Importance of Deubiquitination in Macrophage-Mediated Viral Response and Inflammation


 and
and 
Abstract
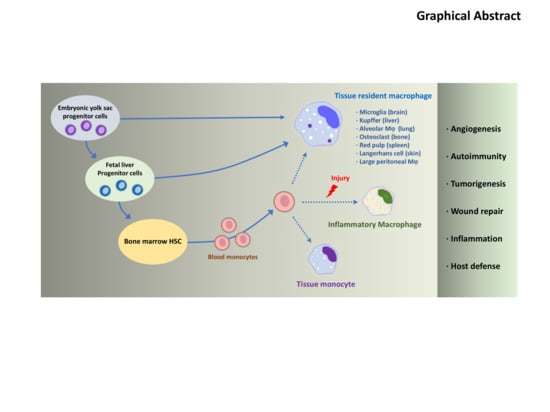
:
1. Introduction
2. Ubiquitination and Deubiquitination in the RLR Pathway
2.1. K63-Linked Ubiquitination: RIG-I Activation and Cytosolic Warriors
2.2. K48-Linked Ubiquitination: RIG-I Degradation
2.3. Deubiquitination: Negative Regulators of RIG-I
2.3.1. CYLD
2.3.2. A20
2.3.3. USP21
2.3.4. USP3
2.3.5. USP15
2.3.6. USP25
2.3.7. USP14
2.3.8. USP18
2.4. Positive Regulators of RIG-I
USP4 and USP17
3. Ubiquitination and Deubiquitination in TLR Signaling
3.1. Ubiquitination of TLR Signaling Molecules: TRAF3, TRAF6, TBK1, RIP1, TAK1
3.2. Deubiquitination of TLR Signaling Molecules
3.2.1. USP2/USP2a
3.2.2. USP4
3.2.3. USP7
3.2.4. USP10
3.2.5. USP18
3.2.6. USP25
3.2.7. USP12
3.2.8. A20
3.2.9. CYLD
3.2.10. MYSM1
4. Ubiquitination in NLR Signaling
4.1. Ubiquitination of NLRP3 and RIP2
4.2. Deubiquitination of NLR Signaling Molecules
4.2.1. A20
4.2.2. BRCC3
4.2.3. USP7 and USP47
5. DUB Inhibitors
5.1. DUB Inhibitor (WP1130) for Bacterial Killing in Macrophages
5.2. DUB Inhibitors in Inflammasome Assembly
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PRR | Pattern recognition receptors |
| PAMPS | Pathogen-associated molecular patterns |
| DAMPS | Danger-associated molecular patterns |
| DUB | Deubiquitinating enzyme |
| KC | Kupffer cell |
| TLR | Toll-like receptors |
| RIG-I | Retinoic acid-inducible gene I |
| RLR | RIG-I- like receptors |
| MDA5 | Melanoma-differentiation-associated gene 5 |
| NOD | Nucleotide-binding oligomerization domain |
| NLR | NOD-like receptors |
| IFN | Interferons |
| IL | Interleukin |
| AIM2 | Absent in melanoma 2 |
| NLRP3 | Nucleotide-binding oligomerization domain leucine-rich repeat and pyrin domain-containing protein 3 |
| ASC | Apoptosis-associated speck-like protein containing a caspase activating and recruitment domain |
| CARD | Caspase activation and recruitment domains |
| VSV | Vesicular stomatitis virus |
| TRIM25 | Tripartite motif containing 25 |
| MAVS | Mitochondrial antiviral-signaling protein |
| NF-κB | Nuclear factor-κB |
| TRAF | Tumor necrosis factor receptor-associated factor |
| IKK | Inhibitor of nuclear factor κB kinase |
| TBK1 | Tank-binding kinase 1 |
| IRF | IFN regulatory factor |
| CHIP | C-terminus of Hsc70 interacting protein |
| RNF | RING-finger protein |
| STING | Stimulator of IFN genes |
| LUBAC | Linear ubiquitin assembly complex |
| TAX1BP1 | Tax1-binding protein 1 |
| ISRE | IFN-stimulated response element |
| MyD88 | Myeloid differentiation primary response 88 |
| TIR | Toll/Interleukin-1 receptor |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| IRAK | IL-1R-associated serine/threonine kinases |
| TNFR1 | TNF receptor-1 |
| EV71 | Enterovirus 71 |
| MNV 1 | Murine norovirus 1 |
| FBXO3 | F-box protein 3 |
| BRCC3 | BRCA1/BRCA2-containing complex 3 |
| BRISC | BRCC36 isopeptidase complex |
| EMPs | Erythromyeloid progenitors |
| HSCs | Hematopoietic stem cells |
| IPS-1 | Interferon-beta promoter stimulator 1 |
| ISG | Interferon stimulating genes |
References
- Gordon, S.; Martinez-Pomares, L. Physiological roles of macrophages. Pflügers Arch. 2017, 469, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Gittes, G.K. Concise Review: New Insights into the Role of Macrophages in β-Cell Proliferation. Stem Cells Transl. Med. 2015, 4, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmacher, Y.; Hiller, K. Biochemistry of proinflammatory macrophage activation. Cell Mol. Life Sci. 2018, 75, 2093–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. New insights into tissue macrophages: From their origin to the development of memory. Immune Netw. 2015, 15, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitina, E.; Larionova, I.; Choinzonov, E.; Kzhyshkowska, J. Monocytes and macrophages as viral targets and reservoirs. Int. J. Mol. Sci. 2018, 19, 2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Recognition of viruses by innate immunity. Immunol. Rev. 2007, 220, 214–224. [Google Scholar] [CrossRef]
