Advanced Methods for Studying Structure and Interactions of Macrolide Antibiotics

Abstract
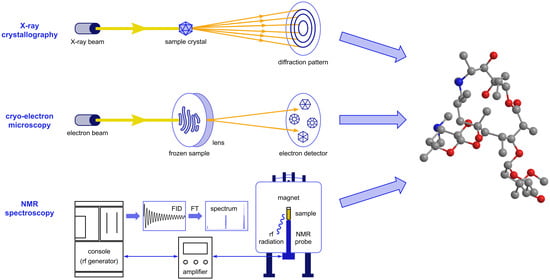
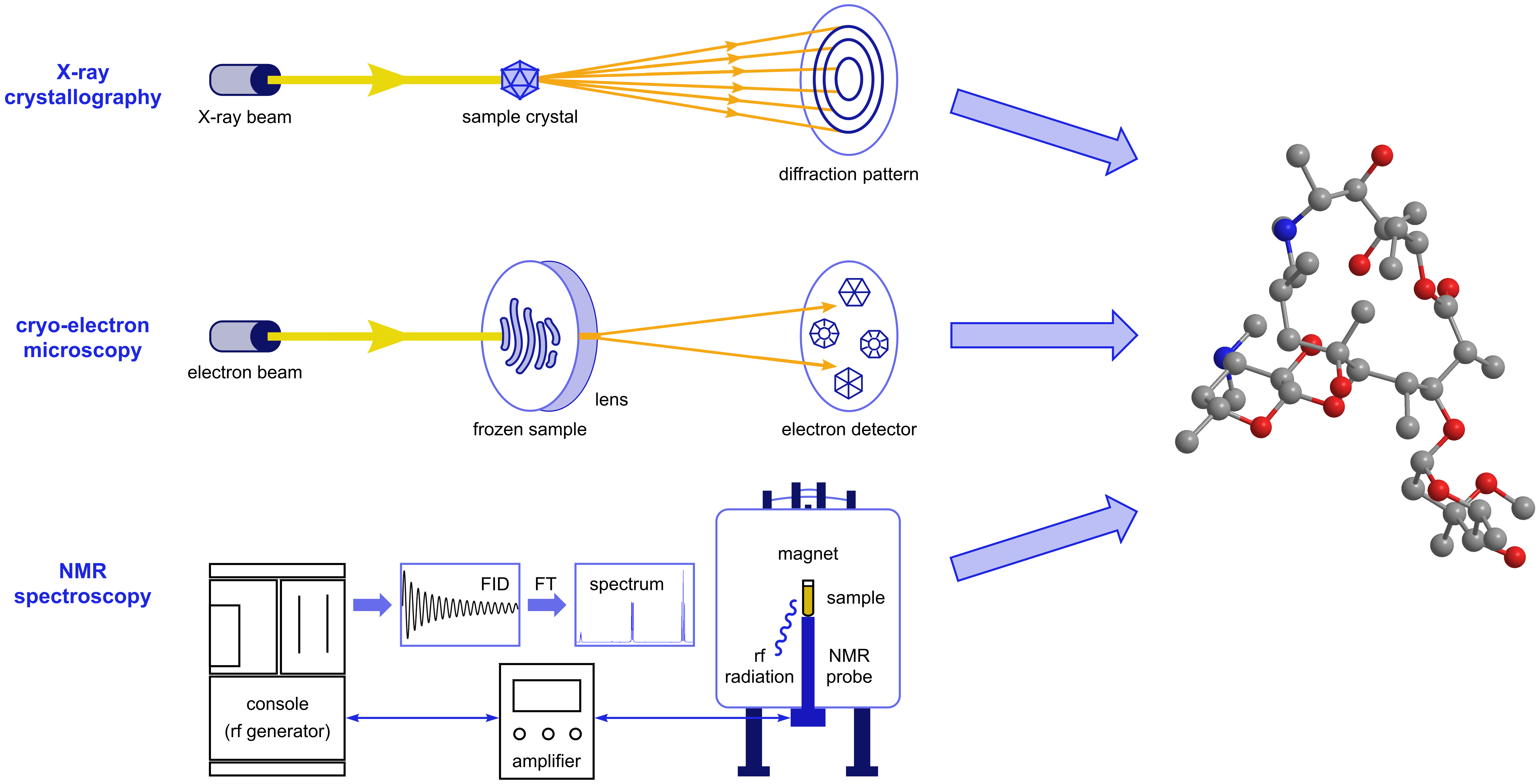
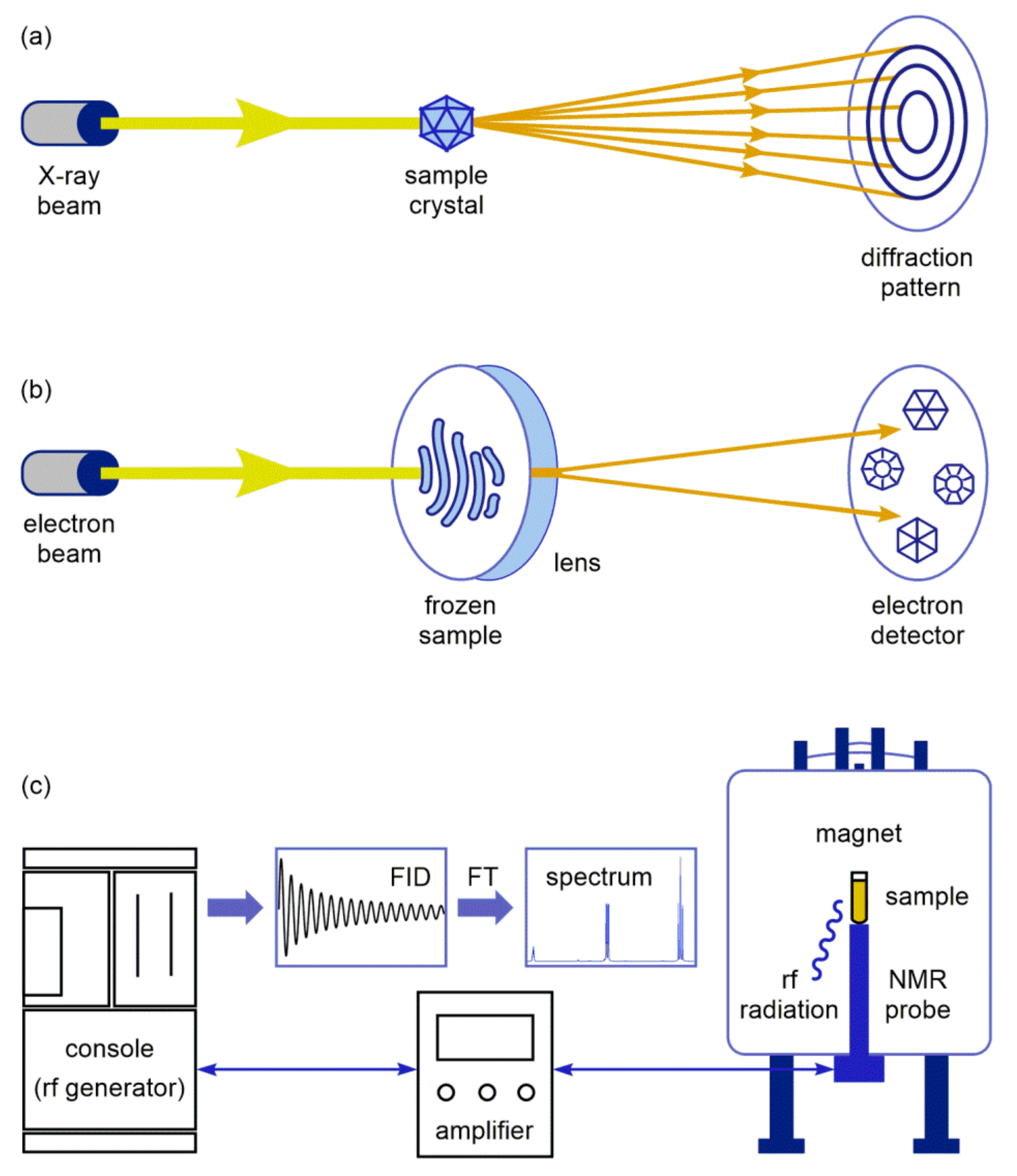
:
1. Introduction
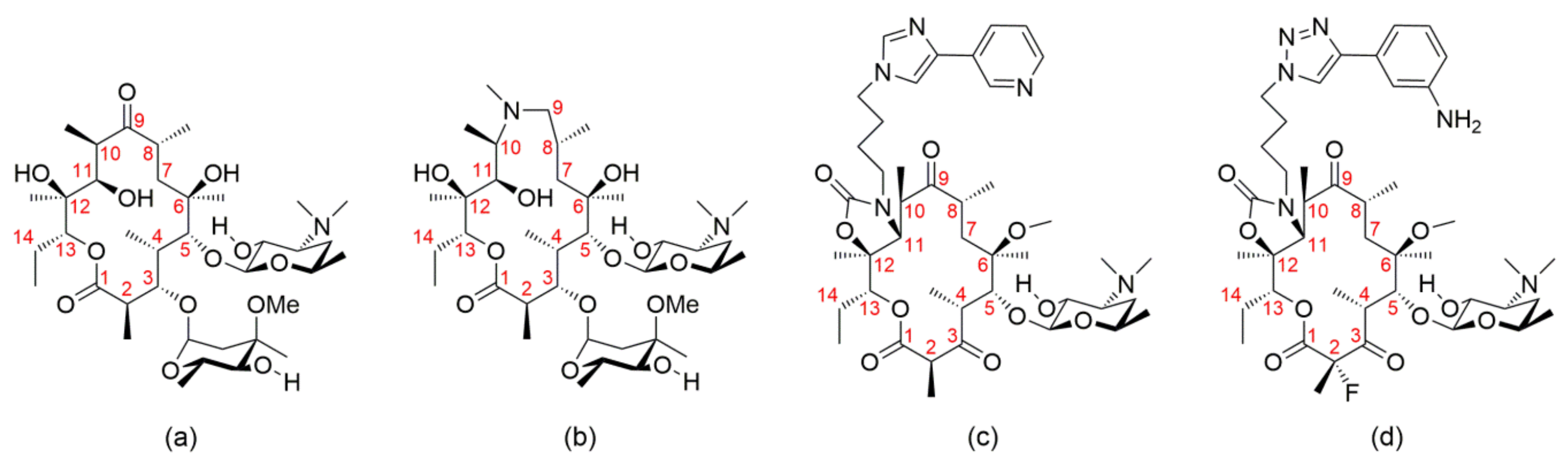
2. X-ray Structure Characterization of Macrolide-Ribosome Complexes
3. Cryo-EM Imaging of Macrolide-Targeted Ribosomes
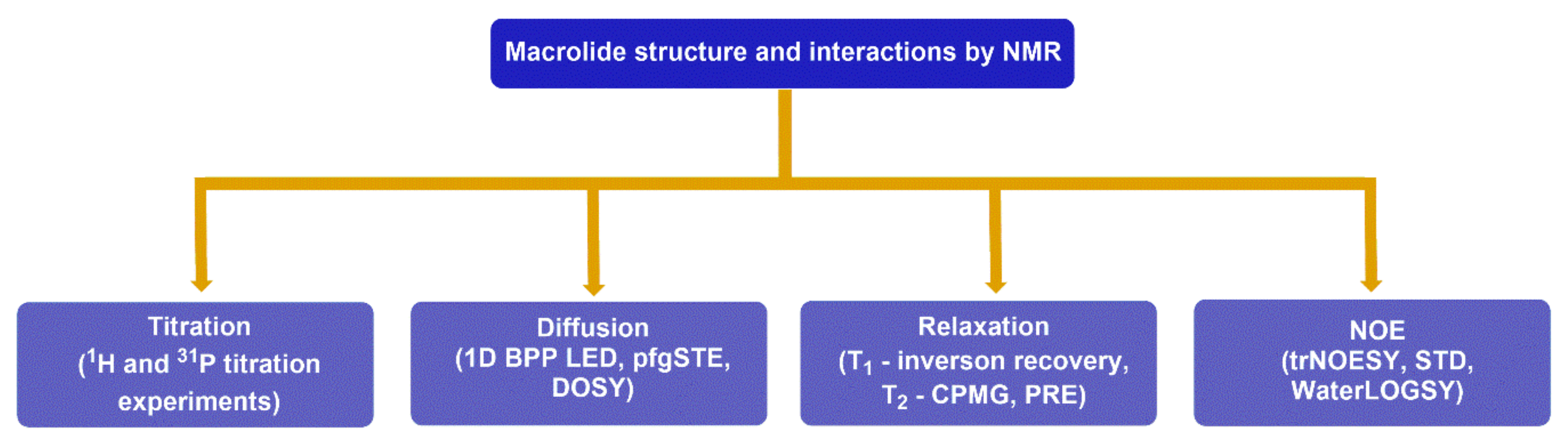
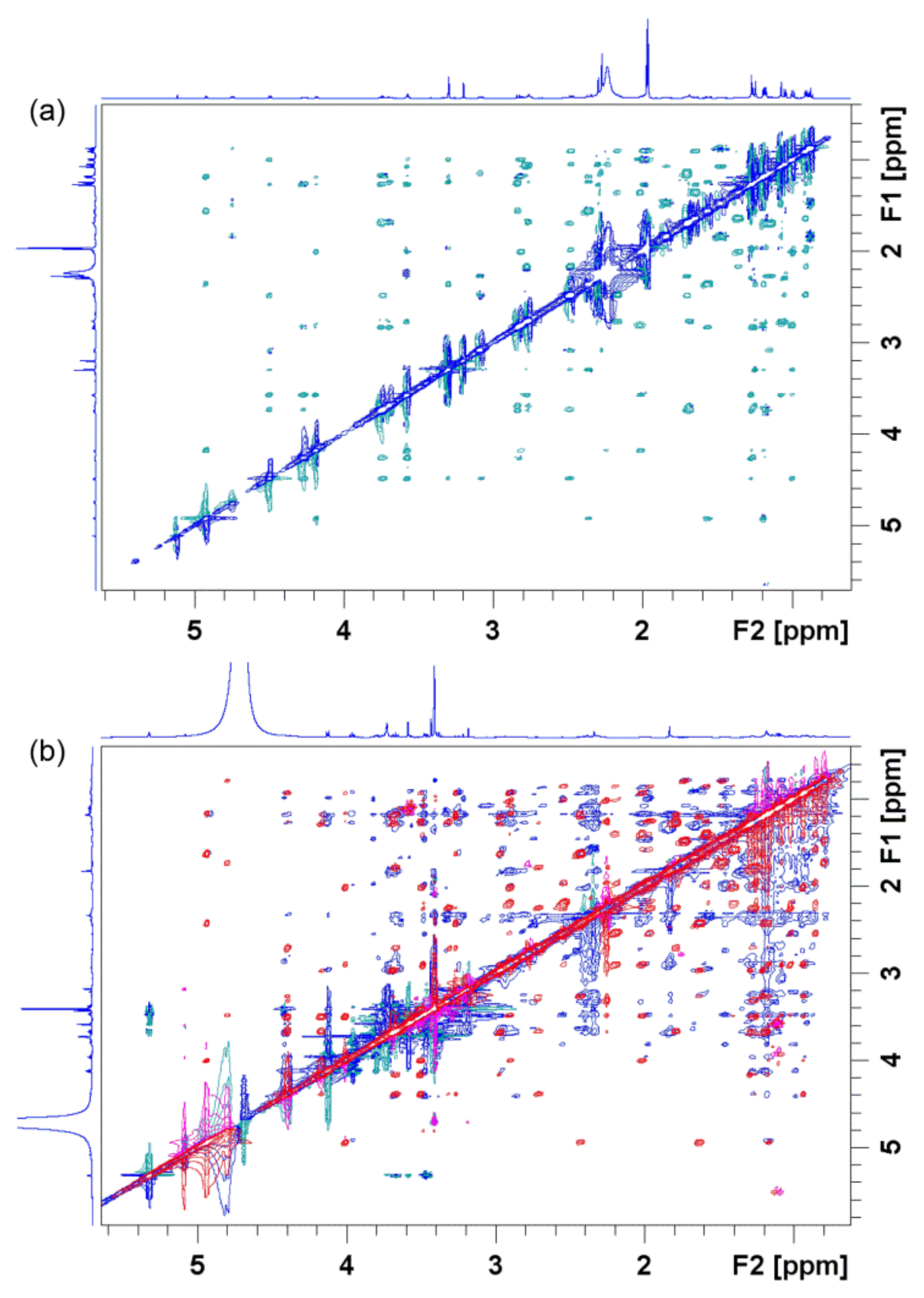
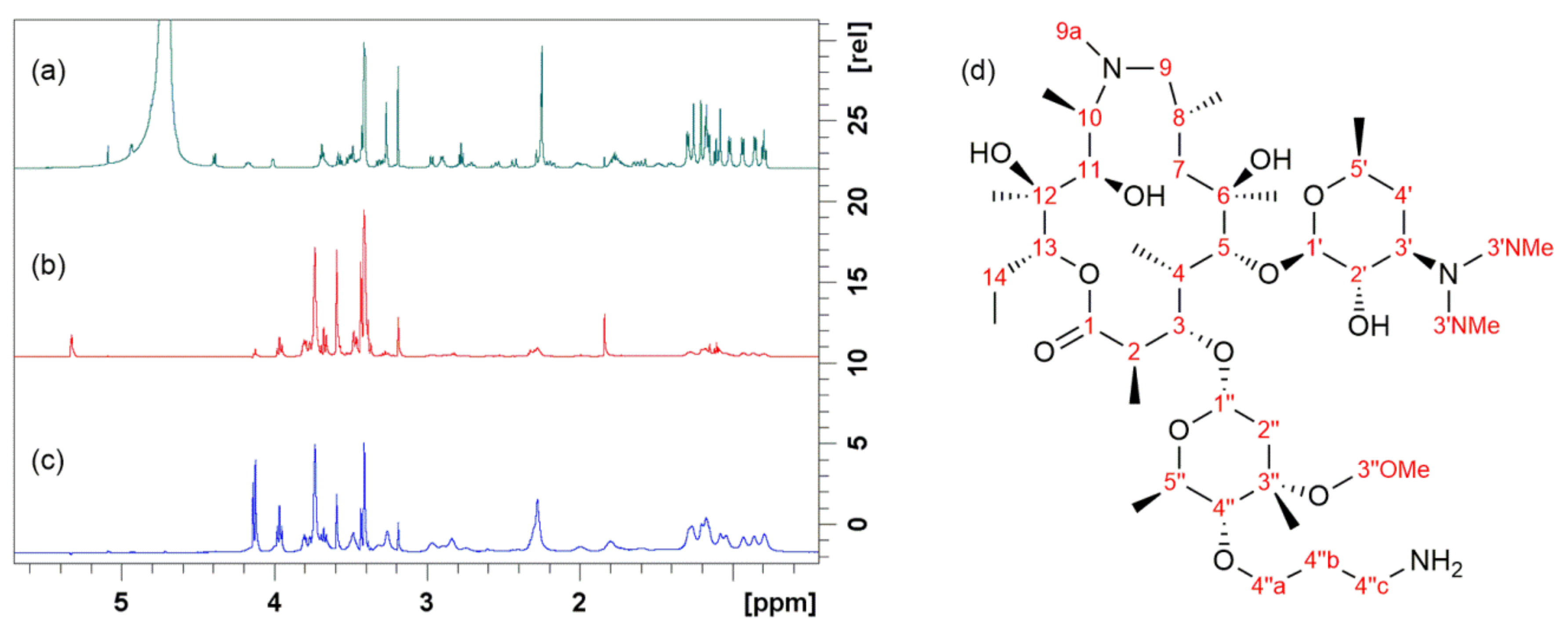
4. Probing Macrolide Interactions by NMR Spectroscopy
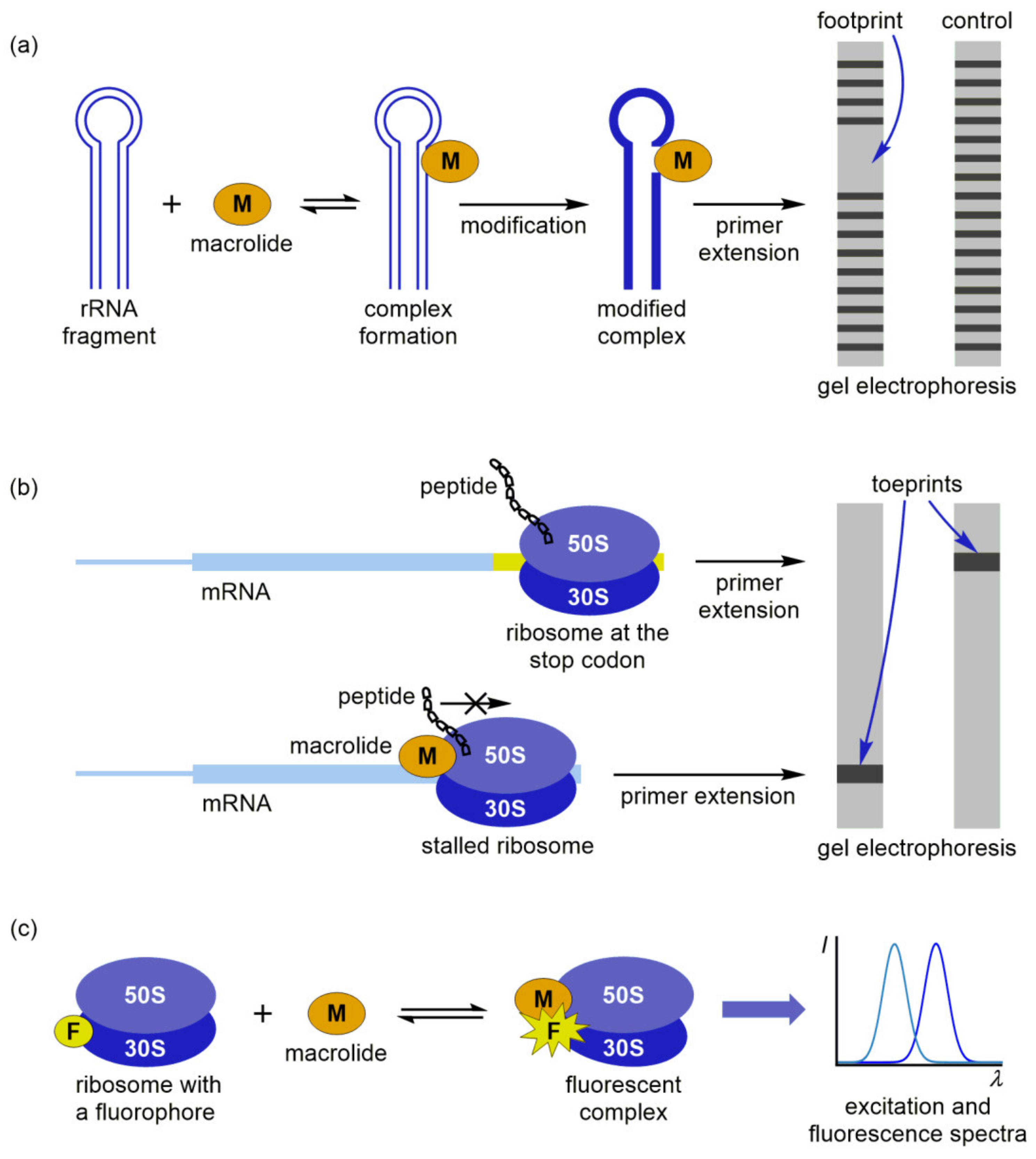
5. Other Methods for Macrolide Binding Studies
6. Computational Simulations of Macrolide Interactions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PTC | peptidyl transferase center |
| FDA | U.S. Food and Drug Administration |
| cryo-EM | cryogenic electron microscopy |
| NMR | nuclear magnetic resonance |
| rf | radiofrequency |
| FID | free induction decay |
| FT | Fourier transformation |
| MD | molecular dynamics |
| NPET | nascent peptide exit tunnel |
| MABP-1 | macrolide antibiotic binding protein-1 |
| SEM | scanning electron microscopy |
| SPA | single-particle analysis |
| cryo-ET | cryogenic electron tomography |
| ErmBL | erythromycin resistance methyltransferase B leader peptide |
| ErmCL | erythromycin resistance methyltransferase C leader peptide |
| ermaa-tRNA | erythromycin resistance methyltransferase geneaminoacyl-tRNA |
| p-tRNA | peptidyl-tRNA |
| TnaCMsrE | tryptophanase leader peptide |
| RSF | macrolide-streptogramin B resistance proteinribosomal silencing factor |
| ABC | ATP-binding cassette |
| NOE | nuclear Overhauser effect |
| STD | saturation transfer difference |
| trNOESY | transferred nuclear Overhauser effect spectroscopy |
| DOSY | diffusion-ordered NMR spectroscopy |
| PREs | paramagnetic relaxation enhancements |
| ROESY | rotating frame nuclear Overhauser effect spectroscopy |
| SDS | sodium dodecylsulphate |
| DPC | dodecylphosphocholine |
| BSA | bovine serum albumin |
| MRSA | methicillin-resistant Staphylococcus aureus |
| MAS | magic angle spinning |
| DOPC | dioleoylphosphatidylcholine |
