Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons


Abstract
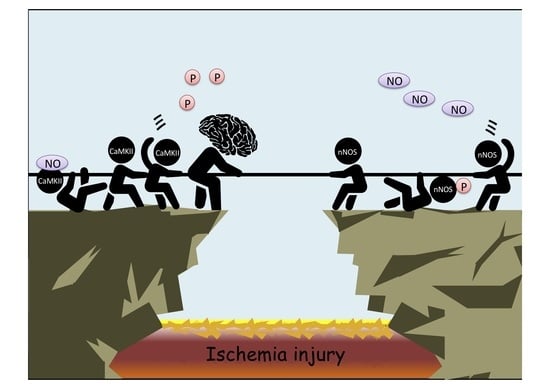
:
1. Introduction
2. Regulation of nNOS by CaMKII
2.1. Molecular Mechanism
2.2. Experimental Models
2.2.1. Cultured Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue/Cell Type | Method | Outcome | Reference |
|---|---|---|---|
| Mouse neostriatal slices | Treatment with calyculin A or okadaic ocid | Inhibition of glutamate/NO signal | [47] |
| Rat hippocampal neurons | Treatment with glutamate (low/high concentration) | Inhibition of NO production | [49] |
| Mouse hippocampal HT22 cells | Treatment of cells exposed CoCl2 with propofol | Neuroprotective effects | [48] |
| CGCs1 cells, PC12 cells (nNOS expressed) | Treatment with nicotine | Neuroprotective effects | [28] |
| Rat hippocampus | Ischemia/reperfusion | Neuroprotective effects | [54] |
| Rat hippocampus | SAH (increasing ICP2) | Neuroprotective effects | [55] |
| Rat hippocampus | Treatment of GABA3 agonists | Neuroprotective effects | [56] |
| Rat hippocampus | Transient ischemic preconditionning | Neuroprotective effects | [57] |
| Rat hippocampus | Ischemia/reperfusion | Neuroprotective effects | [58] |
| Rat hippocampus | Ischemia/reperfusion | Neuroprotective effects | [59] |
| Mouse hippocampus | Hypothermia | Neuroprotective effects | [60] |
| Hamster suprachiasmatic nuclei | Diumal circadian | Photic circadian entrainment | [61] |
| Mouse spinal cords | Peripheral nerve injury | Neuroprotective effects | [62] |
| Rat supraoptic/paraventricular nuclei | Treatment of prolactin | Inhibition of vasopressin/oxytocin secretion | [63] |
| Mouse nucleus intermediolateralis | Spinal cord injury | Autonomic failure | [64] |
| Mouse periaqueductal grey matter | Treatment of morphine | Analgesic tolerance | [65] |
| Rat cerebellum | Hyperammonemia | Inhibition of NO production | [66] |
2.2.2. Animal Models
3. Regulation of CaMKII by nNOS
3.1. Molecular Mechanisms
3.2. Experimental Models
Cultured Cells
4. Mutual Regulation of CaMKII and nNOS
5. Small Molecule Inhibitors of CaMKII, a Further Update
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CaM | calmodulin |
| CaMK | Ca2+/calmodulin-dependent protein kinase |
| FAD | flavin adenine dinucleotide |
| FMN | flavin mononucleotide |
| GABA | γ-aminobutyric acid |
| LTP | long-term potentiation |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NMDA | N-methyl-D-Aspartate |
| NMDAR | NMDA receptor |
| NO | nitric oxide |
| NOS | NO synthase |
| nNOS | neuronal NO synthase |
| PPase | protein phosphatase |
| PP1 | protein phosphatase 1 |
| PP2A | protein phosphatase 2A |
| PSD | post-synaptic density |
| PSD-95 | postsynaptic density 95 kDa |
| RSS | reactive sulfur species |
References
- Erondu, N.E.; Kennedy, M.B. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J. Neurosci. 1985, 5, 3270–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Kim, M.J.; Cheng, D.; Duong, D.M.; Gygi, S.P.; Sheng, M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 2004, 279, 21003–21011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Hoogenraad, C.C.; Rush, J.; Ramm, E.; Schlager, M.A.; Duong, D.M.; Xu, P.; Wijayawardana, S.R.; Hanfelt, J.; Nakagawa, T.; et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteom. 2006, 5, 1158–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao-Cheng, J.H.; Dosemeci, A.; Winters, C.A.; Reese, T.S. Changes in the distribution of calcium calmodulin-dependent protein kinase II at the presynaptic bouton after depolarization. Brain Cell Biol. 2006, 35, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Choi, S.; Lovinger, D.M.; Colbran, R.J. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J. Biol. Chem. 1997, 272, 13467–13470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.; Meyer, T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 1999, 284, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Dosemeci, A.; Tao-Cheng, J.H.; Vinade, L.; Winters, C.A.; Pozzo-Miller, L.; Reese, T.S. Glutamate-induced transient modification of the postsynaptic density. Proc. Natl. Acad. Sci. USA 2001, 98, 10428–10432. [Google Scholar] [CrossRef] [Green Version]
