Immunological Memory of Psoriatic Lesions

 , , and
, , and
Abstract
:1. Introduction
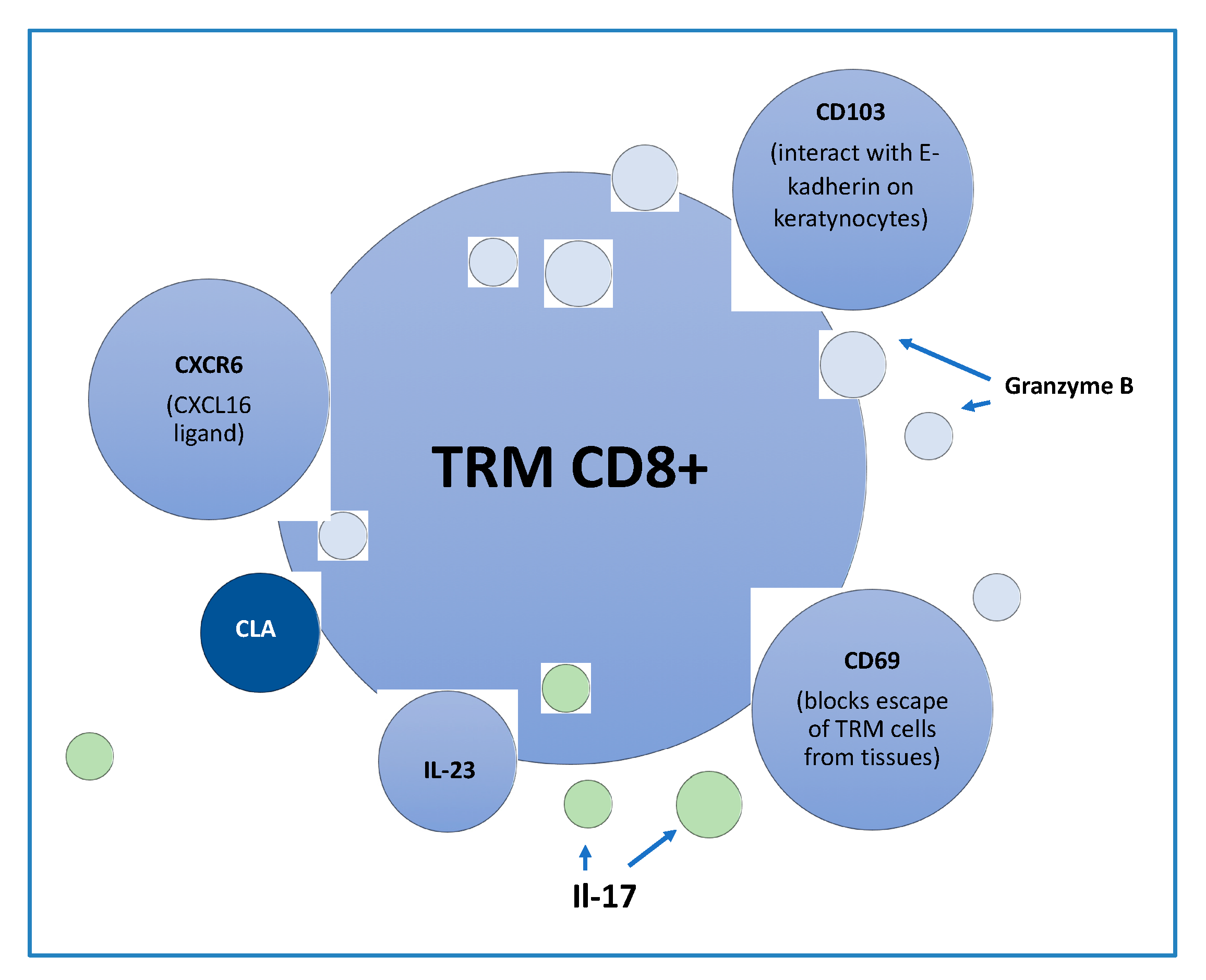
2. Tissue Resident Memory Cells (TRM)
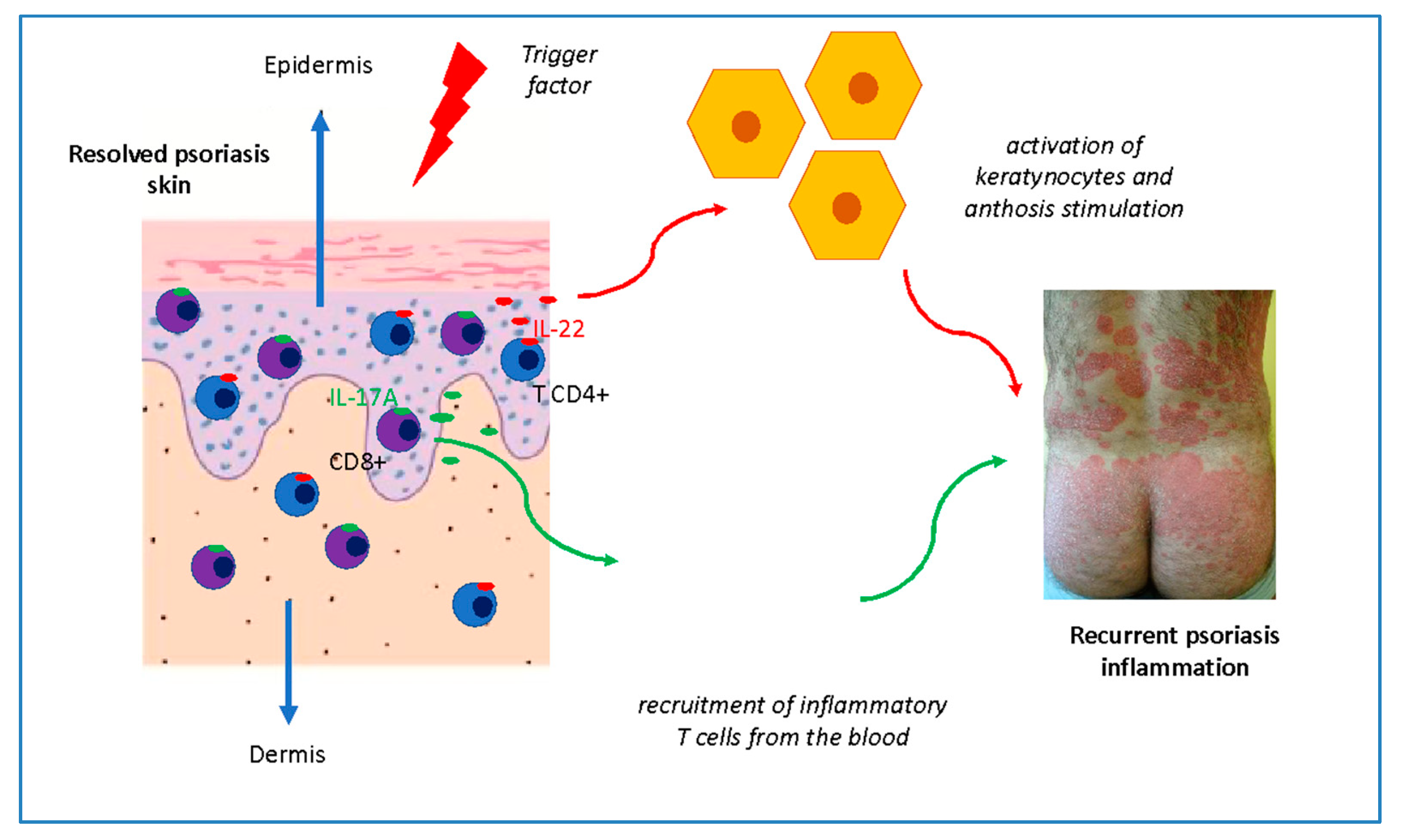
3. Tissue Resident Memory Cells in Psoriasis
4. Tissue Resident Memory Cells in Psoriatic Arthritis (PsA)
5. The Role of Memory γδ T Cells
6. Th22 and Tc17 Lymphocytes
7. Epidermal Langerhans Cells (LCs)
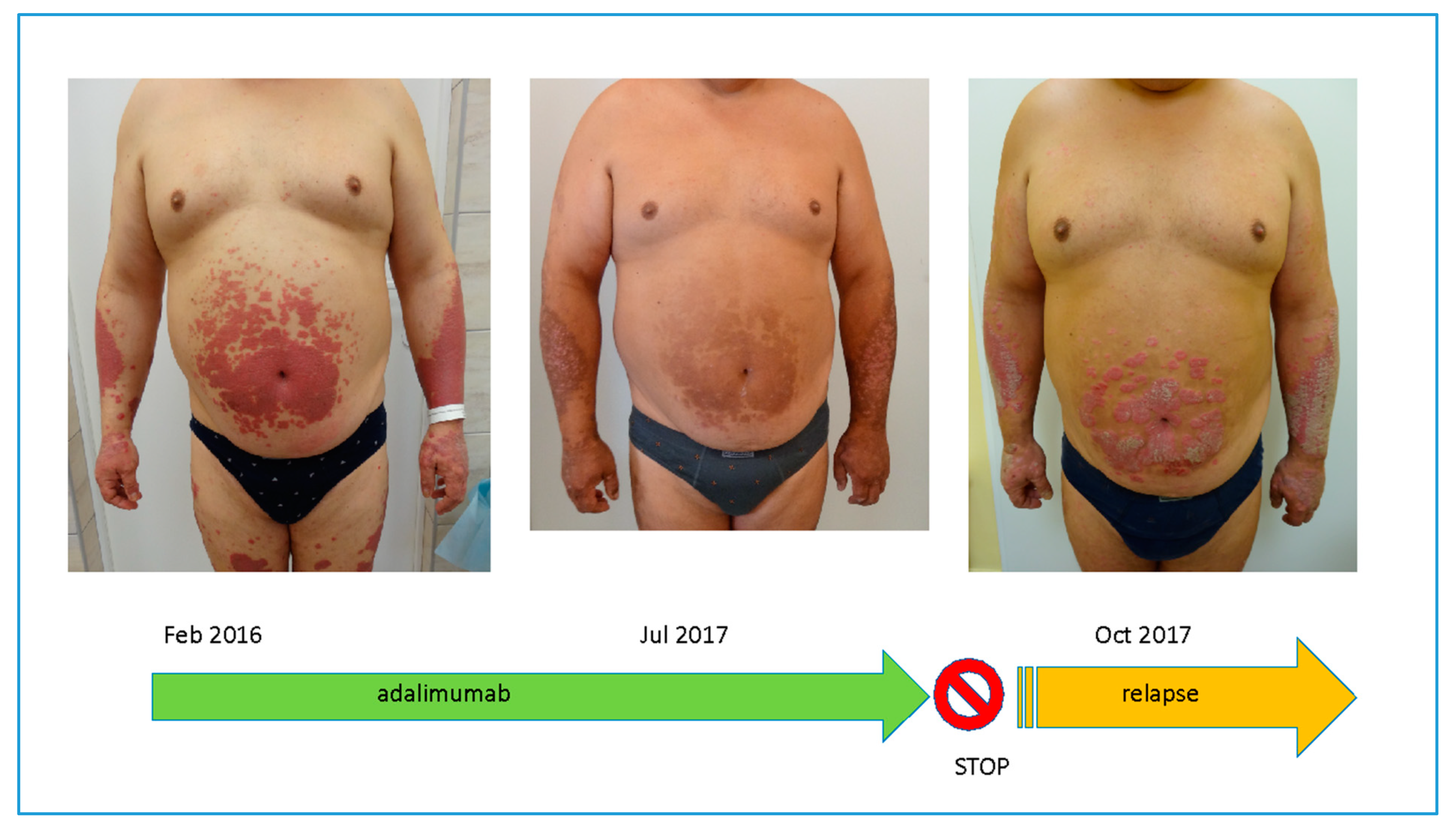
8. Clinical Significance of the Memory of Psoriasis Lesions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Diani, M.; Altomare, G.; Reali, E. T helper cell subsets in clinical manifestations of psoriasis. J. Immunol. Res. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Eberle, F.C.; Brück, J.; Holstein, J.; Hirahara, K.; Ghoreshi, K. Recent advances in understanding psoriasis. F1000 Res. 2016, 5, 770. [Google Scholar] [CrossRef] [Green Version]
- Matos, T.; O’Malley, J.T.; Lowry, E.L.; Hamm, D.; Kirsch, I.R.; Robins, H.S.; Kupper, T.S.; Krueger, J.G.; Clark, R.A. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17–producing αβ T cell clones. J. Clin. Invest. 2017, 127, 4031–4041. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R. Protective and pathogenic roles of resident memory T cells in human skin disorders. J. Dermatol. Sci. 2019, 95, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Cheuk, S.; Wikén, M.; Blomqvist, L.; Nylén, S.; Talme, T.; Stàhle, M.; Eidsmo, L. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J. Immunol. 2014, 192, 3111–3120. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Farinas, M.; Fuentes-Duculan, J.; Lowes, M.A.; Krueger, J.G. Resolved psoriasis lesions retain expression of a subset of disease-related genes. J. Investig. Dermatol. 2011, 131, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Casey, K.A.; Fraser, K.A.; Schenkelm, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef] [Green Version]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Milner, J.J.; Goldrath, A.W. Transcriptional programming of tissue-resident memory CD8+ T cells. Curr. Opin. Immunol. 2018, 51, 162–169. [Google Scholar] [CrossRef]