| ESR | electron spin resonance |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DMPG | 1,2-dimyristoyl-sn-glycero-3-phosphorylglycerol |
| SAR | structure-activity relationship |
| RDC | residual dipolar couplings |
| DMS | dimethyl sulfate |
| CMCT | 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluene sulfonate |
| F | fluorophore |
| FRET | Förster resonance energy transfer |
| QM | quantum mechanical |
| AMBER | Assisted Model Building and Energy Refinement |
| pBLCLZα | erythromycin-inducible ermC-β-galactosidase |
| CHARMM | Chemistry at HARvard Macromolecular Mechanics |
| PES | potential energy surface |
| ffTK | force field toolkit |
| VMD | variable molecular dynamics |
| GCMC | grand canonical Monte Carlo |
| MMFF | Merck Molecular Force Field |
| MOE | MOELowModeMD |
| MC | MacroModel |
References
- Retsema, J.; Fu, W. Macrolides: Structures and microbial targets. Int. J. Antimicrob. Agents 2001, 18, S3–S10. [Google Scholar] [CrossRef]
- Arsić, B.; Barber, J.; Novak, P. The macrolide antibiotics and their semi-synthetic derivatives. In Macrolides: Properties, Synthesis and Applications; Arsić, B., Ed.; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2018; pp. 1–30. [Google Scholar] [CrossRef]
- Sohmen, D.; Chiba, S.; Shimokawa-Chiba, N.; Innis, C.A.; Berninghausen, O.; Beckmann, R.; Ito, K.; Wilson, D.N. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat. Commun. 2015, 6, 6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opron, K.; Burton, Z.F. Ribosome structure, function, and early evolution. Int. J. Mol. Sci. 2019, 20, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsic, B.; Barber, J.; Čikoš, A.; Mladenovic, M.; Stankovic, N.; Novak, P. 16-membered macrolide antibiotics: A review. Int. J. Antimicrob. Agents 2018, 51, 283–298. [Google Scholar] [CrossRef]
- Jelić, D.; Antolović, R. From erythromycin to azithromycin and new potential ribosome-binding antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, R. Mechanisms of resistance to macrolides and lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.; Corey, G.R.; Abrutyn, E. Telithromycin. Ann. Intern. Med. 2006, 144, 447–448. [Google Scholar] [CrossRef]
