1. Introduction
In human proteome, the pleckstrin homology (PH) domain is present in nearly 300 proteins involved in diverse physiological processes. A typical PH domain comprises about 100 amino acid residues structured into a seven-stranded antiparallel β-sheet and a C-terminal α-helix [
1]. The most well-known physiological property of PH domains is recognition of phosphoinositides with high affinity and specificity, which regulate localization of PH domain-containing proteins to and from intracellular membranes [
2]. PH domains recognizing phosphoinositide display polar surface charge distribution around its β1–β2 region for accommodation of negatively charged head groups of phosphoinositides. On the other hand, PH domains also contribute to binding interaction partner proteins, including G-protein coupled receptors (GPCRs), GTPase exchange factors (GEFs), and focal adhesion kinase (FAK) [
3,
4]. Phosphotyrosine-containing peptides and polyproline helixes may bind on pockets created by the β-sheet and the C-terminal α-helix of the PH domain [
5]. Considering its pivotal role in regulating protein subcellular localization, mutations within the PH domains can lead to aberrant cell signaling [
4].
FAM129B (family with sequence similarity 129, member B), also known as MINERVA (melanoma invasion by ERK), is a member of a small family of proteins that includes FAM129A and FAM129C, which are present only in vertebrates. Sequence analysis of the FAM129 family members reveals that they share a common PH domain on their N-terminal region with high sequence similarity of 57% between MINERVA and FAM129A, and 40% between MINERVA and FAM129C [
6], even though the roles of the PH domains in their functions are not well understood. While FAM129A is an endoplasmic reticulum stress-induced protein that is upregulated in renal and thyroid cancer [
7], FAM129C is a B-cell membrane protein that is overexpressed in chronic lymphocytic leukemia [
8].
Following the first identification of MINERVA as a target of ERK phosphorylation [
6], several studies have reported that MINERVA is upregulated in many types of cancer, including breast, kidney, large intestine, lung and endometrial cancers as well as hematopoietic and central nervous system tumors [
9,
10]. Moreover, cancer patients overexpressing MINERVA exhibited bad prognosis and low survival rate [
11,
12]. Overexpression of MINERVA was associated with activation of focal adhesion kinase (FAK) by facilitating phosphorylation of Tyr397 and Tyr925 residues on FAK [
11], which is associated with increased cell migration [
13]. Similar observations were made from MINERVA-deficient mice that exhibited a delayed rate of wound healing, suggesting a critical role of MINERVA in promoting cell motility [
14].
MINERVA is a target of phosphorylation by both epidermal growth factor receptor (EGFR) and ERK, the central kinases in the ERK growth signaling pathway [
6,
15]. EGFR is a receptor tyrosine kinase that phosphorylates MINERVA on its Tyr593 residue in response to extracellular growth signaling molecule epidermal growth factor (EGF). MINERVA phosphorylated on Tyr593 is capable of direct binding to H-Ras and K-Ras for their constitutive activation by reducing association of Ras GTPase-activating proteins (GAPs) to inhibit Ras-bound GTP hydrolysis [
15]. Constitutive activation of Ras proteins in turn would activate downstream ERK for β-catenin-TCF/LEF transcriptional complex-mediated upregulation of cyclin D1, c-Myc, and PKM2 for promotion of Warburg effect, thereby promoting cell proliferation and tumorigenesis [
15]. ERK also phosphorylates multiple serine residues on the C-terminal flexible region of MINERVA as mentioned above, enhancing invasion capabilities of cells presumably via the activation of FAK [
6,
11]. In sparse, exponentially growing HeLa cells, MINERVA is localized in cytoplasm, whereas the protein is translocalized into plasma membrane colocalizing with β-catenin at adherence junctions in growth-inhibited confluent cells [
16]. The localization of MINERVA at the adherence junctions seems to protect β-catenin from proteolytic degradation, delaying onset of apoptosis elicited by the cellular confluency [
16]. Moreover, MINERVA interacts with LATS1 kinase that suppresses nuclear transportation of YAP, suggesting its involvement in the Hippo signaling pathway activated in confluency [
17]. Overall, MINERVA modulates crosstalk among essential cellular signaling pathways for cell proliferation, invasion, and apoptosis.
Sequence and secondary structure analyses of 746 amino acid-long MINERVA protein indicate a PH domain on the N-terminus (residues Asp66–Gly197) and a proline-rich region on the C-terminus (residues Asp575–Phe746) which contains the phosphorylatable serine residues [
6]. PH domains are well-known for binding phosphatidylinositol phosphates (PIPs) for regulating protein localization [
2]. Additionally, MINERVA contains an N-terminal myristoylation motif
1-MGX
3S-
6 [
18], which supports intracellular membrane localization of MINERVA [
16]. Within the C-terminal region, there is a
708-DLGX
7ETGE-
721 motif that interacts with Kelch domain on Kelch-like ECH-associated protein 1 (Keap1), which is involved in Keap1-mediated ubiquitination of nuclear factor-erythroid 2-related factor 2 (Nrf2) for proteasomal degradation [
12]. Nrf2 is a key regulator in cellular response against oxidative stress by regulating transcription of antioxidant gene expression that is targeted for degradation under quiescent conditions [
19,
20]. Hence, MINERVA was suggested to act as an upstream regulator of Nrf2-mediated response to oxidative stress in cancer by hindering interaction of Keap1 and Nrf2 by competing for the Kelch domain through the “DLGX
nETGE” motif [
12].