- Bayer, K.U.; De Koninck, P.; Leonard, A.S.; Hell, J.W.; Schulman, H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 2001, 411, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Colbran, R.J.; Brown, A.M. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr. Opin. Neurobiol. 2004, 14, 318–327. [Google Scholar] [CrossRef]
- Strack, S.; Colbran, R.J. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J. Biol. Chem. 1998, 273, 20689–20692. [Google Scholar] [CrossRef] [Green Version]
- Bayer, K.U.; LeBel, E.; McDonald, G.L.; O’Leary, H.; Schulman, H.; De Koninck, P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 2006, 26, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Merrill, M.A.; Chen, Y.; Strack, S.; Hell, J.W. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol. Sci. 2005, 26, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, B.; Ge, Q.; Wang, Z.W. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J. Neurosci. 2007, 27, 10404–10413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, J. Criteria for identifying the molecular basis of the engram (CaMKII, PKMzeta). Mol. Brain 2017, 10, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacktor, T.C.; Fenton, A.A. What does LTP tell us about the roles of CaMKII and PKMζ in memory? Mol. Brain 2018, 11, 77. [Google Scholar] [CrossRef]
- Vigil, F.A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and memory destabilization: A new role in memory maintenance. J. Neurochem. 2018, 147, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Coultrap, S.J.; Vest, R.S.; Ashpole, N.M.; Hudmon, A.; Bayer, K.U. CaMKII in cerebral ischemia. Acta Pharmacol. Sin. 2011, 32, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, L.; Chen, Y.; Zhan, L.; Gao, Z. Therapeutic Targets for Cerebral Ischemia Based on the Signaling Pathways of the GluN2B C Terminus. Stroke 2015, 46, 2347–2353. [Google Scholar] [CrossRef] [Green Version]
- Ashpole, N.M.; Hudmon, A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol. Cell Neurosci. 2011, 46, 720–730. [Google Scholar] [CrossRef]
- Fukunaga, K.; Muller, D.; Ohmitsu, M.; Bako, E.; DePaoli-Roach, A.A.; Miyamoto, E. Decreased protein phosphatase 2A activity in hippocampal long-term potentiation. J. Neurochem. 2000, 74, 807–817. [Google Scholar] [CrossRef]
- Lisman, J.E.; Zhabotinsky, A.M. A model of synaptic memory: A CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 2001, 31, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Soderling, T.R.; Derkach, V.A. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000, 23, 75–80. [Google Scholar] [CrossRef]
- Garthwaite, J. NO as a multimodal transmitter in the brain: Discovery and current status. Br. J. Pharmacol. 2019, 176, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Lamas, S.; Fang, F.C. Nitrosylation. The prototypic redox-based signaling mechanism. Cell 2001, 106, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Pou, S.; Pou, W.S.; Bredt, D.S.; Snyder, S.H.; Rosen, G.M. Generation of superoxide by purified brain nitric oxide synthase. J. Biol. Chem. 1992, 267, 24173–24176. [Google Scholar] [PubMed]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasamatsu, S.; Watanabe, Y.; Sawa, T.; Akaike, T.; Ihara, H. Redox signal regulation via nNOS phosphorylation at Ser847 in PC12 cells and rat cerebellar granule neurons. Biochem. J. 2014, 459, 251–263. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef] [Green Version]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballarin, B.; Tymianski, M. Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharmacol. Sin. 2018, 39, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; Terbrugge, K.G.; Milot, G.; Clark, W.M.; Macdonald, R.L.; Kelly, M.E.; et al. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [Green Version]