- Owens, B. Solithromycin rejection chills antibiotic sector. Nat. Biotechnol. 2017, 35, 187–188. [Google Scholar] [CrossRef]
- Schlünzen, F.; Harms, J.M.; Franceschi, F.; Hansen, H.A.; Bartels, H.; Zarivach, R.; Yonath, A. Structural basis for the antibiotic activity of ketolides and azalides. Structure 2003, 11, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Tu, D.; Blaha, G.; Moore, P.B.; Steitz, T.A. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 2005, 121, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svetlov, M.S.; Plessa, E.; Chen, C.-W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-resolution crystal structures of ribosome-bound chloramphenicol and erythromycin provide the ultimate basis for their competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Pichkur, E.B.; Paleskava, A.; Tereshchenkov, A.G.; Kasatsky, P.; Komarova, E.S.; Shiriaev, D.I.; Bogdanov, A.A.; Dontsova, O.A.; Osterman, I.A.; Sergiev, P.V.; et al. Insights into the improved macrolide inhibitory activity from the high-resolution cryo-EM structure of dirithromycin bound to the E. coli 70S ribosome. RNA 2020, 26, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Halfon, Y.; Matzov, D.; Eyal, Z.; Bashan, A.; Zimmerman, E.; Kjeldgaard, J.; Ingmer, H.; Yonath, A. Exit tunnel modulation as resistance mechanism of S. aureus erythromycin resistant mutant. Sci. Rep. 2019, 9, 11460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, P.; Banić Tomišić, Z.; Tepeš, P.; Lazarevski, G.; Plavec, J.; Turkalj, G. Conformational analysis of oleandomycin and its 8-methylene-9-oxime derivative by NMR and molecular modeling. Org. Biomol. Chem. 2005, 3, 39–47. [Google Scholar] [CrossRef]
- Novak, P.; Tatić, I.; Tepeš, P.; Koštrun, S.; Barber, J. Systematic approach to understanding macrolide -ribosome interactions: NMR and modeling studies of oleandomycin and its derivatives. J. Phys Chem. A 2006, 110, 580–588. [Google Scholar] [CrossRef]
- Novak, P.; Tepeš, P.; Lazić, V. Epitope mapping of macrolide antibiotics to bovine serum albumin by saturation transfer difference NMR spectroscopy. Croat. Chem. Acta 2007, 80, 211–216. [Google Scholar]