Despite the importance of MINERVA in regulating cell growth, migration, apoptosis, and oxidative stress, no structural information of the protein or other FAM129 family members was available. Here, we report the first crystal structure of MINERVA lacking the C-terminal flexible region, which contains the PH domain and a helix bundle domain with a novel fold. In addition, lipid binding ability of the PH domain is assessed for structural and functional analyses of MINERVA. This study shows structural basis for membrane association of MINERVA and its importance in governing multiple signaling pathways in cancer progression.
3. Discussion
Here, we report the first crystal structure of human MINERVA
ΔC, a protein found upregulated in many types of cancer with an unclear function. Sequence analysis predicted intrinsically disordered C-terminal region containing multiple phosphorylation sites and a Keap1-interaction motif, which was excluded in our construct for structure determination. The structure of MINERVA
ΔC comprises the N-terminal PH domain and an α-helix bundle domain (residues Gly2–Glu49 and Glu205–Arg560;
Figure 2a,d) of a novel fold. Flexible α3–β6 loop within the PH domain was cleaved by trypsin for generation of diffraction-quality crystals. To our surprise, intermolecular interaction analysis by the PISA program suggested that the cleavage allowed better stacking of the MINERVA
ΔC molecules in crystal lattice. Helix α8′ (residues Ser292′–Arg306′) from an adjacent MINERVA
ΔC molecule approaches the helix α4 region exposed by the trypsin cleavage, forming several intermolecular interactions with residues Asp129–Asn137 and Lys260–Gln265 (
Figure S3b, gray dashed ribbon). These residues forming the interactions would have been concealed by the α3–β6 loop before trypsin treatment, explaining poor diffraction quality from crystals of untreated MINERVA
ΔC we had obtained.
Our structure has revealed that the N-terminal myristoylated residue Gly2 is positioned adjacent to the PH domain to constitute a positively charged membrane-interacting face for cooperative action of anchoring MINERVA to lipid membranes (
Figure 2d,e). Previous reports, as well as our observation on the localization of MINERVA, showed that the protein is dispersed in cytoplasm as puncta form in exponentially growing HeLa cells, whereas it was recruited to cell–cell contacts whenever the cells became confluent [
16]. Contact inhibition is the major factor in switching cellular functions from growth and proliferation to migration [
31], and we investigated lipid binding ability of the PH domain on MINERVA to gain insight on the function of the protein. Lipid dot-blot assay indicated that MINERVA
FL predominantly binds to PA, whereas MINERVA
ΔC showed higher affinity towards PI3P (
Figure 3c). We speculate that negatively charged residues (Glu80–Ser82) on the β1–β2 loop at the ligand binding site seem to restrict binding of lipids with multiple phosphate groups, while the side chain of Gln79 only allows binding of lipids with smaller head groups. Although PH domains are best known for their abilities to bind phosphatidylinositols [
2], the lipid dot-blot assay results suggest that PA can also be recognized by the PH domain. The difference in binding affinity between MINERVA
FL and MINERVA
ΔC implies that the C-terminal flexible region contributes to the ligand selectivity of the N-terminal PH domain. Despite the importance of PA in cell signaling [
32], domains or motifs responsible for recognizing PA have not been clearly defined. Studies on PA-binding proteins commonly noticed a short stretch of positively charged peptides responsible for the interaction [