- Saito, T.; Hirai, R.; Loo, Y.-M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101. [Google Scholar] [CrossRef]
- Öhman, T.; Rintahaka, J.; Kalkkinen, N.; Matikainen, S.; Nyman, T.A. Actin and RIG-I/MAVS signaling components translocate to mitochondria upon influenza A virus infection of human primary macrophages. J. Immunol. 2009, 182, 5682–5692. [Google Scholar] [CrossRef] [Green Version]
- Lowe, E.L.; Doherty, T.M.; Karahashi, H.; Arditi, M. Ubiquitination and de-ubiquitination: Role in regulation of signaling by Toll-like receptors. J. Endotoxin Res. 2006, 12, 337–345. [Google Scholar]
- Maelfait, J.; Beyaert, R. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev. 2012, 76, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednash, J.S.; Mallampalli, R.K. Regulation of inflammasomes by ubiquitination. Cell Mol. Immunol. 2016, 13, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef] [Green Version]
- Meduri, G.U.; Headley, S.; Kohler, G.; Stentz, F.; Tolley, E.; Umberger, R.; Leeper, K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS: Plasma IL-1β and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 1995, 107, 1062–1073. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Wanderer, A.A. Inflammasome and IL-1β-mediated disorders. Curr. Allergy Asthma Rep. 2010, 10, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508. [Google Scholar] [CrossRef] [PubMed]
- McClurg, U.L.; Robson, C.N. Deubiquitinating enzymes as oncotargets. Oncotarget 2015, 6, 9657. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Maculins, T.; Fiskin, E.; Bhogaraju, S.; Dikic, I. Bacteria-host relationship: Ubiquitin ligases as weapons of invasion. Cell Res. 2016, 26, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Kattah, M.G.; Malynn, B.A.; Ma, A. Ubiquitin-modifying enzymes and regulation of the inflammasome. J. Mol. Biol. 2017, 429, 3471–3485. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; e Sousa, C.R. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlee, M. Master sensors of pathogenic RNA–RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef]
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 2019, 97, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Arjona, A.; Zhang, Y.; Sultana, H.; Dai, J.; Yang, L.; LeBlanc, P.M.; Doiron, K.; Saleh, M.; Fikrig, E. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat. Immunol. 2010, 11, 912. [Google Scholar] [CrossRef] [Green Version]
- Häcker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef]
- Sun, S.-C. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 2008, 8, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Li, Q.; Mao, A.-P.; Hu, M.-M.; Shu, H.-B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol. 2014, 6, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Kuniyoshi, K.; Takeuchi, O.; Pandey, S.; Satoh, T.; Iwasaki, H.; Akira, S.; Kawai, T. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I–mediated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2014, 111, 5646–5651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Wang, J.; Wang, Y.; Zhou, H.; Wu, X.; Tian, Z.; Sun, B. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J. Immunol. 2013, 190, 3613–3619. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Yang, Y.-K.; Wang, R.-P.; Zhou, X.; Diao, F.-C.; Li, M.-D.; Zhai, Z.-H.; Jiang, Z.-F.; Chen, D.-Y. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto, K.-I.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Han, C.; Xie, B.; Hu, X.; Yu, Q.; Shi, L.; Wang, Q.; Li, D.; Wang, J.; Zheng, P. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 2013, 152, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 ubiquitin ligase TRIM40 attenuates antiviral immune responses by targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, X.; Lee, A.J.; Jin, W.; Chang, M.; Wright, A.; Imaizumi, T.; Sun, S.