- Novak, P.; Barber, J.; Čikoš, A.; Arsić, B.; Plavec, J.; Lazarevski, G.; Tepeš, P.; Košutić-Hulita, N. Free and bound state structures of 6-O-methyl homoerythromycins and epitope mapping of their interactions with ribosomes. Bioorg. Med. Chem. 2009, 17, 5857–5867. [Google Scholar] [CrossRef]
- Kosol, S.; Schrank, E.; Krajačić, M.B.; Wagner, G.E.; Meyer, N.H.; Göbl, C.; Rechberger, G.N.; Zangger, K.; Novak, P. Probing the interactions of macrolide antibiotics with membrane-mimetics by NMR spectroscopy. J. Med. Chem. 2012, 55, 5632–5636. [Google Scholar] [CrossRef]
- Glanzer, S.; Pulido, S.A.; Tutz, S.; Wagner, G.E.; Kriechbaum, M.; Gubensäk, N.; Trifunovic, J.; Markus, D.; Fabian, W.M.F.; Novak, P.; et al. Structural and functional implications of the interaction between macrolide antibiotics and bile acids. Chem. Eur. J. 2015, 21, 4350–4358. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C. X-ray Diffraction in Biology: How Can We See DNA and Proteins in Three Dimensions? IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Carroni, M.; Saibil, H.R. Cryo electron microscopy to determine the structure of macromolecular complexes. Methods 2016, 95, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Novak, P.; Jednačak, T. NMR spectroscopy for studying interactions of bioactive molecules. In Physico-Chemical Methods in Drug Discovery and Development; Mandić, Z., Ed.; IAPC Publishing: Zagreb, Croatia, 2011; pp. 1–62. [Google Scholar] [CrossRef]
- Vangaveti, S.; Ranganathan, S.V.; Chen, A.A. Advances in RNA molecular dynamics: A simulator’s guide to RNA force fields. WIREs RNA 2017, 8, e1396. [Google Scholar] [CrossRef]
- Schlünzen, F.; Zarivach, R.; Harm, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef]
- Hansen, J.L.; Ippolito, J.A.; Ban, N.; Nissen, P.; Moore, P.B.; Steitz, T.A. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell 2002, 10, 117–128. [Google Scholar] [CrossRef]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RCSB Protein Data Bank. Available online: https://www.rcsb.org/structure/1YI2 (accessed on 5 October 2020).
- RCSB Protein Data Bank. Available online: https://www.rcsb.org/structure/1M1K (accessed on 5 October 2020).