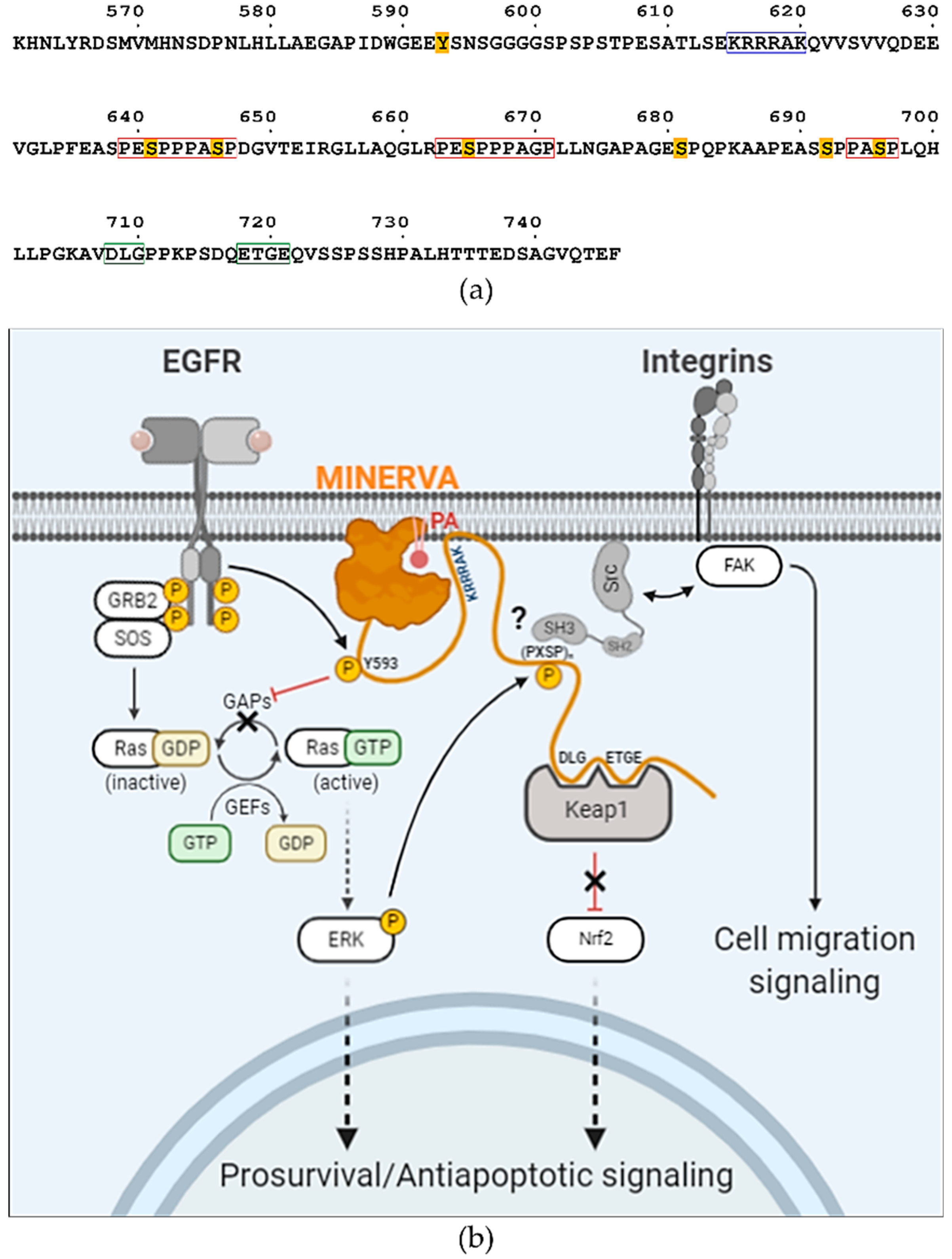
33]. Sequence analysis of the C-terminal flexible region of MINERVA revealed a similar motif
615-KRRRAK-
620 (
Figure 6a), which might be responsible for enhancing ligand specificity of the PH domain on MINERVA
FL towards PA. The facts that MINERVA
ΔC showed higher affinity against PI3P over PA, and that PI3P was recognized as the innate ligand of MINERVA PH domain alone [
29], hint the importance of this positive residue stretch in the C-terminal flexible region for PA recognition. In cells, PI3P is predominantly populated on endosomes, for sorting out protein targets for endocytosis [
34]. Moreover, N-terminal myristoylation on Gly2 residue strongly suggests that cytoplasmic fraction of MINERVA is in fact endosome-associated, and the cooperative action of the N-terminal myristic acid and the PH domain is supported by our structure (
Figure 2d). We speculate that intracellular localization of MINERVA is regulated by the PH domain, depending on the level of cell confluency [
16]. Once cells become confluent, MINERVA would be recycled back to the plasma membrane for activation of cell migratory signaling pathways via, for example, FAK phosphorylation [
11].
The “DLGXnETGE” motif within the C-terminal flexible region was pointed out to be responsible for lifting Keap1-mediated Nrf2 degradation [
12]. Keap1–Nrf2 is a well-known defense system against cellular oxidative stress which is frequently observed in cancer progression. Nrf2 is ubiquitinated and degraded by interaction with Keap1, a scaffold protein for Cul3-containing E3 ubiquitin ligase, whereas under the conditions of oxidative stress, Nrf2 is disrupted from interaction with Keap1 and then is translocated into the nucleus for transcriptions of antioxidant genes [
35,
36]. MINERVA competes with Nrf2 for Keap1-binding through the C-terminal region, resulting in Nrf2 activation [
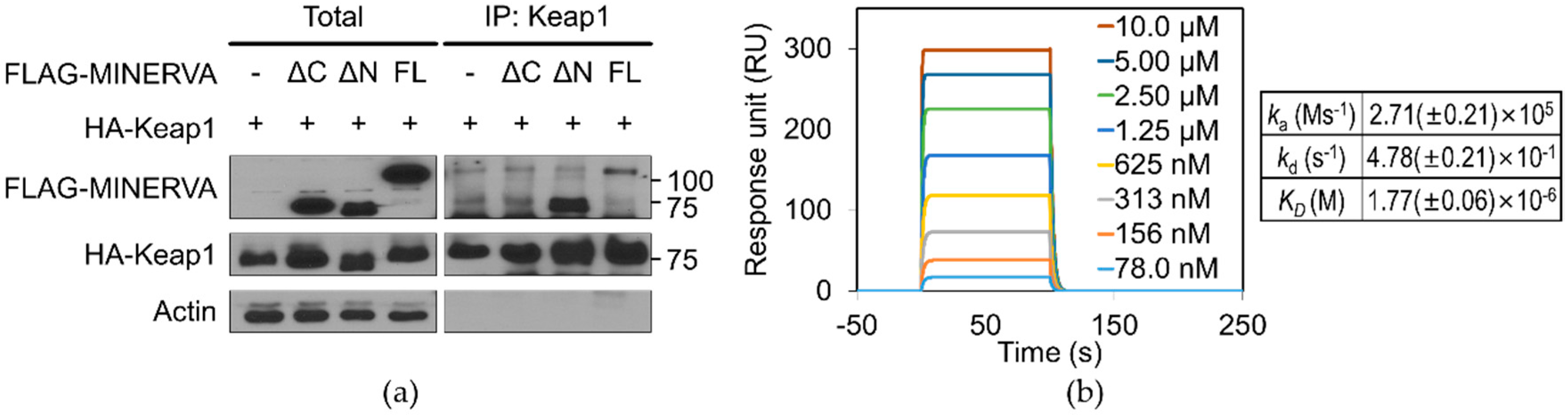
12]. Our SPR analysis of MINERVA
FL and Keap1 provides direct evidence for physical association between the two proteins with micromolar binding affinity (
Figure 5b). Moreover, MINERVA
ΔC failed to interact with Keap1, implying the importance of the C-terminal region of MINERVA for cancer progression through Nrf2 mediated regulation (
Figure 5a). On the other hand, the effect of phosphoserines on MINERVA function was elusive since the first report of MINERVA as a target of ERK phosphorylation [
6]. We noticed that the sequences around these serine residues exhibited a tandem proline-rich consensus motif “(PE
SPP)PA
SP”, where underlined serine residues are the targets of phosphorylation and residues in brackets are less conserved (
Figure 6a). This motif is reminiscent of a Src specific SH3-domain binding motif “(PXXPP)P
ψXP”, where
ψ and X represent hydrophobic and any amino acid residues, respectively [
37,
38]. Thus, it would be interesting to further investigate whether the interaction with Src presents a possible role of MINERVA in regulation of cell motility. Overall, the C-terminal flexible region seems to harbor multiple sequence motifs and PTM sites for interaction with other proteins for regulation of signal transduction (
Figure 6a,b). We can hypothesize that MINERVA recognizing PA or PI3P by the N-terminal PH domain, as well as EGF-mediated ERK phosphorylation of the C-terminal “(PE
SPP)PA
SP” motifs, might allow spatial and temporal regulation of Src activation to promote cell proliferation and migration.
We present the first structure of the globular domain from MINERVA that displays distinct crevices and a cavity shown from surface representations (