-C. Regulation of IκB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J. Biol. Chem. 2008, 283, 18621–18626. [Google Scholar] [CrossRef] [Green Version]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cárdenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lee, A.J.; Wu, X.; Sun, S.-C. Regulation of antiviral innate immunity by deubiquitinase CYLD. Cell Mol. Immunol. 2011, 8, 502–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiley, W.W.; Jin, W.; Lee, A.J.; Wright, A.; Wu, X.; Tewalt, E.F.; Leonard, T.O.; Norbury, C.C.; Fitzpatrick, L.; Zhang, M. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 2007, 204, 1475–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brummelkamp, T.R.; Nijman, S.M.; Dirac, A.M.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Wooten, M.W.; Geetha, T.; Babu, J.R.; Seibenhener, M.L.; Peng, J.; Cox, N.; Diaz-Meco, M.-T.; Moscat, J. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J. Biol. Chem. 2008, 283, 6783–6789. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.S.; Kim, N.; Lee, K.J.; Nam, Y.R.; Lee, U.; Joo, C.H. Lysine 63-linked TANK-binding kinase 1 ubiquitination by mindbomb E3 ubiquitin protein ligase 2 is mediated by the mitochondrial antiviral signaling protein. J. Virol. 2014, 88, 12765–12776. [Google Scholar] [CrossRef] [Green Version]
- Parvatiyar, K.; Barber, G.N.; Harhaj, E.W. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J. Biol. Chem. 2010, 285, 14999–15009. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Kajitani, T.; Togo, S.; Masuko, N.; Ohdan, H.; Hishikawa, Y.; Koji, T.; Matsuyama, T.; Ikura, T.; Muramatsu, M.; et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008, 22, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Urbe, S.; Liu, H.; Hayes, S.D.; Heride, C.; Rigden, D.J.; Clague, M.J. Systematic survey of deubiquitinase localization identifies USP21 as a regulator of centrosome- and microtubule-associated functions. Mol. Biol. Cell 2012, 23, 1095–1103. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yu, Y.; Liu, S.; Shi, Z.; Cheng, J.; Zhang, H.; An, L.; Zhao, Y.; Xu, X.; et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 2014, 211, 313–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, B.; Baek, K.H. Regulatory interplay between deubiquitinating enzymes and cytokines. Cytokine Growth Factor Rev. 2019, 48, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.-F. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014, 24, 400–416. [Google Scholar] [CrossRef] [Green Version]
- Pauli, E.-K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The ubiquitin-specific protease USP15 promotes RIG-I–mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 2014, 7, ra3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, D.; Zhong, H.; Luo, R.; Shang, M.; Liu, D.; Chen, H.; Fang, L.; Xiao, S. Ubiquitin-specific protease 15 negatively regulates virus-induced type I interferon signaling via catalytically-dependent and-independent mechanisms. Sci. Rep. 2015, 5, 11220. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wang, D.; Fang, L.; Zhang, H.; Luo, R.; Shang, M.; Ouyang, C.; Ouyang, H.; Chen, H.; Xiao, S. Ubiquitin-specific proteases 25 negatively regulates virus-induced type I interferon signaling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Liu, X.; Wang, X.; Li, H.; Darnay, B.G.; Lin, X.; Sun, S.C.; Dong, C. Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci. Signal. 2013, 6, ra35. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Zhang, M.; Zhang, M.-X.; Ren, Y.; Jin, J.; Zhao, Q.; Pan, Z.; Wu, M.; Shu, H.-B.; Dong, C. Induction of USP25 by viral infection promotes innate antiviral responses by mediating the stabilization of TRAF3 and TRAF6. Proc. Natl. Acad. Sci. USA 2015, 112, 11324–11329. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, Z.; Ling, J.; Pan, L.; Zhao, X.; Zhu, H.; Yu, J.; Xie, B.; Shen, J.; Chen, W. USP14 promotes K63-linked RIG-I deubiquitination and suppresses antiviral immune responses. Eur. J. Immunol. 