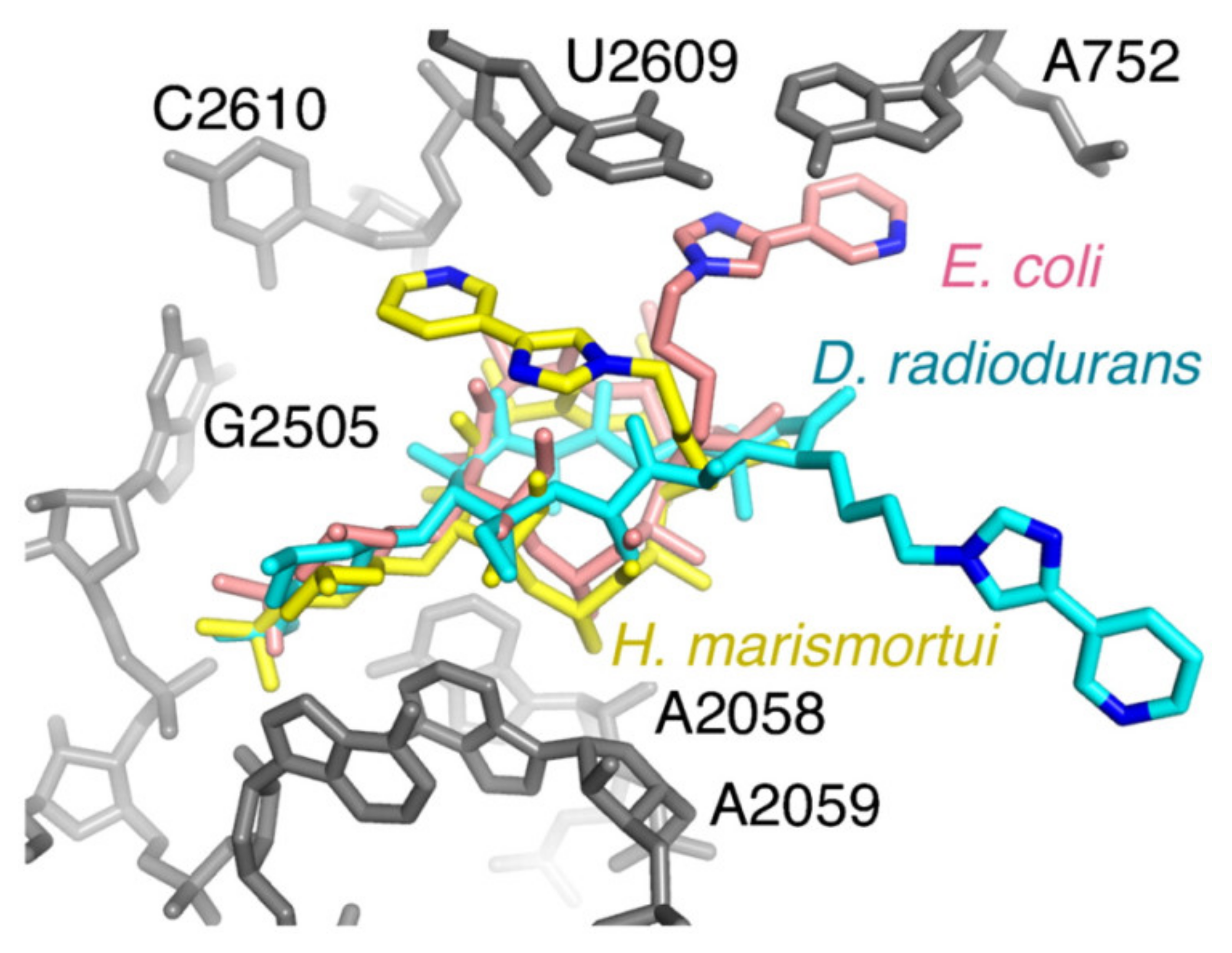
- Kannan, K.; Mankin, A.S. Macrolide antibiotics in the ribosome exit tunnel: Species-specific binding and action. Ann. N. Y. Acad. Sci. 2011, 1241, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, M.M.; Svetlov, M.S.; Hansen, D.A.; Khabibullina, N.F.; Klepacki, D.; Kang, H.-Y.; Sherman, D.H.; Vázquez-Laslop, N.; Polikanov, Y.S.; Mankin, A.S. Co-produced natural ketolides methymycin and pikromycin inhibit bacterial growth by preventing synthesis of a limited number of proteins. Nucleic Acids Res. 2017, 45, 9573–9582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, H.; Liu, X.; Jiang, D.; Zhang, B.; Tian, H.; Yang, C.; Guddat, L.W.; Yang, H.; Mi, K.; et al. Discovery of the first macrolide antibiotic binding protein in Mycobacterium tuberculosis: A new antibiotic resistance drug target. Protein Cell 2018, 9, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, L.K.R.; Edwards, T.A.; O’Neill, A.J. ABC-F proteins mediate antibiotic resistance through ribosomal protection. mBio 2016, 7, e01975. [Google Scholar] [CrossRef] [Green Version]
- Fong, D.H.; Burk, D.L.; Blanchet, J.; Yan, A.Y.; Berghuis, A.M. Structural basis for kinase-mediated macrolide antibiotic resistance. Structure 2017, 25, 750–761.e5. [Google Scholar] [CrossRef] [Green Version]
- Stierle, A.A.; Stierle, D.B.; Decato, D.; Priestley, N.D.; Alverson, J.B.; Hoody, J.; McGrath, K.; Klepacki, D. The berkeleylactones, antibiotic macrolides from fungal coculture. J. Nat. Prod. 2017, 80, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Dai, H.; Makhloufi, G.; Heering, C.; Janiak, C.; Hartmann, R.; Mándi, A.; Kurtán, T.; Müller, W.E.G.; Kassack, M.U.; et al. Cytotoxic 14-membered macrolides from a mangrove-derived endophytic fungus, Pestalotiopsis microspora. J. Nat. Prod. 2016, 79, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-Z.; Li, X.-M.; Yang, S.-Q.; Meng, L.-H.; Wang, B.-G. Thiocladospolides A–D, 12-membered macrolides from the mangrove-derived endophytic fungus Cladosporium cladosporioides MA-299 and structure revision of pandangolide 3. J. Nat. Prod. 2019, 82, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.-L.; Linington, R.G.; Balunas, M.J.; Centeno, A.; Boudreau, P.; Zhang, C.; Engene, N.; Spadafora, C.; Mutka, T.S.; Kyle, D.E.; et al. A potent antimalarial polyhydroxy macrolide from the marine cyanobacterium Okeania hirsuta. J. Org. Chem. 2015, 80, 7849–7855. [Google Scholar] [CrossRef] [PubMed]
- Frank, J. Cryo-electron microscopy as an investigative tool: The ribosome as an example. BioEssays 2001, 23, 725–732. [Google Scholar] [CrossRef]
- Von Loeffelholz, O.; Natchiar, S.K.; Djabeur, N.; Myasnikov, A.G.; Kratzat, H.; Ménétret, J.-F.; Hazemann, I.; Klaholz, B.P. Focused classification and refinement in high-resolution cryo-EM structural analysis of ribosome complexes. Curr. Opin. Struct. Biol. 2017, 46, 140–148. [Google Scholar] [CrossRef]
- Renaud, J.-P.; Chari, A.; Ciferri, C.; Liu, W.-T.; Rémigy, H.-W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef]
- Johnson, R.M.; Higgins, A.J.; Muench, S.P. Emerging role of electron microscopy in drug discovery. Trends Biochem. Sci. 2019, 44, 897–898. [Google Scholar] [CrossRef] [Green Version]
- Ceska, T.; Chung, C.-W.; Cooke, R.; Phillips, C.; Williams, P.A. Cryo-EM in drug discovery. Biochem. Soc. Trans. 2019, 47, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Saur, M.; Hartshorn, M.J.; Dong, J.; Reeks, J.; Bunkoczi, G.; Jhoti, H.; Williams, P.A. Fragment-based drug discovery using cryo-EM. Drug Discov. Today 2020, 25, 485–490. [Google Scholar] [CrossRef]
- Garcia-Nafria, J.; Tate, C.G. Cryo-electron microscopy: Moving beyond X-ray crystal structures for drug receptors and drug development. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 51–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danev, R.; Yanagisawa, H.; Kikkawa, M. Cryo-electron microscopy methodology: Current aspects and future directions. Trends Biochem. Sci. 2019, 44, 837–848. [Google Scholar] [CrossRef]
- Arnold, S.A.; Albiez, S.; Bieri, A.; Syntychaki, A.; Adaixo, R.; McLeod, R.A.; Goldie, K.N.; Stahlberg, H.; Braun, T. Blotting-free and lossless cryo-electron microscopy grid preparation from nanoliter-sized protein samples and single-cell extracts. J. Struct. Biol. 2017, 197, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khusainov, I.; Vicens, Q.; Bochler, A.; Grosse, F.; Myasnikov, A.; Menétrét, J.-F.; Chicher, J.; Marzi, S.; Romby, P.; Yusupova, G.; et al. Structure of the 70S ribosome from human pathogen Staphylococcus aureus. Nucleic Acids Res. 2016, 44, 10491–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentschel, J.; Burnside, C.; Mignot, I.; Leibundgut, M.; Boehringer, D.; Ban, N. The complete structure of the Mycobacterium smegmatis 70S ribosome. Cell Rep. 2017, 20, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojković, V.; Myasnikov, A.G.; Young, I.D.; Frost, A.; Fraser, J.S.; Galonić Fujimori, D. Assessment of the nucleotide modifications in the high-resolution cryo-electron microscopy structure of the Escherichia coli 50S subunit. Nucleic Acids Res. 2020, 48, 2723–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenz, S.; Ramu, H.; Gupta, P.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Mankin, A.S.; Wilson, D.N. Molecular basis for erythromycin-dependent ribosome-stalling during translation of the ErmBL leader peptide. Nat. Commun. 2014, 5, 3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenz, S.; Bock, L.V.; Graf, M.; Innis, C.A.; Beckmann, R.; Grubmüller, H.; Vaiana, A.C.; Wilson, D.N. A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest. Nat. Commun. 2016, 7, 12026. [Google Scholar] [CrossRef]
- Arenz, S.; Meydan, S.; Starosta, A.L.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N. Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol. Cell 2014, 56, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Laslop, N.; Thum, C.; Mankin, A.S. Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell 2008, 30, 190–202. [Google Scholar] [CrossRef]