- Terasaki, Y.; Sasaki, T.; Yagita, Y.; Okazaki, S.; Sugiyama, Y.; Oyama, N.; Omura-Matsuoka, E.; Sakoda, S.; Kitagawa, K. Activation of NR2A receptors induces ischemic tolerance through CREB signaling. J. Cereb. Blood Flow Metab. 2010, 30, 1441–1449. [Google Scholar] [CrossRef]
- Vieira, M.; Yong, X.L.H.; Roche, K.W.; Anggono, V. Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J. Neurochem. 2020, 154, 121–143. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Nishio, M.; Naito, Y.; Yokokura, H.; Nimura, Y.; Hidaka, H.; Watanabe, Y. Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J. Biol. Chem. 1999, 274, 20597–20602. [Google Scholar] [CrossRef] [Green Version]
- Komeima, K.; Hayashi, Y.; Naito, Y.; Watanabe, Y. Inhibition of neuronal nitric-oxide synthase by calcium/calmodulin-dependent protein kinase IIalpha through Ser847 phosphorylation in NG108-15 neuronal cells. J. Biol. Chem. 2000, 275, 28139–28143. [Google Scholar]
- Song, T.; Hatano, N.; Kambe, T.; Miyamoto, Y.; Ihara, H.; Yamamoto, H.; Sugimoto, K.; Kume, K.; Yamaguchi, F.; Tokuda, M.; et al. Nitric oxide-mediated modulation of calcium/calmodulin-dependent protein kinase II. Biochem. J. 2008, 412, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Iyanagi, T. Calmodulin activates intramolecular electron transfer between the two flavins of neuronal nitric oxide synthase flavin domain. Biochim. Biophys. Acta 1999, 1473, 345–355. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Hatano, N.; Horii, M.; Tokumitsu, H.; Yamaguchi, F.; Tokuda, M.; Watanabe, Y. Calcium/calmodulin-dependent protein kinase I inhibits neuronal nitric-oxide synthase activity through serine 741 phosphorylation. FEBS Lett. 2004, 570, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Song, T.; Sugimoto, K.; Horii, M.; Araki, N.; Tokumitsu, H.; Tezuka, T.; Yamamoto, T.; Tokuda, M. Post-synaptic density-95 promotes calcium/calmodulin-dependent protein kinase II-mediated Ser847 phosphorylation of neuronal nitric oxide synthase. Biochem. J. 2003, 372, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Komeima, K.; Watanabe, Y. Dephosphorylation of nNOS at Ser(847) by protein phosphatase 2A. FEBS Lett. 2001, 497, 65–66. [Google Scholar] [CrossRef] [Green Version]
- Takai, A.; Sasaki, K.; Nagai, H.; Mieskes, G.; Isobe, M.; Isono, K.; Yasumoto, T. Inhibition of specific binding of okadaic acid to protein phosphatase 2A by microcystin-LR, calyculin-A and tautomycin: Method of analysis of interactions of tight-binding ligands with target protein. Biochem. J. 1995, 306, 657–665. [Google Scholar] [CrossRef]
- Nishi, A.; Watanabe, Y.; Higashi, H.; Tanaka, M.; Nairn, A.C.; Greengard, P. Glutamate regulation of DARPP-32 phosphorylation in neostriatal neurons involves activation of multiple signaling cascades. Proc. Natl. Acad. Sci. USA 2005, 102, 1199–1204. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, W.; Lin, C.; Wang, J.; Zhu, M.; Chen, J.; Miao, C. The protective effects of propofol against CoCl(2)-induced HT22 cell hypoxia injury via PP2A/CAMKIIα/nNOS pathway. BMC Anesthesiol. 2017, 17, 32. [Google Scholar] [CrossRef] [Green Version]
- Rameau, G.A.; Chiu, L.Y.; Ziff, E.B. Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-D-aspartate receptor. J. Biol. Chem. 2004, 279, 14307–14314. [Google Scholar] [CrossRef] [Green Version]
- Colbran, R.J. Protein phosphatases and calcium/calmodulin-dependent protein kinase II-dependent synaptic plasticity. J. Neurosci. 2004, 24, 8404–8409. [Google Scholar] [CrossRef]