- Patra, V.; Laoubi, L.; Nicolas, J.F.; Vocanson, M.; Wolf, P. A perspective on the interplay of ultraviolet-radiation, skin microbiome and skin resident memory TCRαβ+ cells. Front. Med. (Lausanne) 2018, 5, 166. [Google Scholar] [CrossRef]
- Wu, H.; Liao, W.; Li, Q.; Long, H.; Yin, H.; Zhao, M.; Chan, V.; Lau, C.S.; Lu, Q. Pathogenic role of tissue-resident memory T cells in autoimmune diseases. Autoimmun. Rev. 2018, 17, 906–911. [Google Scholar] [CrossRef]
- Clark, R.A. Resident memory T cells in human health and diseases. Sci. Transl. Med. 2015, 7, 269rv1. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, K.; Fujiyama, T.; Phadungsaksawasdi, P.; Ito, T.; Tokura, Y. Significance of IL-17A-producing CD8+CD103+ skin resident memory T cells in psoriasis lesion and their possible relationship to clinical course. J. Dermatol. Sci. 2019, 95, 21–27. [Google Scholar] [CrossRef]
- Rosato, P.C.; Beura, L.K.; Masopust, D. Tissue resident memory T cells and viral immunity. Curr. Opin. Virol. 2017, 22, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Iwasaki, A. Tissue-resident memory T cells. Immunol. Rev. 2013, 255, 165–181. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Srivastava, R.; Chentoufi, A.A.; Kritzer, E.; Chilukuri, S.; Garg, S.; Yu, D.C.; Vahed, H.; Huang, L.; Syed, S.A.; et al. Bolstering the number and function of HSV-1-specific CD8(+) effector memory T cells and tissue-resident memory T cells in latently infected trigeminal ganglia reduces recurrent ocular herpes infection and disease. J. Immunol. 2017, 199, 186–203. [Google Scholar] [CrossRef]
- Pan, Y.; Kupper, T.S. Metabolic reprogramming and longevity of tissue-resident memory T cells. Front. Immunol. 2018, 9, 1347. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.A.; Borges da Silva, H.; Beura, L.K.; Peng, C.; Hamilton, S.E.; Masopust, D.; Jameson, S.C. The functional requirement for CD69 in establishment of resident memory CD8+ T cells varies with tissue location. J. Immunol. 