- Seidelt, B.; Innis, C.A.; Wilson, D.N.; Gartmann, M.; Armache, J.-P.; Villa, E.; Trabuco, L.G.; Becker, T.; Mielke, T.; Schulten, K.; et al. Structural insight into nascent polypeptide chain-mediated translational stalling. Science 2009, 326, 1412–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, M.; Zavialov, A.; Li, W.; Stagg, S.M.; Sengupta, J.; Nielsen, R.C.; Nissen, P.; Harvey, S.C.; Ehrenberg, M.; Frank, J. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat. Struct. Biol. 2003, 10, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.E.; Huang, W.; Rudin, S.D.; Taylor, D.J.; Kirby, J.E.; Bonomo, R.A.; Yu, E.W. Cryo-electron microscopy structure of the Acinetobacter baumannii 70S ribosome and implications for new antibiotic development. mBio 2020, 11, e03117–e03119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Sun, Q.; Jiang, C.; Yang, K.; Hung, L.-W.; Zhang, J.; Sacchettini, J.C. Structure of ribosomal silencing factor bound to Mycobacterium tuberculosis ribosome. Structure 2015, 23, 1858–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Kumar, V.; Ding, Y.; Ero, R.; Serra, A.; Lee, B.S.T.; Wong, A.S.W.; Shi, J.; Sze, S.K.; Yang, L.; et al. Ribosome protection by antibiotic resistance ATP-binding cassette protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5157–5162. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef]
- Janas, A.; Przybylski, P. 14- and 15-membered lactone macrolides and their analogues and hybrids: Structure, molecular mechanism of action and biological activity. Eur. J. Med. Chem. 2019, 182, 111662. [Google Scholar] [CrossRef]
- Everett, J.R.; Tyler, J.W. The conformational analysis of erythromycin A. J. Chem. Soc. Perkin Trans. 1987, 2, 1659–1667. [Google Scholar] [CrossRef]
- Arsic, B.; Awan, A.; Brennan, R.J.; Aguilar, J.A.; Ledder, R.; McBain, A.J.; Regan, A.C.; Barber, J. Theoretical and experimental investigation on clarithromycin, erythromycin A and azithromycin and descladinosyl derivatives of clarithromycin and azithromycin with 3-O substitution as anti-bacterial agents. Med. Chem. Commun. 2014, 5, 1347–1354. [Google Scholar] [CrossRef]
- Novak, P. Interactions of macrolides with their biological targets. In Macrolides: Properties, Synthesis and Applications; Arsić, B., Ed.; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2018; pp. 63–77. [Google Scholar] [CrossRef]
- Grgičević, I.; Mikulandra, I.; Bukvić, M.; Banjanac, M.; Radovanović, V.; Habinovec, I.; Bertoša, B.; Novak, P. Discovery of macrozones, new antimicrobial thiosemicarbazone-based azithromycin conjugates: Design, synthesis and in vitro biological evaluation. Int. J. Antimicrob. Agents 2020, 56, 106147. [Google Scholar] [CrossRef]
- Yuan, G.; Xu, L.; Xu, X.; Li, P.; Zhong, Q.; Xia, H.; Hu, Y.; Li, P.; Song, X.; Li, J.; et al. Azalomycin F5a, a polyhydroxy macrolide binding to the polar head of phospholipid and targeting to lipoteichoic acid to kill methicillin-resistant Staphylococcus aureus. Biomed. Pharmacother. 2019, 109, 1940–1950. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, F.; Griffin, D.C.; Loraine, J.; Rittig, M.; Delves-Broughton, J.; Bonev, B.B. Recognition of membrane sterols by polyene antifungals amphotericin B and natamycin, a 13C MAS NMR study. Front. Cell Dev. Biol. 2016, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Debouzy, J.C.; Mehenni, L.; Crouzier, D.; Lahiani-Skiba, M.; Nugue, G.; Skiba, M. NMR and ESR study of amphotericin B interactions with various binary phosphatidylcholine/phosphatidylglycerol membranes. Int. J. Pharm. 2017, 521, 384–394. [Google Scholar] [CrossRef]
- Alferova, V.A.; Shuvalova, M.V.; Novikov, R.A.; Trenin, A.S.; Dezhenkova, L.G.; Gladkikh, E.G.; Lapchinskaya, O.A.; Kulyaeva, V.V.; Bychkova, O.P.; Mirchinka, E.P.; et al. Structure-activity studies of irumamycin type macrolides from Streptomyces sp. INA-Ac-5812. Tetrahedron Lett. 2019, 16, 1448–1451. [Google Scholar] [CrossRef]
- Fan, B.Z.; Hiasa, H.; Lv, W.; Brody, S.; Yang, Z.Y.; Aldrich, C.; Cushman, M.; Liang, J.H. Design, synthesis and structure-activity relationships of novel 15-membered macrolides: Quinolone/quinoline-containing sidechains tethered to the C-6 position of azithromycin acylides. Eur. J. Med. Chem. 2020, 193, 112222. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Li, Y.; Ratnayake, R.; Luo, D.; Lo, J.; Reibenspies, J.H.; Xu, Z.; Clare-Salzler, M.J.; Ye, T.; Paul, V.J.; et al. Discovery, total synthesis and key structural elements for the immunosuppressive activity of cocosolide, a symmetrical glycosylated macrolide dimer from marine cyanobacteria. Chem. Eur. J. 2016, 22, 8158–8166. [Google Scholar] [CrossRef] [Green Version]
- Carlomagno, T. NMR in natural products: Understanding conformation, configuration and receptor interactions. Nat. Prod. Rep. 2012, 29, 536–554. [Google Scholar] [CrossRef]
- Nakano, H.; Sugawara, A.; Hirose, T.; Gouda, H.; Hirono, S.; Ōmura, S.; Sunazuka, T. An architectonic macrolide library based on a C2-symmetric macrodiolide toward pharmaceutical compositions. Tetrahedron 2015, 71, 6569–6579. [Google Scholar] [CrossRef]
- Arsić, B.; Aguilar, J.A.; Bryce, R.A.; Barber, J. Conformational study of tylosin A in water and full assignments of 1H and 13C spectra of tylosin A in D2O and tylosin B in CDCl3. Magn. Reson. Chem. 2017, 55, 367–373. [Google Scholar] [CrossRef]
- Wang, W.; Song, T.; Chai, W.; Chen, L.; Chen, L.; Lian, X.Y.; Zhang, Z. Rare polyene-polyol macrolides from mangrove-derived Streptomyces sp. ZQ4BG. Sci. Rep. 2017, 7, 1703. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.; Schleissner, C.; Fernández, R.; Rodríguez, P.; Reyes, F.; Zuñiga, P.; Calle, F.; Cuevas, C. PM100117 and PM100118, new antitumor macrolides produced by a marine Streptomyces caniferus GUA-06-05-006A. J. Antibiot. 2016, 69, 388–394. [Google Scholar] [CrossRef]
- Kim, J.; Shin, D.; Kim, S.H.; Park, W.; Shin, Y.; Kim, W.K.; Lee, S.K.; Oh, K.B.; Shin, J.; Oh, D.C. Borrelidins C-E: New antibacterial macrolides from a saltern-derived halophilic Nocardiopsis sp. Mar. Drugs. 2017, 15, 166. [Google Scholar] [CrossRef] [Green Version]
- Fuwa, H.; Yamagata, N.; Okuaki, Y.; Ogata, Y.; Saito, A.; Sasaki, M. Total synthesis and complete stereostructure of a marine macrolide glycoside, (−)-Lyngbyaloside B. Chem. Eur. J. 2016, 22, 6815–6829. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Hatano, M.; Umekita, M.; Hayashi, C.; Wada, S.; Nagayoshi, M.; Sawa, R.; Kubota, Y.; Kawada, M.; Igarashi, M.; et al. ATP depletion assay led to the isolation of new 36-membered polyol macrolides deplelides A and B from Streptomyces sp. MM581-NF15. Org. Lett. 2017, 19, 4207–4210. [Google Scholar] [CrossRef]
- Okabe, M.; Sugita, T.; Kinoshita, K.; Koyama, K. Macrolides from a marine-derived fungus, Penicillium meleagrinum var. viridiflavum, showing synergistic effects with fluconazole against azole-resistant Candida albicans. J. Nat. Prod. 2016, 79, 1208–1212. [Google Scholar] [CrossRef]
- Inahashi, Y.; Iwatsuki, M.; Ishiyama, A.; Matsumoto, A.; Hirose, T.; Oshita, J.; Sunazuka, T.; Panbangred, W.; Takahashi, Y.; Kaiser, M.; et al. Actinoallolides A-E, new anti-trypanosomal macrolides, produced by an endophytic actinomycete, Actinoallomurus fulvus MK10-036. Org. Lett. 2015, 17, 864–867. [Google Scholar] [CrossRef]