2019, 49, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, M.-X.; Zhang, Q.; Zhu, G.-F.; Yuan, L.; Zhang, D.-E.; Zhu, Q.; Yao, J.; Shu, H.-B.; Zhong, B. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016, 26, 1302–1319. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, L.; Zhong, B.; Tan, B.; Liu, Y.; Shu, H.-B. The ubiquitin-specific protease 17 is involved in virus-triggered type I IFN signaling. Cell Res. 2010, 20, 802–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, W.; Zhang, M.; Wang, P.; Zhao, K.; Zhao, X.; Yang, S.; Gao, C. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J. Virol. 2013, 87, 4507–4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Park, J.S.; Kim, J.H.; Jung, S.M.; Lee, J.Y.; Kim, S.-J.; Park, S.H. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-β1-induced degradation of MyD88 in TLR4 signalling. Nat. Commun. 2011, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, T.; Zhang, J.; Yang, M.; Li, N.; Xu, X.; Cao, X. The E3 ubiquitin ligase Nrdp1’preferentially’promotes TLR-mediated production of type I interferon. Nat. Immunol. 2009, 10, 744. [Google Scholar] [CrossRef]
- Davis, M.E.; Gack, M.U. Ubiquitination in the antiviral immune response. Virology 2015, 479, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Fearns, C.; Pan, Q.; Mathison, J.C.; Chuang, T.-H. Triad3A regulates ubiquitination and proteasomal degradation of RIP1 following disruption of Hsp90 binding. J. Biol. Chem. 2006, 281, 34592–34600. [Google Scholar] [CrossRef] [Green Version]
- Nakhaei, P.; Mesplede, T.; Solis, M.; Sun, Q.; Zhao, T.; Yang, L.; Chuang, T.-H.; Ware, C.F.; Lin, R.; Hiscott, J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Li, Y.; Li, C.; Liu, L.-J.; Zhang, X.-D.; Liu, Y.; Shu, H.-B. USP2a negatively regulates IL-1β-and virus-induced NF-κB activation by deubiquitinating TRAF6. J. Mol. Cell Biol. 2013, 5, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Ishino, T.; Shimamoto, Y.; Okabe, J.; Miyamoto, T.; Takahashi, E.; Miyoshi, I. Ubiquitin-specific protease 2 modulates the lipopolysaccharide-elicited expression of proinflammatory cytokines in macrophage-like HL-60 cells. Mediators Inflamm. 2017, 2017, 6909415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Yu, Y.; Mao, R.; Tan, X.; Xu, G.; Zhang, H.; Lu, X.; Fu, S.; Yang, J. USP4 targets TAK1 to downregulate TNF α-induced NF-κ B activation. Cell Death Differ. 2011, 18, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Li, H.; Luo, J.; Wang, R.; Chen, H.; Chen, J.; Wang, P. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFα-induced cancer cell migration. Biochem. J. 2012, 441, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Peng, Y.; Zhang, Q.; Xu, X.-P.; Kong, X.-M.; Shi, W.-F. USP4 positively regulates RLR-induced NF-κB activation by targeting TRAF6 for K48-linked deubiquitination and inhibits enterovirus 71 replication. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Meredith, M.; Orr, A.; Cross, A.; Kathoria, M.; Parkinson, J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997, 16, 566–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, R.D.; Meredith, M.; Orr, A. The ability of herpes simplex virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expression and stimulation of virus replication. J. Virol. 1999, 73, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Holowaty, M.N.; Sheng, Y.; Nguyen, T.; Arrowsmith, C.; Frappier, L. Protein interaction domains of the ubiquitin-specific protease, USP7/HAUSP. J. Biol. Chem. 2003, 278, 47753–47761. [Google Scholar] [CrossRef] [Green Version]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar] [CrossRef] [Green Version]