Figure 3e). We suggest that these characteristic crevices and the cavity provide a binding platform for MINERVA interacting partners. The widths and lengths of the crevices seem adequate for accommodating 20–30 amino acid long loops each. Although a certain region within the C-terminal flexible region may be the inherent occupant of the crevices, we excluded such possibility as crystallization attempts with trypsin-digested MINERVA
FL produced mostly the same structure model with no additional electron densities at the crevices. However, it should be noted that the presented structure of MINERVA
ΔC might not necessarily represent the in vivo structure since it lacks the C-terminal flexible region harboring post-translational modifications sites and motifs for interaction with other proteins. Binding of interaction partners often elicits conformational changes required for protein function [
39]. Interactome analysis indicates that interacting partners of MINERVA includes not only signaling molecules discussed above (EGFR, H-Ras, ERK, and Keap1), but also proteins responsible for endosomal sorting, such as SNX2 and SNX6, and VPS26A, VPS29, and VPS35 (
https://thebiogrid.org/122328) [
40]. The fact that E-cadherin was suggested as another binding partner of MINERVA hints at the role of MINERVA in cell migration upon confluency [
41]. These interacting partners may exploit the crevices and cavity present on the structured domain of MINERVA
ΔC, since the binding regions of the other signaling molecules are focused on the C-terminal flexible region. Regarding its possible influence over multiple signaling pathways regulating cell growth, proliferation, motility, oxidative stress, and survival and apoptosis, the structural elucidation of MINERVA would present valuable information on cancer progression and therapy.
4. Materials and Methods
4.1. Cell Lines
All human cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and were maintained at 5% CO2 and 37 °C in Dulbecco’s modified Eagle’s medium (DMEM), Roswell Park Memorial Institute (RPMI) 1640 or Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Waltham, MA, USA).
4.2. Antibodies and Reagents
The primary antibody against MINERVA (5122) was purchased from Cell Signaling Technology (Danvers, MA, USA) or from NOVUSBIO (NBP1-88783) for western blot or immunofluorescence; those against β-actin (A300-491A) and HA (A190-108A) were purchased from Bethyl Laboratories (Montgomery, TX, USA); those against FLAG M2 (F1804) and FLAG (F7425) were purchased from Sigma-Aldrich (St. Louis, MO, USA) for western blot; those against FLAG (D6W5B) were purchased from Cell Signaling Technology (Danvers, MA, USA) for immunofluorescence. The secondary antibodies, horseradish peroxidase (HRP)-linked anti-rabbit (A120-101P) and anti-mouse (A90-116P), were purchased from Bethyl Laboratories (Montgomery, TX, USA). Lipofectamine 2000 (11668019) and RNAiMAX (13778150) were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
4.3. DNA Construct and siRNA
For the construction of the FLAG-MINERVA, a PCR-amplified DNA fragment encoding MINERVA was inserted between the NotI and XbaI sites of the pcDNA3 or pcDNA3.1 vector. The deletion mutants of the FLAG-MINERVA (MINERVAΔC; residues Met1–Ser574 and MINERVAΔN; residues Thr307–Phe746) were constructed with NotI and XbaI sites of the pcDNA. siRNA targeting the respective genes of interest and negative control siRNA (non-targeting pool) were purchased from Genolution Inc. (Seoul, Korea). The following siRNA sequences were used for the indicated target genes; siMINERVA_#1 5′-UCACGGACAUGAACCUGAACGUCAU-3′ and siMINERVA_#2 5′-CAGUAUGGCGUGGCUCUCUUCAACA-3′.