- Klüppel, A.; Gille, A.; Karayel, C.E.; Hiersemann, M. Synthesis of a diastereomer of the marine macrolide Lytophilippine, A. Org. Lett. 2019, 21, 2421–2425. [Google Scholar] [CrossRef]
- Che, Q.; Li, T.; Liu, X.; Yao, T.; Li, J.; Gu, Q.; Li, D.; Li, W.; Zhu, T. Genome scanning inspired isolation of reedsmycins A–F, polyene-polyol macrolides from Streptomyces sp. CHQ-64. RSC Adv. 2015, 5, 2277–22782. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, L.; Yang, J.; Zhang, F.; Sun, Y.; Yu, M.; Yan, Y.; Ma, Y.-T.; Huang, S.-X. A new antifungal macrolide from Streptomyces sp. KIB-H869 and structure revision of halichomycin. Tetrahedron Lett. 2016, 57, 1375–1378. [Google Scholar] [CrossRef]
- Gjerde, D.T.; Hoang, L.; Hornby, D. RNA Purification and Analysis: Sample Preparation, Extraction, Chromatography; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 127–145. [Google Scholar] [CrossRef]
- Xiong, L.; Korkhin, Y.; Mankin, A.S. Binding site of the bridged macrolides in the Escherichia coli ribosome. Antimicrob. Agents Chemother. 2005, 49, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, S.M.; Karlsson, M.; Johansson, L.B.; Vester, B. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 2001, 41, 1091–1099. [Google Scholar] [CrossRef]
- Petropoulos, A.D.; Kouvela, E.C.; Starosta, A.L.; Wilson, D.N.; Dinos, G.P.; Kalpaxis, D.L. Time-resolved binding of azithromycin to Escherichia coli ribosomes. J. Mol. Biol. 2009, 385, 1179–1192. [Google Scholar] [CrossRef]
- Nilsen, T.W. Toeprinting. Cold Spring Harb. Protoc. 2013, 2013, 896–899. [Google Scholar] [CrossRef]
- Koch, M.; Willi, J.; Pradère, U.; Hall, J.; Polacek, N. Critical 23S rRNA interactions for macrolide-dependent ribosome stalling on the ErmCL nascent peptide chain. Nucleic Acids Res. 2017, 45, 6717–6728. [Google Scholar] [CrossRef]
- Sothiselvam, S.; Neuner, S.; Rigger, L.; Klepacki, D.; Micura, R.; Vázquez-Laslop, N.; Mankin, A.S. Binding of macrolide antibiotics leads to ribosomal selection against specific substrates based on their charge and size. Cell Rep. 2016, 16, 1789–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.; Chen, J.; Tsai, A.; Kornberg, G.; Puglisi, J.D. Sequence-dependent elongation dynamics on macrolide-bound ribosomes. Cell Rep. 2014, 7, 1534–1546. [Google Scholar] [CrossRef] [Green Version]
- Tereshchenkov, A.G.; Shishkina, A.V.; Karpenko, V.V.; Chertkov, V.A.; Konevega, A.L.; Kasatsky, P.S.; Bogdanov, A.A.; Sumbatyan, N.V. New fluorescent macrolide derivatives for studying interactions of antibiotics and their analogs with the ribosomal exit tunnel. Biochem. Mosc. 2016, 81, 1163–1172. [Google Scholar] [CrossRef]
- Mortier, J.; Rakers, C.; Bermudez, M.; Murgueitio, M.S.; Riniker, S.; Wolber, G. The impact of molecular dynamics on drug design: Applications for the characterization of ligand-macromolecule complexes. Drug Discov. Today 2015, 20, 686–702. [Google Scholar] [CrossRef]
- Makarov, G.I.; Makarova, T.M.; Sumbatyan, N.V.; Bogdanov, A.A. Investigation of ribosomes using molecular dynamics simulation methods. Biochem. Mosc. 2016, 81, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Perilla, J.R.; Goh, B.C.; Cassidy, C.K.; Liu, B.; Bernardi, R.C.; Rudack, T.; Yu, H.; Wu, Z.; Schulten, K. Molecular dynamics simulations of large macromolecular complexes. Curr. Opin. Struct. Biol. 2015, 31, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerenberg, P.S.; Head-Gordon, T. New developments in force fields for biomolecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauber-Osguthorpe, P.; Hagler, A.T. Biomolecular force fields: Where have we been, where are we now, where do we need to go and how do we get there? J. Comput. Aided Mol. Des. 2019, 33, 133–203. [Google Scholar] [CrossRef]
- Dans, P.D.; Gallego, D.; Balaceanu, A.; Darré, L.; Gómez, H.; Orozco, M. Modeling, simulations, and bioinformatics at the service of RNA structure. Chem 2019, 5, 51–73. [Google Scholar] [CrossRef] [Green Version]
- Šponer, J.; Krepl, M.; Banáš, P.; Kührová, P.; Zgarbová, M.; Jurečka, P.; Havrila, M.; Otyepka, M. How to understand atomistic molecular dynamics simulations of RNA and protein–RNA complexes? WIREs RNA 2017, 8, e1405. [Google Scholar] [CrossRef]
- Bock, L.V.; Kolář, M.H.; Grubmüller, H. Molecular simulations of the ribosome and associated translation factors. Curr. Opin. Struct. Biol. 2018, 49, 27–35. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Case, D.A.; Singh, U.C.; Ghio, C.; Alagona, G.; Profeta, S.; Weiner, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar] [CrossRef]
- Schauperl, M.; Nerenberg, P.S.; Jang, H.; Wang, L.-P.; Bayly, C.I.; Mobley, D.L.; Gilson, M.K. Non-bonded force field model with advanced restrained electrostatic potential charges (RESP2). Commun. Chem. 2020, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Liu, B.; Klepacki, D.; Gupta, V.; Schulten, K.; Mankin, A.S.; Vázquez-Laslop, N. Nascent peptide assists the ribosome in recognizing chemically distinct small molecules. Nat. Chem. Biol. 2016, 12, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Makarov, G.I.; Golovin, A.V.; Sumbatyan, N.V.; Bogdanov, A.A. Molecular dynamics investigation of a mechanism of allosteric signal transmission in ribosomes. Biochem. Mosc. 2015, 80, 1047–1056. [Google Scholar] [CrossRef]
- Feng, T.; Zhang, Y.; Ding, J.-N.; Fan, S.; Han, J.-G. Insights into resistance mechanism of the macrolide biosensor protein MphR(A) binding to macrolide antibiotic erythromycin by molecular dynamics simulation. J. Comput. Aided Mol. Des. 2015, 29, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta 2015, 1850, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, A.; Gumbart, J.C. Parametrization of macrolide antibiotics using the force field toolkit. J. Comput. Chem. 2015, 36, 2052–2063. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, A.; Parks, J.M.; Oyelere, A.K.; Gumbart, J.C. Toward the rational design of macrolide antibiotics to combat resistance. Chem. Biol. Drug Des. 2017, 90, 641–652. [Google Scholar] [CrossRef]
- Small, M.C.; Lopes, P.; Andrade, R.B.; MacKerell, A.D., Jr. Impact of ribosomal modification on the binding of the antibiotic telithromycin using a combined grand canonical Monte Carlo/molecular dynamics simulation approach. PLoS Comput. Biol. 2013, 9, e1003113. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- CHARMM-GUI. Effective Simulation Input Generator and More: Merck Molecular Force Field (MMFF94). Available online: http://www.charmm-gui.org/charmmdoc/mmff.html (accessed on 8 October 2020).
