S.; Santoni, F.A.; Gimelli, S.; Guipponi, M.; Pelte, M.F.; Bena, F.S.; et al. Modelling and rescuing neurodevelopmental defect of D own syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 2013, 6, 259–277. [Google Scholar] [CrossRef]
- Kurabayashi, N.; Nguyen, M.D.; Sanada, K. DYRK 1A overexpression enhances STAT activity and astrogliogenesis in a Down syndrome mouse model. EMBO Rep. 2015, 16, 1548–1562. [Google Scholar] [CrossRef]
- Lorenzi, H.A.; Reeves, R.H. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res. 2006, 1104, 153–159. [Google Scholar] [CrossRef]
- Contestabile, A.; Fíla, T.; Cappellini, A.; Bartesaghi, R.; Ciani, E. Widespread impairment of cell proliferation in the neonate Ts65Dn mouse, a model for Down syndrome. Cell Prolif. 2009, 42, 171–181. [Google Scholar] [CrossRef]
- Anderson, A.J.; Stoltzner, S.; Lai, F.; Su, J.; Nixon, R.A. Morphological and biochemical assessment of DNA damage and apoptosis in Down syndrome and Alzheimer disease, and effect of postmortem tissue archival on TUNEL. Neurobiol. Aging 2000, 21, 511–524. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.-H.; Haydon, P.G. Synaptic Islands Defined by the Territory of a Single Astrocyte. J. Neurosci. 2007, 27, 6473–6477. [Google Scholar] [CrossRef] [Green Version]
- Colombo, J.A.; Reisin, H.D.; Jones, M.; Bentham, C. Development of interlaminar astroglial processes in the cerebral cortex of control and Down’s syndrome human cases. Exp. Neurol. 2005, 193, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; Wang, H.-K.; Ye, C.-Q.; Ge, W.; Chen, Y.; Jiang, Z.-L.; Wu, C.-P.; Poo, M.-M.; Duan, S. ATP Released by Astrocytes Mediates Glutamatergic Activity-Dependent Heterosynaptic Suppression. Neuron 2003, 40, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.-Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic Purinergic Signaling Coordinates Synaptic Networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Dunwiddie, T.V.; Masino, S.A. The Role and Regulation of Adenosine in the Central Nervous System. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, B.E.; Shuttleworth, C.W. Adenosine receptor activation is responsible for prolonged depression of synaptic transmission after spreading depolarization in brain slices. Neuroscience 2012, 223, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Risser, D.; Lubec, G.; Cairns, N.; Herrera-Marschitz, M. Excitatory amino acids and monoamines in parahippocampal gyrus and frontal cortical pole of adults with down syndrome. Life Sci. 1997, 60, 1231–1237. [Google Scholar] [CrossRef]
- Reynolds, G.P.; Warner, C.E. Amino acid neurotransmitter deficits in adult Down’s syndrome brain tissue. Neurosci. Lett. 1988, 94, 224–227. [Google Scholar] [CrossRef]
- Begni, B.; Brighina, L.; Fumagalli, L.; Andreoni, S.; Castelli, E.; Francesconi, C.; Del Bo, R.; Bresolin, N.; Ferrarese, C. Altered glutamate uptake in peripheral tissues from Down Syndrome patients. Neurosci. Lett. 2003, 343, 73–76. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014, 5, 4430. [Google Scholar] [CrossRef] [Green Version]
- Belichenko, P.V.; Kleschevnikov, A.M.; Masliah, E.; Wu, C.; Takimoto-Kimura, R.; Salehi, A.; Mobley, W.C. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2009, 512, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Harashima, C.; Jacobowitz, D.M.; Stoffel, M.; Chakrabarti, L.; Haydar, T.F.; Siarey, R.J.; Galdzicki, Z. Elevated Expression of the G-Protein-Activated Inwardly Rectifying Potassium Channel 2 (GIRK2) in Cerebellar Unipolar Brush Cells of a Down Syndrome Mouse Model. Cell. Mol. Neurobiol. 2006, 26, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Best, T.K.; Cramer, N.P.; Chakrabarti, L.; Haydar, T.F.; Galdzicki, Z. Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp. Neurol. 2011, 233, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.C.; Grybko, M.J. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: A model of Down syndrome. Neurosci. Lett. 2005, 382, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Kurt, M.; Davies, D.C.; Kidd, M.; Dierssen, M.; Flórez, J. Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res. 2000, 858, 191–197. [Google Scholar] [CrossRef]
- Oka, A.; Takashima, S. The up-regulation of metabotropic glutamate receptor 5 (mGluR5) in Down’s syndrome brains. Acta Neuropathol. 1999, 97, 275–278. [Google Scholar] [CrossRef]