2019, 203, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Behr, F.M.; Chuwonpad, A.; Stark, R.; van Gisbergen, K. Armed and ready: Transcriptional regulation of Tissue-Resident Memory CD8 T cells. Front. Immunol. 2018, 9, 1770. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Cheuk, S.; Schlums, H.; Gallais Sérézal, I.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; van Wijk, F. CD8(+) T cells in human autoimmune arthritis: The unusual suspects. Nat. Rev. Rheumatol. 2016, 12, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Ritchlin, C. Tissue-resident T cells: Sequestered immune sensors and effectors of inflammation in spondyloarthritis. Arthritis Rheumatol. 2019, 17. [Google Scholar] [CrossRef]
- Chen, L.; Shen, Z. Tissue-resident memory T cells and their biological characteristics in the recurrence of inflammatory skin disorders. Cell. Mol. Immunol. 2019, 8. [Google Scholar] [CrossRef]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef]
- Arakawa, A.; Siewert, K.; Stöhr, J.; Besgen, P.; Kim, S.M.; Rühl, G.; Nickel, J.; Vollmer, S.; Thomas, P.; Krebs, S.; et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 2015, 212, 2203–2212. [Google Scholar] [CrossRef]
- Adachi, T.; Kobayashi, T.; Sugihara, E.; Yamada, T.; Ikuta, K.; Pittaluga, S.; Saya, H.; Amagai, M.; Nagao, K. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med. 2015, 21, 1272–1279. [Google Scholar] [CrossRef]
- Diani, M.; Galasso, M.; Cozzi, C.; Sgambelluri, F.; Altomare, A.; Cigni, C.; Frigerio, E.; Drago, L.; Volinia, S.; Granucci, F.; et al. Blood to skin recirculation of CD4+ memory T cells associates with cutaneous and systemic manifestations of psoriatic disease. Clin. Immunol. 2017, 180, 84–94. [Google Scholar] [CrossRef]
- Di Meglio, P.; Villanova, F.; Navarini, A.A.; Mylonas, A.; Tosi, I.; Nestle, F.O.; Conrad, C. Targeting CD8(+) T cells prevents psoriasis development. J. Allergy Clin. Immunol. 2016, 138, 274–276. [Google Scholar] [CrossRef] [Green Version]
- Farber, D.L.; Yudanin, N.A.; Restifo, N.P. Human memory T cells: Generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 2014, 14, 24–35. [Google Scholar] [CrossRef]