- Wahl, J.; Freyss, J.; von Korff, M.; Sander, T. Accuracy evaluation and addition of improved dihedral parameters for the MMFF94s. J. Cheminform. 2019, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Koštrun, S.; Munic Kos, V.; Matanović Škugor, M.; Palej Jakopović, I.; Malnar, I.; Dragojević, S.; Ralić, J.; Alihodžić, S. Around the macrolide—Impact of 3D structure of macrocycles on lipophilicity and cellular accumulation. Eur. J. Med. Chem. 2017, 133, 351–364. [Google Scholar] [CrossRef]
- Poongavanam, V.; Danelius, E.; Peintner, S.; Alcaraz, L.; Caron, G.; Cummings, M.D.; Wlodek, S.; Erdelyi, M.; Hawkins, P.C.D.; Ermondi, G.; et al. Conformational sampling of macrocyclic drugs in different environments: Can we find the relevant conformations? ACS Omega 2018, 3, 11742–11757. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
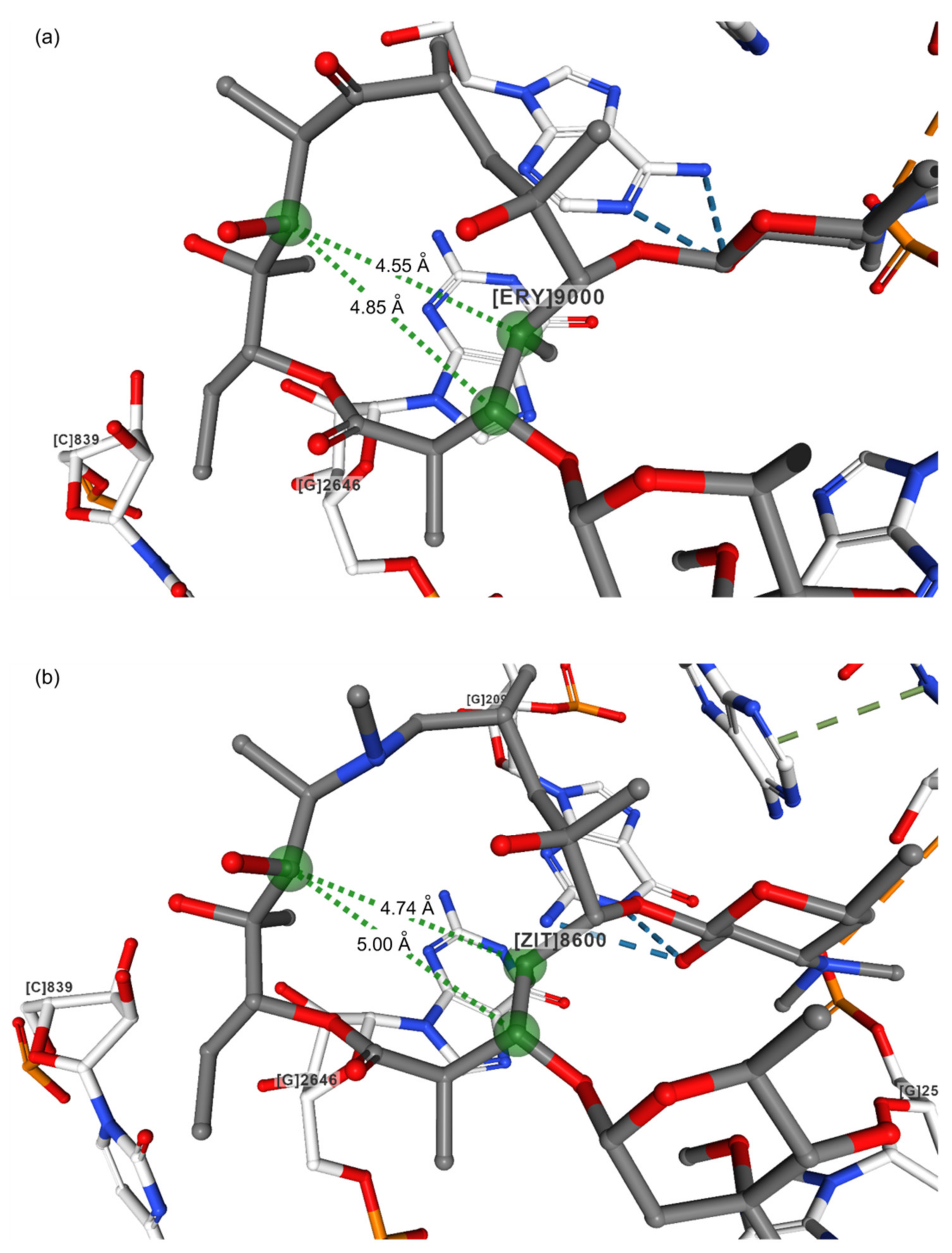
| Compound | Code | Resolution/Å | Distance/Å | |||||
|---|---|---|---|---|---|---|---|---|
| C3–C11 | C4–C11 | C4–C6Me | C5–C6Me | C3–C8 | C8–C11 | |||
| Erythromycin, folded out 1 | NAVTAF | 0.85 | 4.69 | 4.33 | 3.90 | 2.52 | 5.50 | 3.65 |
| Azithromycin, folded out 1 | GEGJAD | 0.82 | 4.86 | 4.60 | 3.92 | 2.50 | 5.74 | 4.80 |
| Erythromycin bound to the H. marismortui 50S subunit [11] 2 | 1YI2 | 2.65 | 4.85 | 4.55 | 3.99 | 2.61 | 5.64 | 3.59 |
| Azithromycin bound to the H. marismortui 50S subunit [11] 2 | 1YHQ | 2.40 | 4.80 | 5.01 | 4.03 | 2.59 | 5.76 | 4.62 |
| Azithromycin bound to the H. marismortui 50S subunit [27] 2 | 1M1K | 3.20 | 5.00 | 4.74 | 3.92 | 2.55 | 5.82 | 4.84 |
| Erythromycin bound to the T. thermpohilus ribosome [28] 2 | 4V7X | 3.00 | 4.85 | 4.55 | 3.99 | 2.61 | 5.64 | 3.59 |
| Azithromycin bound to the T. thermpohilus ribosome [28] 2 | 4V7Y | 3.00 | 4.80 | 5.01 | 4.03 | 2.59 | 5.76 | 4.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jednačak, T.; Mikulandra, I.; Novak, P. Advanced Methods for Studying Structure and Interactions of Macrolide Antibiotics. Int. J. Mol. Sci. 2020, 21, 7799. https://doi.org/10.3390/ijms21207799
Jednačak T, Mikulandra I, Novak P. Advanced Methods for Studying Structure and Interactions of Macrolide Antibiotics. International Journal of Molecular Sciences. 2020; 21(20):7799. https://doi.org/10.3390/ijms21207799
Chicago/Turabian StyleJednačak, Tomislav, Ivana Mikulandra, and Predrag Novak. 2020. "Advanced Methods for Studying Structure and Interactions of Macrolide Antibiotics" International Journal of Molecular Sciences 21, no. 20: 7799. https://doi.org/10.3390/ijms21207799