- Song, T.; Hatano, N.; Sugimoto, K.; Horii, M.; Yamaguchi, F.; Tokuda, M.; Miyamoto, Y.; Kambe, T.; Watanabe, Y. Nitric oxide prevents phosphorylation of neuronal nitric oxide synthase at serine1412 by inhibiting the Akt/PKB and CaM-K II signaling pathways. Int. J. Mol. Med. 2012, 30, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adak, S.; Santolini, J.; Tikunova, S.; Wang, Q.; Johnson, J.D.; Stuehr, D.J. Neuronal nitric-oxide synthase mutant (Ser-1412 --> Asp) demonstrates surprising connections between heme reduction, NO complex formation, and catalysis. J. Biol. Chem. 2001, 276, 1244–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, T.; Zaki, M.H.; Okamoto, T.; Akuta, T.; Tokutomi, Y.; Kim-Mitsuyama, S.; Ihara, H.; Kobayashi, A.; Yamamoto, M.; Fujii, S.; et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3’,5’-cyclic monophosphate. Nat. Chem. Biol. 2007, 3, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Osuka, K.; Watanabe, Y.; Usuda, N.; Nakazawa, A.; Fukunaga, K.; Miyamoto, E.; Takayasu, M.; Tokuda, M.; Yoshida, J. Phosphorylation of neuronal nitric oxide synthase at Ser847 by CaM-KII in the hippocampus of rat brain after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2002, 22, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, K.; Osuka, K.; Watanabe, Y.; Usuda, N.; Hara, M.; Aoyama, M.; Takayasu, M.; Wakabayashi, T. Increased ICP promotes CaMKII-mediated phosphorylation of neuronal NOS at Ser⁸⁴⁷ in the hippocampus immediately after subarachnoid hemorrhage. Brain Res. 2015, 1616, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, C.; Yu, H.M.; Zhang, F.; Han, D.; Zhang, G.Y. Neuroprotection of gamma-aminobutyric acid receptor agonists via enhancing neuronal nitric oxide synthase (Ser847) phosphorylation through increased neuronal nitric oxide synthase and PSD95 interaction and inhibited protein phosphatase activity in cerebral ischemia. J. Neurosci. Res. 2008, 86, 2973–2983. [Google Scholar] [PubMed]
- Wang, M.; Qi, D.S.; Zhou, C.; Han, D.; Li, P.P.; Zhang, F.; Zhou, X.Y.; Han, M.; Di, J.H.; Ye, J.S.; et al. Ischemic preconditioning protects the brain against injury via inhibiting CaMKII-nNOS signaling pathway. Brain Res. 2016, 1634, 140–149. [Google Scholar] [CrossRef]
- Qu, Z.W.; Miao, W.Y.; Hu, S.Q.; Li, C.; Zhuo, X.L.; Zong, Y.Y.; Wu, Y.P.; Zhang, G.Y. N-methyl-D-aspartate receptor-dependent denitrosylation of neuronal nitric oxide synthase increase the enzyme activity. PLoS ONE 2012, 7, e52788. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.M.; Zhang, T.Y.; Yin, X.H.; Yang, Q.; Lu, F.; Yan, J.Z.; Li, C. Denitrosylation of nNOS induced by cerebral ischemia-reperfusion contributes to nitrosylation of CaMKII and its inhibition of autophosphorylation in hippocampal CA1. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7674–7683. [Google Scholar]
- Hayashi, S.; Osuka, K.; Watanabe, Y.; Yasuda, M.; Takayasu, M.; Wakabayashi, T. Hypothermia enhances the colocalization of calmodulin kinase IIα with neuronal nitric oxide synthase in the hippocampus following cerebral ischemia. Neurosci. Lett. 2011, 505, 228–232. [Google Scholar] [CrossRef]
- Agostino, P.V.; Ferreyra, G.A.; Murad, A.D.; Watanabe, Y.; Golombek, D.A. Diurnal, circadian and photic regulation of calcium/calmodulin-dependent kinase II and neuronal nitric oxide synthase in the hamster suprachiasmatic nuclei. Neurochem. Int. 2004, 44, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.R.; Roh, D.H.; Yoon, S.Y.; Choi, H.S.; Kang, S.Y.; Han, H.J.; Beitz, A.J.; Lee, J.H. Astrocyte D-serine modulates the activation of neuronal NOS leading to the development of mechanical allodynia in peripheral neuropathy. Mol. Pain 2019, 15, 1744806919843046. [Google Scholar] [CrossRef]