- Boyman, O.; Hefti, H.P.; Conrad, C.; Nickoloff, B.J.; Suter, M.; Nestle, F.O. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J. Exp. Med. 2004, 199, 731–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosè, F.; Petti, L.; Diani, M.; Moscheni, C.; Molteni, S.; Altomare, A.; Rossi, R.L.; Talarico, D.; Fontana, R.; Russo, V.; et al. Inhibition of CCR7/CCL19 axis in lesional skin is a critical event for clinical remission induced by TNF blockade in patients with psoriasis. Am. J. Pathol. 2013, 183, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Sgambelluri, F.; Diani, M.; Altomare, A.; Frigerio, E.; Drago, L.; Granucci, F.; Banfi, G.; Altomare, G.; Reali, E. A role for CCR5(+)CD4 T cells in cutaneous psoriasis and for CD103(+) CCR4(+) CD8 Teff cells in the associated systemic inflammation. J. Autoimmun. 2016, 70, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Boyman, O.; Tonel, G.; Tun-Kyi, A.; Laggner, U.; de Fougerolles, A.; Kotelianski, V.; Gardner, H.; Nestle, F.O. Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat. Med. 2007, 13, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Gallais Sérézal, I.; Classon, C.; Cheuk, S.; Barrientos-Somarribas, M.; Wadman, E.; Martini, E.; Chang, D.; Xu Landén, N.; Ehrström, M.; Nylén, S.; et al. Resident T cells in resolved psoriasis steer tissue responses that stratify clinical outcome. J. Invest. Dermatol. 2018, 138, 1754–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, S.; Watanabe, R.; Koguchi-Yoshioka, H.; Matsumura, Y.; Ishitsuka, Y.; Nakamura, Y.; Okiyama, N.; Fujisawa, Y.; Fujimoto, M. CD8 resident memory T cells with interleukin 17A producing potential are accumulated in disease-naïve nonlesional sites of psoriasis possibly in correlation with disease duration. Br. J. Dermatol. 2019, 181, 410–412. [Google Scholar] [CrossRef]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: A biologic rationale for their distinct clinical behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef]