- Colleran, A.; Collins, P.E.; O’Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of NF-κB by Ubiquitin-Specific Protease-7 promotes transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Guan, J.; Li, S.; Zhang, X.; Zheng, X. HSCARG downregulates NF-κ B signaling by interacting with USP7 and inhibiting NEMO ubiquitination. Cell Death Dis. 2014, 5, e1229. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Huang, X.; Xin, H.-B.; Fu, M.; Xue, A.; Wu, Z.-H. TRAF family member-associated NF-κB activator (TANK) inhibits genotoxic nuclear factor κB activation by facilitating deubiquitinase USP10-dependent deubiquitination of TRAF6 ligase. J. Biol. Chem. 2015, 290, 13372–13385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, J.; Shi, Y.; Xue, J.; Miao, R.; Huang, S.; Wang, T.; Wu, J.; Fu, M.; Wu, Z.H. USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 2013, 32, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xian, H.; Hu, J.; Tian, S.; Qin, Y.; Wang, R.-F.; Cui, J. USP18 negatively regulates NF-κB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci. Rep. 2015, 5, 12738. [Google Scholar] [CrossRef] [Green Version]
- Blount, J.R.; Burr, A.A.; Denuc, A.; Marfany, G.; Todi, S.V. Ubiquitin-specific protease 25 functions in Endoplasmic Reticulum-associated degradation. PLoS ONE 2012, 7, e36542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Bai, H.; Chen, N.; Zhang, W.; Zhu, X.; Li, P.; Gong, J. USP25 promotes endotoxin tolerance via suppressing K48-linked ubiquitination and degradation of TRAF3 in Kupffer cells. Mol. Immunol. 2019, 106, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Dixit, V.M. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2011, 21, 22–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, T.K.S.; Alamuru-Yellapragada, N.P.; Parsa, K.V. Deubiquitinase USP12 promotes LPS induced macrophage responses through inhibition of IκBα. Biochem. Biophys. Res. Commun. 2017, 483, 69–74. [Google Scholar] [CrossRef]
- Parvatiyar, K.; Harhaj, E.W. Regulation of inflammatory and antiviral signaling by A20. Microbes Infect. 2011, 13, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Oshima, N.; Ishihara, S.; Rumi, M.; Aziz, M.; Mishima, Y.; Kadota, C.; Moriyama, I.; Ishimura, N.; Amano, Y.; Kinoshita, Y. A20 is an early responding negative regulator of Toll-like receptor 5 signalling in intestinal epithelial cells during inflammation. Clin. Exp. Immunol. 2010, 159, 185–198. [Google Scholar] [CrossRef]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Ahmad, R.-C.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004, 5, 1052–1060. [Google Scholar] [CrossRef]
- Turer, E.E.; Tavares, R.M.; Mortier, E.; Hitotsumatsu, O.; Advincula, R.; Lee, B.; Shifrin, N.; Malynn, B.A.; Ma, A. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J. Exp. Med. 2008, 205, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-C.; Miyata, M.; Lim, J.H.; Li, J.-D. Deubiquitinase CYLD acts as a negative regulator for bacterium NTHi-induced inflammation by suppressing K63-linked ubiquitination of MyD88. Proc. Natl. Acad. Sci. USA 2016, 113, E165–E171. [Google Scholar] [CrossRef] [Green Version]
- Legarda, D.; Justus, S.J.; Ang, R.L.; Rikhi, N.; Li, W.; Moran, T.M.; Zhang, J.; Mizoguchi, E.; Zelic, M.; Kelliher, M.A. CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. 2016, 15, 2449–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S.; Nilsson, J.A.; Gekara, N.O. Deubiquitinase MYSM1 regulates innate immunity through inactivation of TRAF3 and TRAF6 complexes. Immunity 2015, 43, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Lear, T.B.; Jerome, J.A.; Rajbhandari, S.; Snavely, C.A.; Gulick, D.L.; Gibson, K.F.; Zou, C.; Chen, B.B.; Mallampalli, R.K. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J. Biol. Chem. 2015, 290, 18124–18133. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.; Zhang, Q.; Liu, W.; Jia, Y.; Ai, S.; Wang, T.; Wang, W.; Pan, P.; Yang, G.; Xiang, Q. Cullin1 binds and promotes NLRP3 ubiquitination to repress systematic inflammasome activation. FASEB J. 2019, 33, 5793–5807. [Google Scholar] [CrossRef]