- Vega, C.; Moreno-Carranza, B.; Zamorano, M.; Quintanar-Stephano, A.; Mendez, I.; Thebault, S.; Martinez de la Escalera, G.; Clapp, C. Prolactin promotes oxytocin and vasopressin release by activating neuronal nitric oxide synthase in the supraoptic and paraventricular nuclei. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1701–R1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuka, K.; Watanabe, Y.; Usuda, N.; Atsuzawa, K.; Aoshima, C.; Yamauchi, K.; Takayasu, M.; Yoshida, J. Phosphorylation of neuronal nitric oxide synthase at Ser847 in the nucleus intermediolateralis after spinal cord injury in mice. Neuroscience 2007, 145, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Blazquez, P.; Rodriguez-Munoz, M.; Garzon, J. Mu-opioid receptors transiently activate the Akt-nNOS pathway to produce sustained potentiation of PKC-mediated NMDAR-CaMKII signaling. PLoS ONE 2010, 5, e11278. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Pastor, A.; Llansola, M.; Felipo, V. Extracellular Protein Kinase A Modulates Intracellular Calcium/Calmodulin-Dependent Protein Kinase II, Nitric Oxide Synthase, and the Glutamate-Nitric Oxide-cGMP Pathway in Cerebellum. Differential Effects in Hyperammonemia. ACS Chem. Neurosci. 2016, 7, 1753–1759. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Almeida, A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1411, 415–436. [Google Scholar] [CrossRef] [Green Version]
- Dawson, V.L.; Kizushi, V.M.; Huang, P.L.; Snyder, S.H.; Dawson, T.M. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J. Neurosci. 1996, 16, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Kitao, Y.; Hori, O. Ischemia-induced neuronal cell death and stress response. Antioxid. Redox Signal. 2007, 9, 573–587. [Google Scholar] [CrossRef]
- Shi, Z.Q.; Sunico, C.R.; McKercher, S.R.; Cui, J.; Feng, G.S.; Nakamura, T.; Lipton, S.A. S-nitrosylated SHP-2 contributes to NMDA receptor-mediated excitotoxicity in acute ischemic stroke. Proc. Natl. Acad. Sci. USA 2013, 110, 3137–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.; Kiuchi, K.; Ishikawa, T.; Ishii, A.; Hagiwara, M.; Nagatsu, T.; Hidaka, H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 1991, 181, 968–975. [Google Scholar] [CrossRef]
- Takenouchi, T.; Sugiura, Y.; Morikawa, T.; Nakanishi, T.; Nagahata, Y.; Sugioka, T.; Honda, K.; Kubo, A.; Hishiki, T.; Matsuura, T.; et al. Therapeutic hypothermia achieves neuroprotection via a decrease in acetylcholine with a concurrent increase in carnitine in the neonatal hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 794–805. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Liu, M.; Liu, Y.Y.; Ma, L.L.; Jiang, Y.; Chen, X.H. Ischemic preconditioning protects against ischemic brain injury. Neural Regen. Res. 2016, 11, 765–770. [Google Scholar]
- Tokumitsu, H.; Chijiwa, T.; Hagiwara, M.; Mizutani, A.; Terasawa, M.; Hidaka, H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1990, 265, 4315–4320. [Google Scholar]
- Takata, T.; Kimura, J.; Tsuchiya, Y.; Naito, Y.; Watanabe, Y. Calcium/calmodulin-dependent protein kinases as potential targets of nitric oxide. Nitric Oxide 2011, 25, 145–152. [Google Scholar] [CrossRef]
- Takamasa Tobimatsu, H.F. Tissue-specific Expression of Four Types of Rat Calmodulin-dependent Protein Kinase I1 mRNAs. J. Biol. Chem. 1989, 264, 17907–17912. [Google Scholar]
- Coultrap, S.J.; Bayer, K.U. Nitric oxide induces Ca2+-independent activity of the Ca2+/calmodulin-dependent protein kinase II (CaMKII). J. Biol. Chem. 2014, 289, 19458–19465. [Google Scholar] [CrossRef] [Green Version]
- Reiner, D.J.; Newton, E.M.; Tian, H.; Thomas, J.H. Diverse behavioural defects caused by mutations in Caenorhabditis elegans unc-43 CaM kinase II. Nature 1999, 402, 199–203. [Google Scholar] [CrossRef]
- Cheung, B.H.; Arellano-Carbajal, F.; Rybicki, I.; de Bono, M. Soluble guanylate cyclases act in neurons exposed to the body fluid to promote C. elegans aggregation behavior. Curr. Biol. 2004, 14, 1105–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.R. Mechanisms of CaMKII Activation in the Heart. Front. Pharmacol. 2014, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.R.; Pereira, L.; Wang, L.; Han, G.; Ferguson, A.; Dao, K.; Copeland, R.J.; Despa, F.; Hart, G.W.; Ripplinger, C.M.; et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013, 502, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Pellicena, P.; Schulman, H. CaMKII inhibitors: From research tools to therapeutic agents. Front. Pharmacol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Vest, R.S.; O’Leary, H.; Coultrap, S.J.; Kindy, M.S.; Bayer, K.U. Effective post-insult neuroprotection by a novel Ca(2+)/ calmodulin-dependent protein kinase II (CaMKII) inhibitor. J. Biol. Chem. 2010, 285, 20675–20682. [Google Scholar] [CrossRef] [Green Version]