- Gallais Sérézal, I.; Hoffer, E.; Ignatov, B.; Martini, E.; Zitti, B.; Ehrström, M.; Eidsmo, L. A skewed pool of resident T cells triggers psoriasis-associated tissue responses in never-lesional skin from patients with psoriasis. J. Allergy Clin. Immunol. 2019, 143, 1444–1454. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 9, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmaeili, B.; Mansouri, P.; Doustimotlagh, A.H.; Izad, M. Redox imbalance and IL-17 responses in memory CD4+ T cells from patients with psoriasis. Scand. J. Immunol. 2019, 89, e12730. [Google Scholar] [CrossRef] [PubMed]
- Steel, K.J.A.; Srenathan, U.; Ridley, M.; Durham, L.E.; Wu, S.Y.; Ryan, S.E.; Hughes, C.D.; Chan, E.; Kirkham, B.W.; Taams, L.S. Synovial IL-17A+ CD8+ T cells display a polyfunctional, pro-inflammatory and tissue-resident memory phenotype and function in psoriatic arthritis. Arthritis Rheumatol. 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beura, L.K.; Wijeyesinghe, S.; Thompson, E.A.; Macchietto, M.G.; Rosato, P.C.; Pierson, M.J.; Schenkel, J.M.; Mitchell, J.S.; Vezys, V.; Fife, B.T.; et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T Cells. Immunity 2018, 48, 327–338e5. [Google Scholar] [CrossRef] [PubMed]
- Guggino, G.; Rizzo, A.; Mauro, D.; Macaluso, F.; Ciccia, F. Gut-derived CD8+ tissue-resident memory T cells are expanded in the peripheral blood and synovia of SpA patients. Ann. Rheum. Dis. 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Lalor, S.J.; McLoughlin, R.M. Memory γδ T cells-newly appreciated protagonists in infection and immunity. Trends Immunol. 2016, 37, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Eidsmo, L.; Martini, E. Human langerhans cells with pro-inflammatory features relocate within psoriasis lesions. Front. Immunol. 2018, 22, 300. [Google Scholar] [CrossRef]
- Martini, E.; Wikén, M.; Cheuk, S.; Gallais Sérézal, I.; Baharom, F.; Ståhle, M.; Smed Sörensen, A.; Eidsmo, L. Dynamic changes in resident and infiltrating epidermal dendritic cells in active and resolved psoriasis. J. Invest. Dermatol. 2017, 137, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Cumberbatch, M.; Singh, M.; Dearman, R.J.; Young, H.S.; Kimber, I.; Griffiths, C.E. Impaired Langerhans cell migration in psoriasis. J. Exp. Med. 2006, 203, 953–960. [Google Scholar] [CrossRef]