- Kawashima, A.; Karasawa, T.; Tago, K.; Kimura, H.; Kamata, R.; Usui-Kawanishi, F.; Watanabe, S.; Ohta, S.; Funakoshi-Tago, M.; Yanagisawa, K.; et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J. Immunol. 2017, 199, 3614–3622. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013, 49, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Humphries, F.; Bergin, R.; Jackson, R.; Delagic, N.; Wang, B.; Yang, S.; Dubois, A.V.; Ingram, R.J.; Moynagh, P.N. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-W.; Chen, C.-H.; Chang, J.-N.; Chen, C.-H.; Hsu, Y.-H. Far-infrared promotes burn wound healing by suppressing NLRP3 inflammasome caused by enhanced autophagy. J. Mol. Med. 2016, 94, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, M.A.; Bowman, J.W.; Fujita, H.; Orazio, N.; Shi, M.; Liang, Q.; Amatya, R.; Kelly, T.J.; Iwai, K.; Ting, J. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 2014, 211, 1333–1347. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Lamkanfi, M.; Kanneganti, T.-D. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J. Immunol. 2015, 194, 2064–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.-M.T.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Cui, J.; Wang, H.Y.; Zhu, L.; Matsueda, S.; Wang, Q.; Yang, X.; Hong, J.; Songyang, Z.; Chen, Z.J. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity 2011, 34, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Walle, L.V.; Jordao, M.J.C. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Palazón-Riquelme, P.; Worboys, J.D.; Green, J.; Valera, A.; Martín-Sánchez, F.; Pellegrini, C.; Brough, D.; López-Castejón, G. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018, 19, e44766. [Google Scholar] [CrossRef]
- Imaizumi, T.; Hayakari, R.; Watanabe, S.; Aizawa, T.; Matsumiya, T.; Yoshida, H.; Tsuruga, K.; Kawaguchi, S.; Tanaka, H. Cylindromatosis (CYLD), a Deubiquitinase, Attenuates Inflammatory Signaling Pathways by Activating Toll-Like Receptor 3 in Human Mesangial Cells. Kidney Blood Press. Res. 2017, 42, 942–950. [Google Scholar] [CrossRef]
- Hrdinka, M.; Fiil, B.K.; Zucca, M.; Leske, D.; Bagola, K.; Yabal, M.; Elliott, P.R.; Damgaard, R.B.; Komander, D.; Jost, P.J.; et al. CYLD Limits Lys63- and Met1-Linked Ubiquitin at Receptor Complexes to Regulate Innate Immune Signaling. Cell Rep. 2016, 14, 2846–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, S.; Pagano, J.S. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 2010, 84, 6130–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, H.; Zhong, B.; Blonska, M.; Gorjestani, S.; Yan, M.; Tian, Q.; Zhang, D.E.; Lin, X.; Dong, C. USP18 inhibits NF-κB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J. Exp. Med. 2013, 210, 1575–1590. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; de Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874. [Google Scholar] [CrossRef] [Green Version]
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J. US Food and Drug Administration approval: Carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Hari, P.; Matous, J.V.; Voorhees, P.M.; Shain, K.H.; Obreja, M.; Frye, J.; Fujii, H.; Jakubowiak, A.J.; Rossi, D.; Sonneveld, P. Oprozomib in patients with newly diagnosed multiple myeloma. Blood Cancer J. 2019, 9, 1–4. [Google Scholar] [CrossRef]
- Dhakal, B.; D’Souza, A.; Hamadani, M.; Arce-Lara, C.; Schroeder, K.; Chhabra, S.; Shah, N.N.; Gauger, K.; Keaton, T.; Pasquini, M. Phase I/II trial of bendamustine, ixazomib, and dexamethasone in relapsed/refractory multiple myeloma. Blood Cancer J. 2019, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, A.C.; Bauer, T.M.; Aggarwal, C.; Lee, C.B.; Harvey, R.D.; Cohen, R.B.; Sedarati, F.; Nip, T.K.; Faessel, H.; Dash, A.B. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Investig. New Drugs. 2019, 37, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.; Rubinstein, L.; Arbuck, S.G.; Christian, M.C.; Freidlin, B.; Collins, J. Accelerated titration designs for phase I clinical trials in oncology. J. Natl. Cancer Inst. 1997, 89, 1138–1147. [Google Scholar] [CrossRef]