4.4. Immunoprecipitation
MINERVA and its truncated forms were tagged with a FLAG epitope and Keap1 was tagged with a human influenza HA epitope. Each combination of the proteins was co-expressed in HEK293T cells. HEK293T cells were rinsed in ice-cold PBS and lysed in lysis buffer composed of 1% NP-40, 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 2 mM ethylenediaminetetraacetic acid (EDTA), 10 mM NaF, 1 mM Na3O4V, and 0.2 mM phenylmethylsulfonyl fluoride (PMSF), including a protease inhibitor cocktail (11836153001; Roche Applied Bioscience, Penzberg, Germany) and 1% phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Next, 0.5 mg lysates for co-immunoprecipitation in cells overexpressing the above proteins were incubated with 2 μg primary antibodies for anti-FLAG M2 (Sigma-Aldrich, St. Louis, MO, USA) or anti-HA antibody (Bethyl Laboratories, Montgomery, TX, USA), or rabbit IgG (Sigma-Aldrich, St. Louis, MO, USA) at 4 °C with overnight shaking after adding 50 μL protein A agarose beads (GenDEPOT, Katy, TX, USA). Immunoprecipitates were washed three times with wash buffer and then eluted by boiling in sodium dodecyl sulfate (SDS) sample buffer with β-mercaptoethanol for 5 min. Then, the immunoprecipitate complex was immunoblotted with the indicated antibodies.
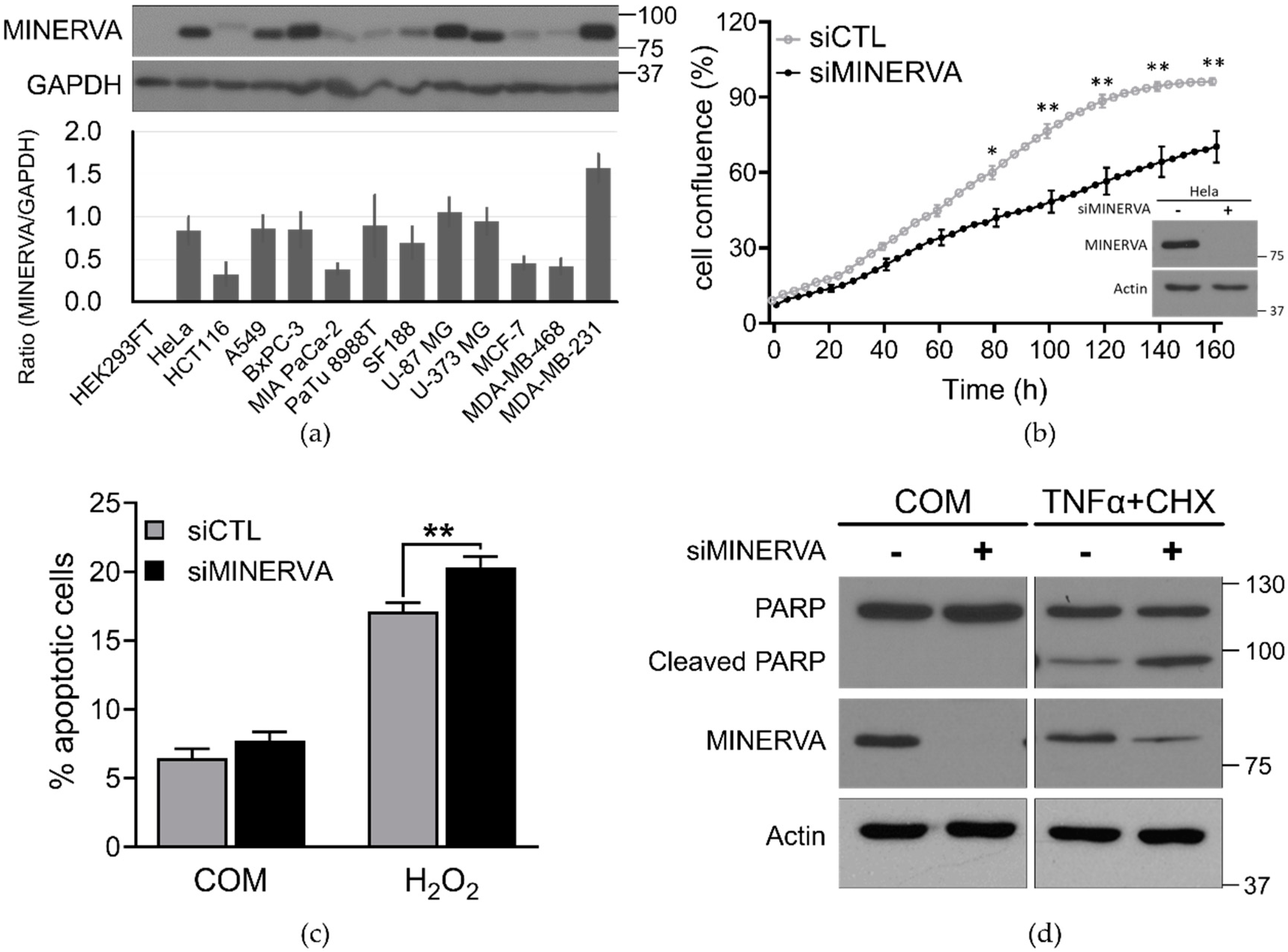
4.5. Cell Proliferation and Viability Assays
siRNA-transfected cells were seeded at 4 × 103 per well in a 96-well plate, which was placed in the IncuCyte ZOOM™ system (Essen BioScience, Ann Arbor, MI, USA). After incubation for the indicated times live-cell images were obtained using a 10× objective lens (four images per well) within the instrument, and cell density was analyzed using IncuCyte ZOOM 2016B software.