- Shaw, F.L.; Mellody, K.T.; Ogden, S.; Dearman, R.J.; Kimber, I.; Griffiths, C. Treatment-related restoration of Langerhans cell migration in psoriasis. J. Investig. Dermatol. 2014, 134, 268–271. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, J.; Beura, L.K.; Bobr, A.; Astry, B.; Chicoine, B.; Kashem, S.W.; Welty, N.E.; Igyártó, B.Z.; Wijeyesinghe, S.; Thompson, E.A.; et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat. Immunol. 2016, 17, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A. Gone but not forgotten: Lesional memory in psoriatic skin. J. Invest. Dermatol. 2011, 131, 283–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Type of Cells | Psoriasis Lesions | Resolved Psoriasis Skin | |
|---|---|---|---|
| TRM | CD4+ | A few in dermis CD103- and a low number in epidermis CD103+ | A few |
| CD8+ | A lot of in epidermis CD103+: CD8+CD49a+ producing perforin, IFN-γ CD8+CD49a- producing IL-17 IL-22, IFN-γ, a few in dermis CD103- and a low number CD103+ | A few in epidermis | |
| Dendritic Cells | LC | In epidermis producing a lot of IL-23 | In epidermis producing IL-23 |
| eDC | In epidermis producing of IL-1β, IL-23, TNFα | Absent | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owczarczyk-Saczonek, A.; Krajewska-Włodarczyk, M.; Kasprowicz-Furmańczyk, M.; Placek, W. Immunological Memory of Psoriatic Lesions. Int. J. Mol. Sci. 2020, 21, 625. https://doi.org/10.3390/ijms21020625
Owczarczyk-Saczonek A, Krajewska-Włodarczyk M, Kasprowicz-Furmańczyk M, Placek W. Immunological Memory of Psoriatic Lesions. International Journal of Molecular Sciences. 2020; 21(2):625. https://doi.org/10.3390/ijms21020625
Chicago/Turabian StyleOwczarczyk-Saczonek, Agnieszka, Magdalena Krajewska-Włodarczyk, Marta Kasprowicz-Furmańczyk, and Waldemar Placek. 2020. "Immunological Memory of Psoriatic Lesions" International Journal of Molecular Sciences 21, no. 2: 625. https://doi.org/10.3390/ijms21020625