- Mattern, M.R.; Wu, J.; Nicholson, B. Ubiquitin-based anticancer therapy: Carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim. Biophys. Acta 2012, 1823, 2014–2021. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Luo, H. The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 2006, 84, 5–14. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Nagai, A.; Kadowaki, H.; Maruyama, T.; Takeda, K.; Nishitoh, H.; Ichijo, H. USP14 inhibits ER-associated degradation via interaction with IRE1alpha. Biochem. Biophys. Res. Commun. 2009, 379, 995–1000. [Google Scholar] [CrossRef]
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N.J. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010, 70, 9265–9276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkholder, K.M.; Perry, J.W.; Wobus, C.E.; Donato, N.J.; Showalter, H.D.; Kapuria, V.; O’Riordan, M.X. A small molecule deubiquitinase inhibitor increases localization of inducible nitric oxide synthase to the macrophage phagosome and enhances bacterial killing. Infect. Immun. 2011, 79, 4850–4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.W.; Ahmed, M.; Chang, K.O.; Donato, N.J.; Showalter, H.D.; Wobus, C.E. Antiviral activity of a small molecule deubiquitinase inhibitor occurs via induction of the unfolded protein response. PLoS Pathog. 2012, 8, e1002783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charbonneau, M.E.; Gonzalez-Hernandez, M.J.; Showalter, H.D.; Donato, N.J.; Wobus, C.E.; O’Riordan, M.X. Small molecule deubiquitinase inhibitors promote macrophage anti-infective capacity. PLoS ONE 2014, 9, e104096. [Google Scholar] [CrossRef]
- Passalacqua, K.D.; Charbonneau, M.E.; Donato, N.J.; Showalter, H.D.; Sun, D.; Wen, B.; He, M.; Sun, H.; O’Riordan, M.X.; Wobus, C.E. Anti-infective Activity of 2-Cyano-3-Acrylamide Inhibitors with Improved Drug-Like Properties against Two Intracellular Pathogens. Antimicrob. Agents Chemother. 2016, 60, 4183–4196. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.F.; Hibbs, J.B., Jr. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 1991, 3, 65–70. [Google Scholar] [CrossRef]
- Hornung, V.; Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 2010, 10, 123–130. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Luheshi, N.M.; Compan, V.; High, S.; Whitehead, R.C.; Flitsch, S.; Kirov, A.; Prudovsky, I.; Swanton, E.; Brough, D. Deubiquitinases regulate the activity of caspase-1 and interleukin-1beta secretion via assembly of the inflammasome. J. Biol. Chem. 2013, 288, 2721–2733. [Google Scholar] [CrossRef] [Green Version]
- Cross, B.C.; McKibbin, C.; Callan, A.C.; Roboti, P.; Piacenti, M.; Rabu, C.; Wilson, C.M.; Whitehead, R.; Flitsch, S.L.; Pool, M.R.; et al. Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J. Cell Sci. 2009, 122, 4393–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKibbin, C.; Mares, A.; Piacenti, M.; Williams, H.; Roboti, P.; Puumalainen, M.; Callan, A.C.; Lesiak-Mieczkowska, K.; Linder, S.; Harant, H.; et al. Inhibition of protein translocation at the endoplasmic reticulum promotes activation of the unfolded protein response. Biochem. J. 2012, 442, 639–648. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknas, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef]
- Park, Y.; Jin, H.-S.; Aki, D.; Lee, J.; Liu, Y.-C. The ubiquitin system in immune regulation. Adv. Immunol. 2014, 124, 17–66. [Google Scholar] [PubMed]
- Liu, S.; Chen, Z.J. Expanding role of ubiquitination in NF-κB signaling. Cell Res. 2011, 21, 6–21. [Google Scholar] [CrossRef]
- Temmerman, S.T.; Ma, C.A.; Borges, L.; Kubin, M.; Liu, S.; Derry, J.M.; Jain, A. Impaired dendritic-cell function in ectodermal dysplasia with immune deficiency is linked to defective NEMO ubiquitination. Blood 2006, 108, 2324–2331. [Google Scholar] [CrossRef]