Cell viability was determined by annexin V and PI staining following standard protocols at the indicated time periods (556547, BD Biosciences, San Jose, CA, USA). Cells negative for both annexin V and PI were considered live cells. The proportion of dead cells was measured based on the number of annexin V and PI single and both-stained cells. The fluorescence of stained cells was detected using the LSRFortessa fluorescence activated cell sorting (FACS) analyzer (BD Biosciences).
4.6. Western Blotting
Cells were harvested in ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, and 1 mM EDTA) containing protease inhibitor cocktail (Roche Applied Bioscience, Penzberg, Germany) and phosphatase inhibitor (Sigma-Aldrich, St. Louis, MO, USA). Soluble lysate fractions were isolated by centrifugation at 20,000× g, for 20 min at 4 °C and quantified using the Pierce bicinchoninic acid (BCA) Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using equal concentrations of protein and transferred to PVDF membranes. The membranes were blocked with 5% skim milk and then probed with the indicated primary and secondary antibodies following standard protocols. Immunoblotted proteins were quantified using ImageJ software (NIH, Bethesda, MD, USA). Data are expressed as the mean ± standard deviation (SD) or standard error of the mean (SEM) which are from at least three independent experiments. Statistical significance was calculated using Student’s t-test in GraphPad Prism. A value of p < 0.05 was considered statistically significant (* p < 0.05; ** p < 0.01).
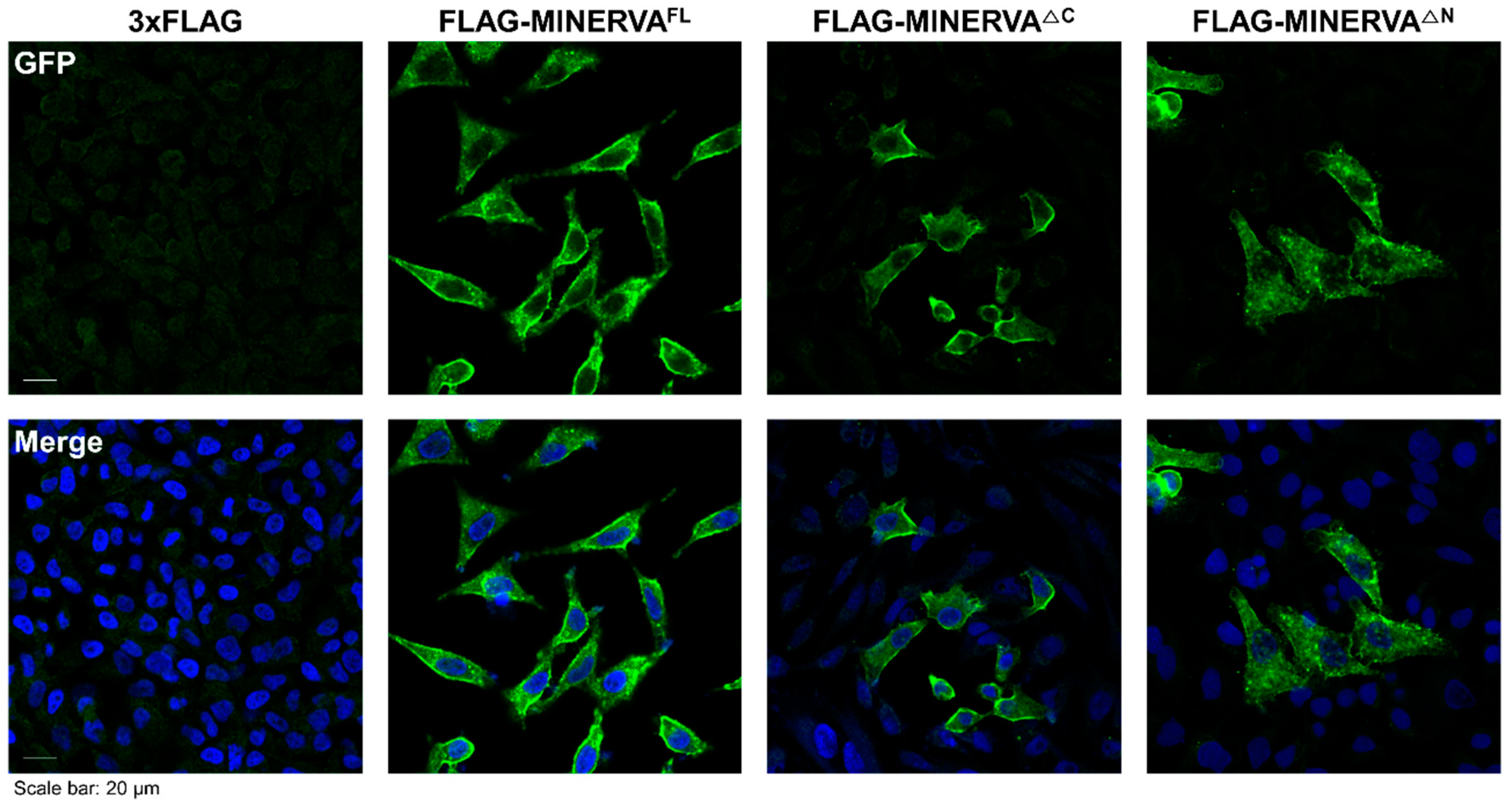
4.7. Immunofluorescence Analysis
To determine the localization of MINERVA, immunostaining against MINERVA was performed using Hela cells. After they were plated onto glass coverslips in complete medium for 18 h, cells were incubated in control (3 × FLAG only) and MINERVA domain construct mixture treatment for 48 h. For immunostaining, cells were rinsed with ice-cold PBS, fixed with 3.7% formaldehyde for 20 min, and permeabilized with 0.3% Triton X-100 in PBS (PBS-T) for 5 min. After rinsing with PBS-T, cells were blocked with blocking buffer (5% BSA in PBS-T) for 1 h, incubated with primary antibodies against MINERVA or FLAG in blocking buffer for 16 h in 4 °C, washed three times with PBS-T and then incubated with secondary antibodies conjugated with fluorescent probe in PBS-T for 1 h at room temperature. After three PBS-T washes, cells were stained with DAPI (4′,6-diamidino-2-phenylindole) for 10 min and mounted on microscope slides. A confocal fluorescence microscope (Zeiss LSM 780, Oberkochen, Germany) was used for imaging and analysis.