- Ledoux, J.; Chartier, D.; Leblanc, N. Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J. Pharmacol. Exp. Ther. 1999, 290, 1165–1174. [Google Scholar]
- Wong, M.H.; Samal, A.B.; Lee, M.; Vlach, J.; Novikov, N.; Niedziela-Majka, A.; Feng, J.Y.; Koltun, D.O.; Brendza, K.M.; Kwon, H.J.; et al. The KN-93 Molecule Inhibits Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII) Activity by Binding to Ca(2+)/CaM. J. Mol. Biol. 2019, 431, 1440–1459. [Google Scholar] [CrossRef]
- Marley, P.D.; Thomson, K.A. The Ca++/calmodulin-dependent protein kinase II inhibitors KN62 and KN93, and their inactive analogues KN04 and KN92, inhibit nicotinic activation of tyrosine hydroxylase in bovine chromaffin cells. Biochem. Biophys. Res. Commun. 1996, 221, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Yanagihara, N.; Fukunaga, K.; Minami, K.; Nakashima, Y.; Kuroiwa, A.; Miyamoto, E.; Izumi, F. Ca(2+)/calmodulin-dependent protein kinase II inhibitor KN-62 inhibits adrenal medullary chromaffin cell functions independent of its action on the kinase. J. Neurochem. 1996, 66, 2517–2522. [Google Scholar] [CrossRef] [PubMed]
- Shirakura, T.; Han, F.; Shiota, N.; Moriguchi, S.; Kasahara, J.; Sato, T.; Shirasaki, Y.; Fukunaga, K. Inhibition of nitric oxide production and protein tyrosine nitration contribute to neuroprotection by a novel calmodulin antagonist, DY-9760e, in the rat microsphere embolism. Biol. Pharm. Bull. 2005, 28, 1658–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, L.; Lu, Z.; Zhu, X.; Xu, W.; Li, L.; Li, X.; Chen, S.; Sun, W.; Xu, E. Hypoxic preconditioning attenuates necroptotic neuronal death induced by global cerebral ischemia via Drp1-dependent signaling pathway mediated by CaMKIIα inactivation in adult rats. FASEB J. 2019, 33, 1313–1329. [Google Scholar] [CrossRef]
- Waxham, M.N.; Grotta, J.C.; Silva, A.J.; Strong, R.; Aronowski, J. Ischemia-induced neuronal damage: A role for calcium/calmodulin-dependent protein kinase II. J. Cereb. Blood Flow Metab. 1996, 16, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Devarie-Baez, N.O.; Zhang, D.; Li, S.; Whorton, A.R.; Xian, M. Direct methods for detection of protein S-nitrosylation. Methods 2013, 62, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.; Fu, M.; Stokes, E.; Wu, L.; Yang, G. H(2)S-Mediated Protein S-Sulfhydration: A Prediction for Its Formation and Regulation. Molecules 2017, 22, 1334. [Google Scholar] [CrossRef]
- Araki, S.; Takata, T.; Tsuchiya, Y.; Watanabe, Y. Reactive sulfur species impair Ca(2+)/calmodulin-dependent protein kinase II via polysulfidation. Biochem. Biophys. Res. Commun. 2019, 508, 550–555. [Google Scholar] [CrossRef]
- Takata, T.; Araki, S.; Tsuchiya, Y.; Watanabe, Y. Persulfide Signaling in Stress-Initiated Calmodulin Kinase Response. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araki, S.; Osuka, K.; Takata, T.; Tsuchiya, Y.; Watanabe, Y. Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons. Int. J. Mol. Sci. 2020, 21, 7997. https://doi.org/10.3390/ijms21217997
Araki S, Osuka K, Takata T, Tsuchiya Y, Watanabe Y. Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons. International Journal of Molecular Sciences. 2020; 21(21):7997. https://doi.org/10.3390/ijms21217997
Chicago/Turabian StyleAraki, Shoma, Koji Osuka, Tsuyoshi Takata, Yukihiro Tsuchiya, and Yasuo Watanabe. 2020. "Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons" International Journal of Molecular Sciences 21, no. 21: 7997. https://doi.org/10.3390/ijms21217997