- Hu, H.; Sun, S.-C. Ubiquitin signaling in immune responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronski, M.A.; Boulter, J.M.; Moskophidis, D.; Nguyen, L.T.; Holmberg, K.; Elford, A.R.; Deenick, E.K.; Kim, H.O.; Penninger, J.M.; Odermatt, B. TCR affinity and negative regulation limit autoimmunity. Nat. Med. 2004, 10, 1234–1239. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Machner, M.P. Exploitation of the host cell ubiquitin machinery by microbial effector proteins. J. Cell Sci. 2017, 130, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Charbonneau, M.-E.; Passalacqua, K.D.; Hagen, S.E.; Showalter, H.D.; Wobus, C.E.; O’Riordan, M.X. Perturbation of ubiquitin homeostasis promotes macrophage oxidative defenses. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, P.; Wang, X.; Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54. [Google Scholar] [CrossRef] [Green Version]
- Osinalde, N.; Duarri, A.; Ramirez, J.; Barrio, R.; de Nanclares, G.P.; Mayor, U. Impaired proteostasis in rare neurological diseases. Semin. Cell Dev. Biol. 2019, 93, 164–177. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Edelmann, M.J. Deubiquitinases: Novel therapeutic targets in immune surveillance? Mediators Inflamm. 2016, 2016, 3481371. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Pathway | DUBs | Target | Mechanism | Reference |
|---|---|---|---|---|
| RLR | CYLD | RIG-I, MAVS, TBK1, IKKε, TRAF2, TRAF3, TRAF6 | Deubiquitinates K63-Ub chain | [41,44,90,110,111] |
| A20 | TBK1, IKKi, TRAF6, IRF7 | Deubiquitinates K63-Ub chain | [47,90,112] | |
| USP21 | RIG-I, MDA5 | Deubiquitinates K63-Ub chain, Inhibits TRIM25 and RNF135 | [51,52] | |
| USP3 | RIG-I, MDA5 | Deubiquitinates K63-Ub chain | [53] | |
| USP15 | RIG-I | Deubiquitinates K63-Ub chain | [54,55] | |
| USP25 | RIG-I, TRAF2, TRAF3, TRAF6 | Deubiquitinates K63-Ub chain from RIG-I and TRAF6, Deubiquitinates K48-Ub chain from TRAF3 | [57,58] | |
| USP14 | RIG-I | Deubiquitinates K63-Ub chain | [59] | |
| USP18 | TAK1, NEMO | Recruits USP20 to deconjugate K48-Ub chain from STING | [60] | |
| USP4 | RIG-I | Deubiquitinates K63-Ub chain | [61,62] | |
| USP47 | RIG-I | Deubiquitinates K63-Ub chain | [62] | |
| TLR | USP2/USP2a | TRAF6 | Deubiquitinates K63-Ub chain | [70,71] |
| USP4 | TAK1, TRAF2, TRAF6 | Deubiquitinates K63-Ub from TAK1 and TRAF6, Deubiquitinates K48-Ub from TRAF6, rescues IκBα from degradation | [72,73,74] | |
| USP7 | NF-κB, NEMO, TRAF5 | Deubiquitination | [75,76,77,78,79,80] | |
| USP10 | NEMO, TRAF6 | Deubiquitinates K63-Ub from TRAF6, Decrease M1-linked Ub from NEMO | [81,82] | |
| USP18 | TAK1, NEMO | Deubiquitinates K63-Ub from TAK1, masks ubiquitin sites of NEMO | [83,113] | |
| USP25 | TRAF3 | Deubiquitinates K48-Ub | [57,84,85] | |
| USP12 | NF-κB | promotes LPS induced signaling through the dephosphorylation of IκBα. | [86,87] | |
| A20 | TRAF6, RIPK1, NEMO | Deubiquitinates K63-Ub from RIPK1, attaches K48-Ub to RIPK1, inhibits ubiquitination of TRAF6, blocks NF-κB activation by binding to NEMO | [88,89,90,91] | |
| CYLD | MyD88, RIPK1, TRAF2, NEMO | Deubiquitinates K63-Ub from MyD88, RIPK1, TRAF2 and linear-Ub from NEMO | [43,92,93] | |
| MYSM1 | TRAF3, TRAF6 | Deubiquitinates K63-Ub | [94] | |
| NLR | A20 | RIPK2, RIPK3 | Deubiquitinates RIPK2 | [107,108] |
| BRCC3 | NLRP3 | Deubiquitinates K63-Ub | [99,100] | |
| USP7 | NF-κB-p65, NLRP3 | Deubiquitinates K48-Ub from NF-κB-p65, regulates NLRP3 directly or indirectly | [21,79] | |
| USP47 | NLRP3 | Regulates NLRP3 directly or indirectly | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasaei, R.; Sarodaya, N.; Kim, K.-S.; Ramakrishna, S.; Hong, S.-H. Importance of Deubiquitination in Macrophage-Mediated Viral Response and Inflammation. Int. J. Mol. Sci. 2020, 21, 8090. https://doi.org/10.3390/ijms21218090
Rasaei R, Sarodaya N, Kim K-S, Ramakrishna S, Hong S-H. Importance of Deubiquitination in Macrophage-Mediated Viral Response and Inflammation. International Journal of Molecular Sciences. 2020; 21(21):8090. https://doi.org/10.3390/ijms21218090
Chicago/Turabian StyleRasaei, Roya, Neha Sarodaya, Kye-Seong Kim, Suresh Ramakrishna, and Seok-Ho Hong. 2020. "Importance of Deubiquitination in Macrophage-Mediated Viral Response and Inflammation" International Journal of Molecular Sciences 21, no. 21: 8090. https://doi.org/10.3390/ijms21218090