- Gibson, J.R.; Bartley, A.F.; Hays, S.A.; Huber, K.M. Imbalance of Neocortical Excitation and Inhibition and Altered UP States Reflect Network Hyperexcitability in the Mouse Model of Fragile X Syndrome. J. Neurophysiol. 2008, 100, 2615–2626. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Paluszkiewicz, S.M.; Martin, B.S.; Kaufmann, W.E.; Corbin, J.G.; Huntsman, M.M. Defective GABAergic Neurotransmission and Pharmacological Rescue of Neuronal Hyperexcitability in the Amygdala in a Mouse Model of Fragile X Syndrome. J. Neurosci. 2010, 30, 9929–9938. [Google Scholar] [CrossRef] [Green Version]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Byerly, M.J.; Sweeney, J.A. Reduced habituation of auditory evoked potentials indicate cortical hyper-excitability in Fragile X Syndrome. Transl. Psychiatry 2016, 6, e787. [Google Scholar] [CrossRef]
- Iyer, A.M.; Van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Lim, D.; Genazzani, A.A.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. Metabotropic Glutamate Receptor 5 in Down’s Syndrome Hippocampus During Development: Increased Expression in Astrocytes. Curr. Alzheimer Res. 2014, 11, 694–705. [Google Scholar] [CrossRef]
- Piers, T.M.; Kim, D.H.; Kim, B.C.; Regan, P.; Whitcomb, D.J.; Cho, K. Translational Concepts of mGluR5 in Synaptic Diseases of the Brain. Front. Pharm. 2012, 3, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi-Yamaguchi, Y.; Furuichi, T. The Homer family proteins. Genome Biol. 2007, 8, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, H.; Lee, H.-R.; Gee, H.Y.; Mah, W.; Kim, J.-I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nat. Cell Biol. 2012, 486, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.J.; Dong, E.N.; Meiler, J.; Conn, P.J. Allosteric modulation of metabotropic glutamate receptors: Structural insights and therapeutic potential. Neuropharmacology 2011, 60, 66–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, K.M. Role for Rapid Dendritic Protein Synthesis in Hippocampal mGluR-Dependent Long-Term Depression. Science 2000, 288, 1254–1256. [Google Scholar] [CrossRef] [Green Version]
- Lepannetier, S.; Gualdani, R.; Tempesta, S.; Schakman, O.; Seghers, F.; Kreis, A.; Yerna, X.; Slimi, A.; De Clippele, M.; Tajeddine, N.; et al. Activation of TRPC1 Channel by Metabotropic Glutamate Receptor mGluR5 Modulates Synaptic Plasticity and Spatial Working Memory. Front. Cell. Neurosci. 2018, 12, 318. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Lipton, J.O.; Boyle, L.M.; Madsen, J.R.; Goldenberg, M.C.; Pascual-Leone, A.; Sahin, M.; Rotenberg, A. Direct current stimulation induces mGluR5-dependent neocortical plasticity. Ann. Neurol. 2016, 80, 233–246. [Google Scholar] [CrossRef]
- Mor-Shaked, H.; Eiges, R. Reevaluation of FMR1 Hypermethylation Timing in Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; McCarter, M.; Lian, G.; Esposito, G.; Capoccia, E.; Delli-Bovi, L.C.; Hecht, J.; Sheen, V. Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet. 2016, 25, 1714–1727. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Lee, Y.K.; Lim, Y.C.; Zheng, Z.; Lin, X.M.; Ng, D.P.Y.; Holbrook, J.D.; Law, H.Y.; Kwek, K.Y.C.; Yeo, G.S.H.; et al. Global DNA Hypermethylation in Down Syndrome Placenta. PLoS Genet. 2013, 9, e1003515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laufer, B.I.; Hwang, H.; Ciernia, A.V.; Mordaunt, C.E.; LaSalle, J.M. Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights. Epigenetics 2019, 14, 672–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Toma, I.; Ortega, M.; Catuara-Solarz, S.; Sierra, C.; Sabidó, E.; Dierssen, M. Re-establishment of the epigenetic state and rescue of kinome deregulation in Ts65Dn mice upon treatment with green tea extract and environmental enrichment. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Blanco, Á.; Dierssen, M. Rethinking Intellectual Disability from Neuro- to Astro-Pathology. Int. J. Mol. Sci. 2020, 21, 9039. https://doi.org/10.3390/ijms21239039
Fernández-Blanco Á, Dierssen M. Rethinking Intellectual Disability from Neuro- to Astro-Pathology. International Journal of Molecular Sciences. 2020; 21(23):9039. https://doi.org/10.3390/ijms21239039
Chicago/Turabian StyleFernández-Blanco, Álvaro, and Mara Dierssen. 2020. "Rethinking Intellectual Disability from Neuro- to Astro-Pathology" International Journal of Molecular Sciences 21, no. 23: 9039. https://doi.org/10.3390/ijms21239039