4.8. Cloning, Expression, and Purification of Recombinant MINERVA Constructs for Structure Determination and Affinity Assay
The full length MINERVAFL (residues Met1–Phe746) and C-terminally truncated human MINERVAΔC (residues Met1–Ser574) were amplified using PCR and cloned into pET-28a(+) and pET-21a(+) vectors (Novagen, Burlington, MA, USA) to each contain an N-terminal or a C-terminal His6-tag, respectively. The plasmids containing the MINERVA constructs were each transformed into Rosetta 2(DE3)pLysS or BL21-CodonPlus (DE3)-RIPL Escherichia coli strain, respectively.
The transformed cells were grown for 16 h in Luria Broth media without induction to be used as seeds for the scale-up culture. The uninduced E. coli seeds containing MINERVA plasmids were inoculated into a scale-up culture media at a ratio of 1:50. The cells in scale-up culture were grown in Luria Broth media and induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside, followed by further incubation at 20 °C for 16 h. Harvested cells were lysed by a cell sonicator (SONICS) in a lysis buffer containing 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, 35 mM imidazole, and 1 mM PMSF. Cell debris was removed by centrifugation at 35,000× g for 50 min at 4 °C, then the supernatant was filtered with 0.22 μm filter to remove cell debris and any aggregated proteins. The filtered supernatant was applied onto a HiTrap Chelating HP column (GE Healthcare, Chicago, IL, USA) equilibrated with the lysis buffer for affinity chromatography. Unbound or weakly bound proteins were removed from the HiTrap Chelating HP column with six column volume elutions of the washing buffer containing 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 50 mM imidazole. The retained proteins were eluted with a gradually increasing addition of a buffer containing 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 500 mM imidazole. The eluted protein samples were further purified by size-exclusion chromatography with HiLoad 16/600 Superdex200 pg column (GE Healthcare, Chicago, IL, USA) equilibrated with a buffer containing 20 mM Tris-HCl (pH 8.0) and 200 mM NaCl. The purification steps for SeMet-substituted MINERVAΔC were as above except that the buffer for the final size exclusion chromatography additionally contained 1 mM tris(2-carboxyethyl)phosphine (TCEP).
4.9. Cloning, Expression, and Purification of Keap1321–609 for Affinity Assay
N- and C-terminally truncated Keap1 protein (residues Ala321–Thr609) containing the Kelch domain was amplified using PCR and cloned into the pET-28a(+) vector to contain an N-terminal His6-tag. The plasmid containing Keap1321–609 was transformed into Rosetta 2(DE3)pLysS E. coli strain. The purification steps for Keap1321–609 were as above except that the buffer for the final size exclusion chromatography contained 20 mM Tris-HCl (pH 7.5) and 5 mM dithiothreitol (DTT).
4.10. Limited Trypsin Digestion of MINERVA Proteins
Native and SeMet-substituted MINERVAΔC were subjected to digestion by trypsin (Sigma-Aldrich, St. Louis, MO, USA) by incubating purified MINERVAΔC at a 5000:1 trypsin ratio at 4 °C for 20 h prior to crystallization in order to improve diffraction quality of crystals. Digested proteins were further purified by size-exclusion chromatography with HiLoad 16/600 Superdex200 pg column equilibrated with a buffer containing 20 mM Tris-HCl (pH 8.0) and 200 mM NaCl. The purified trypsin-treated MINERVAΔC proteins were concentrated to 4.8 mg mL−1 for crystallization.
4.11. Crystllization, Data Collection, and Structure Determination
Crystals of the trypsin-treated SeMet-substituted MINERVA
ΔC were grown at 22 °C by the hanging drop vapor diffusion method by mixing equal volumes of the protein at 4.8 mg mL
−1 and a crystallization solution containing 0.2 M sodium tartrate and 20% (
w/v) polyethylene glycol (PEG) 3350. Crystals of the trypsin treated native MINERVA
ΔC were grown at 22 °C by the same methods with a crystallization solution containing 0.2 M lithium acetate and 20% (
w/v) PEG 3350. Crystals of diffraction quality were cryoprotected with the reservoir solution containing additional 25% (
v/v) glycerol and flash-cooled in a nitrogen gas stream at 100 K. Diffraction data from the SeMet-substituted and the native crystals were collected up to resolutions of 2.39 Å and 1.65 Å, respectively. Raw X-ray diffraction data were indexed and scaled using the HKL-2000 program suite or the XDS program package [
42,
43]. Single-wavelength anomalous diffraction (SAD) phases of the SeMet-substituted MINERVA
ΔC were initially calculated with AUTOSOL in the PHENIX software suite [
44] and further improved by the automatic model building program RESOLVE [
45], resulting in an initial model. The initial model was further refined to the final model using iterative cycles of model building with Coot [
46] and subsequent refinement with Refmac5 in the CCP4 suite [
47] and phenix.refine [
48]. The crystal structure of the native MINERVA
ΔC was determined by molecular replacement with the MolRep program [
49], using the refined structure of SeMet-substituted MINERVA
ΔC as a phasing model. Validation of crystal structures was implemented with MolProbity [
50] and the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) validation server. Statistics for the data collection and refinement are summarized in
Table 1. The coordinate and structure factor of the native MINERVA
ΔC have been deposited in the PDB (
http://www.rcsb.org) under accession code 7CTP.
4.12. Size-Exclusion Chromatography with Multi-Angle Light Scattering (SEC–MALS)
The oligomeric states of the recombinant MINERVAFL and MINERVAΔC proteins were assessed by SEC–MALS experiments using an ÄKTA fast protein liquid chromatography (FPLC) system (GE Healthcare, Chicago, IL, USA) connected to a Wyatt DAWN Heleos II MALS instrument and a Wyatt Optilab T-rEX differential refractometer. A Superdex 200 Increase 10/300 GL gel-filtration column (GE Healthcare, Chicago, IL, USA), which was pre-equilibrated with buffer A containing 20 mM Tris-HCl (pH 8.0) and 200 mM NaCl, was normalized using ovalbumin. The proteins (2 mg) were injected at a flow rate of 0.5 mL min−1. Data were analyzed using the Zimm model for fitting static light-scattering data and were graphed using Easy Analytic Software Inc. (EASI) graph with a UV peak in the ASTRA 6 software (Wyatt, Santa Barbara, CA, USA).
4.13. In Silico Docking Experiment
Induced fit docking (IFD) experiment (Schrödinger Suite Induced Fit Docking protocol; Glide version 7.4, Prime version 4.7, Schrödinger, LLC, New York, NY, USA) was implemented in order to predict the binding mode of lipids to the PH domain of MINERVA. The MINERVAΔC structure was prepared for docking calculations using the Protein Preparation Wizard implemented in Maestro (Schrödinger, LLC, New York, NY, USA). The structure of MINERVAΔC was built in Maestro 8.5, and the possible conformations of the lipids were generated using LigPrep (version 4.1, Schrödinger, LLC, New York, NY, USA). The head groups of lipids were docked onto MINERVAΔC using the following steps. (i) The receptor grid of MINERVAΔC was defined as an enclosing box at the centroid of the key amino acid residues in the binding pocket. (ii) In the initial Glide docking stage, a potential docking with van der Waals radius scaling of 0.5 for the protein and ligand was performed retaining a maximum number of 20 poses per ligand. (iii) Residues within 5.0 Å of ligand poses were kept free to move in the Prime refinement step, and the side chains were further optimized. (iv) Poses within 30 kcal mol−1 of the energy cutoff in the previous step were re-docked using Glide XP. (v) The binding free energy (ΔGbind) for each output pose was computed using Prime-MMGBSA (version 3.0).
4.14. Lipid Dot Blot Assay
The PIP strips (Echelon Biosciences, Salt Lake City, UT, USA) were blocked with phosphate-buffered saline containing 0.5% (v/v) Tween-20 (Amersham Biosciences, UK) and 3% (w/v) bovine serum albumin (BSA; Bovogen, East Keilor, Australia) for 1 h and incubated with MINERVAFL or MINERVAΔC for 1 h. MINERVAFL bound strips were treated with α-MINERVA rabbit antibody (5122; Cell Signaling Technology, Danvers, MA, USA) for 1 h then with HRP-conjugated α-rabbit IgG goat antibody (A120-101P; Bethyl Laboratories, Montgomery, TX, USA) for 1 h. MINERVAΔC bound strips were treated with HRP-conjugated α-His6-tag antibody (Santa Cruz Biotechnology, Dallas, TX, USA). The strips were washed with the blocking buffer for 5 min three times between each step. The strips were visualized with an ECL kit (Amersham Biosciences, UK).
4.15. Surface Plasmone Resonance (SPR) Analysis of MINERVA
SPR experiments for the interaction of MINERVAFL with Keap1321–609 were carried out in a Biacore T200 system (GE Healthcare, Chicago, IL, USA) using a buffer comprising 10 mM HEPES and 150 mM NaCl at pH 7.4. MINERVAFL in 10 mM sodium acetate at pH 5.0 was immobilized by the amine coupling method on a CM5 sensor chip according to the manufacturer’s protocol (GE Healthcare, Chicago, IL, USA). A reference flow cell was equally treated but without protein as a control and the response by the control was subtracted from each sample. Keap1321–609 at concentrations of 78.1, 156, 313, 625, 1250, 2500, 5000, and 10,000 nM were injected over the MINERVAFL chip at a rate of 30 μL min−1. Biacore T200 evaluation software was used to estimate the affinity and kinetics fittings using the steady state affinity model and 1:1 binding model, respectively, to calculate the kinetics data. Average and standard deviation were calculated from two separate kinetics’ data measured.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}