Campylobacter concisus Impairs Sodium Absorption in Colonic Epithelium via ENaC Dysfunction and Claudin-8 Disruption

 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
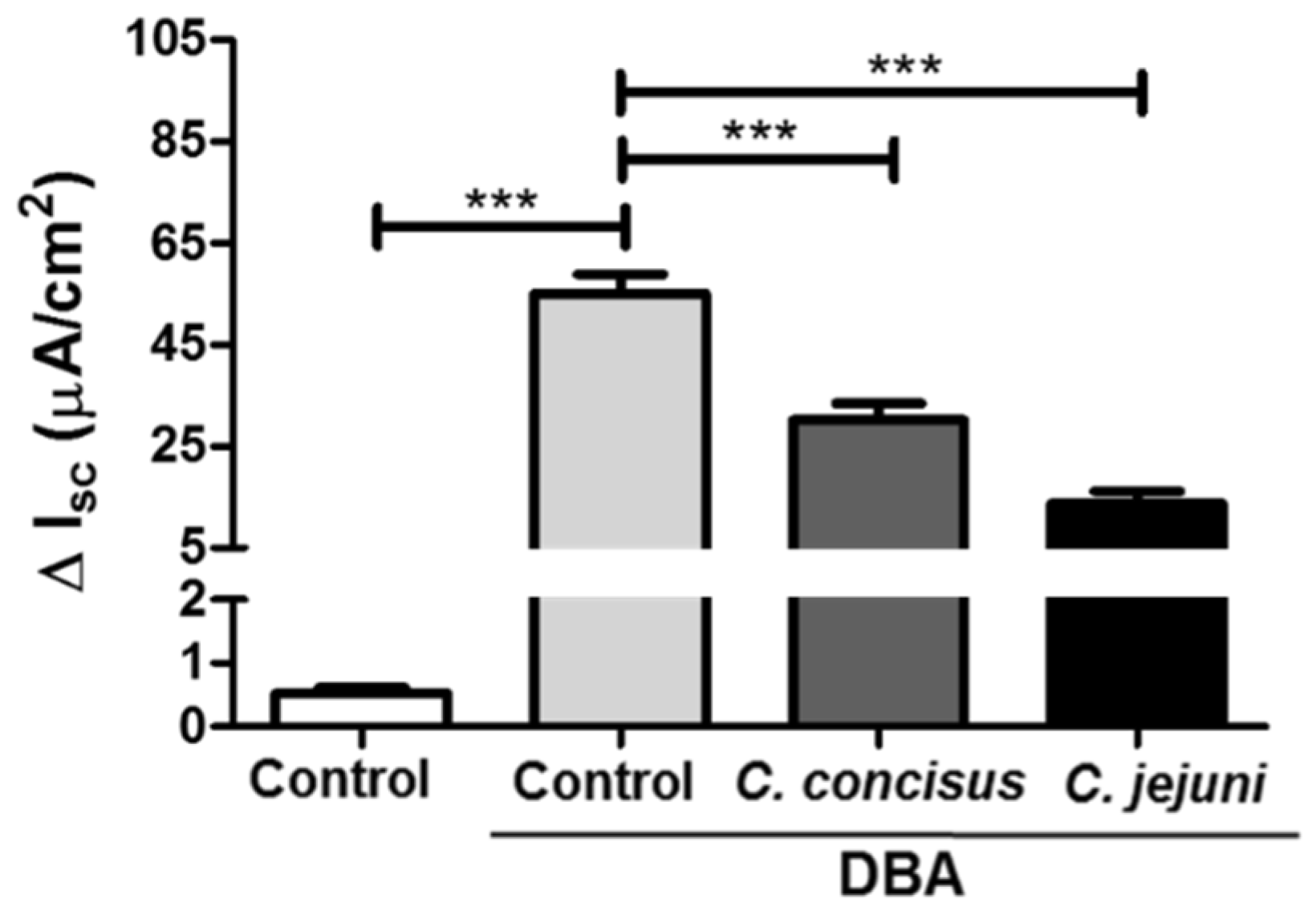
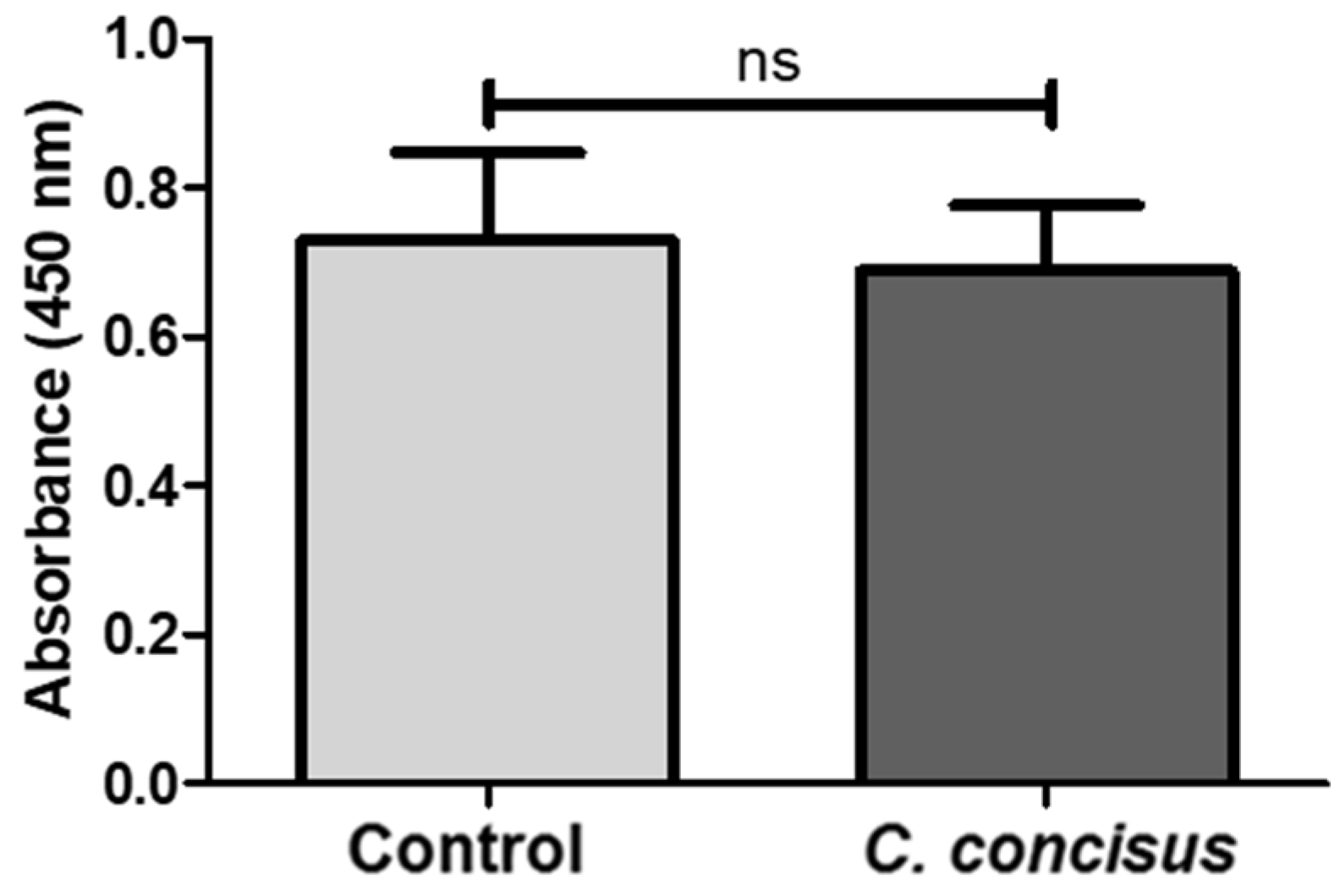
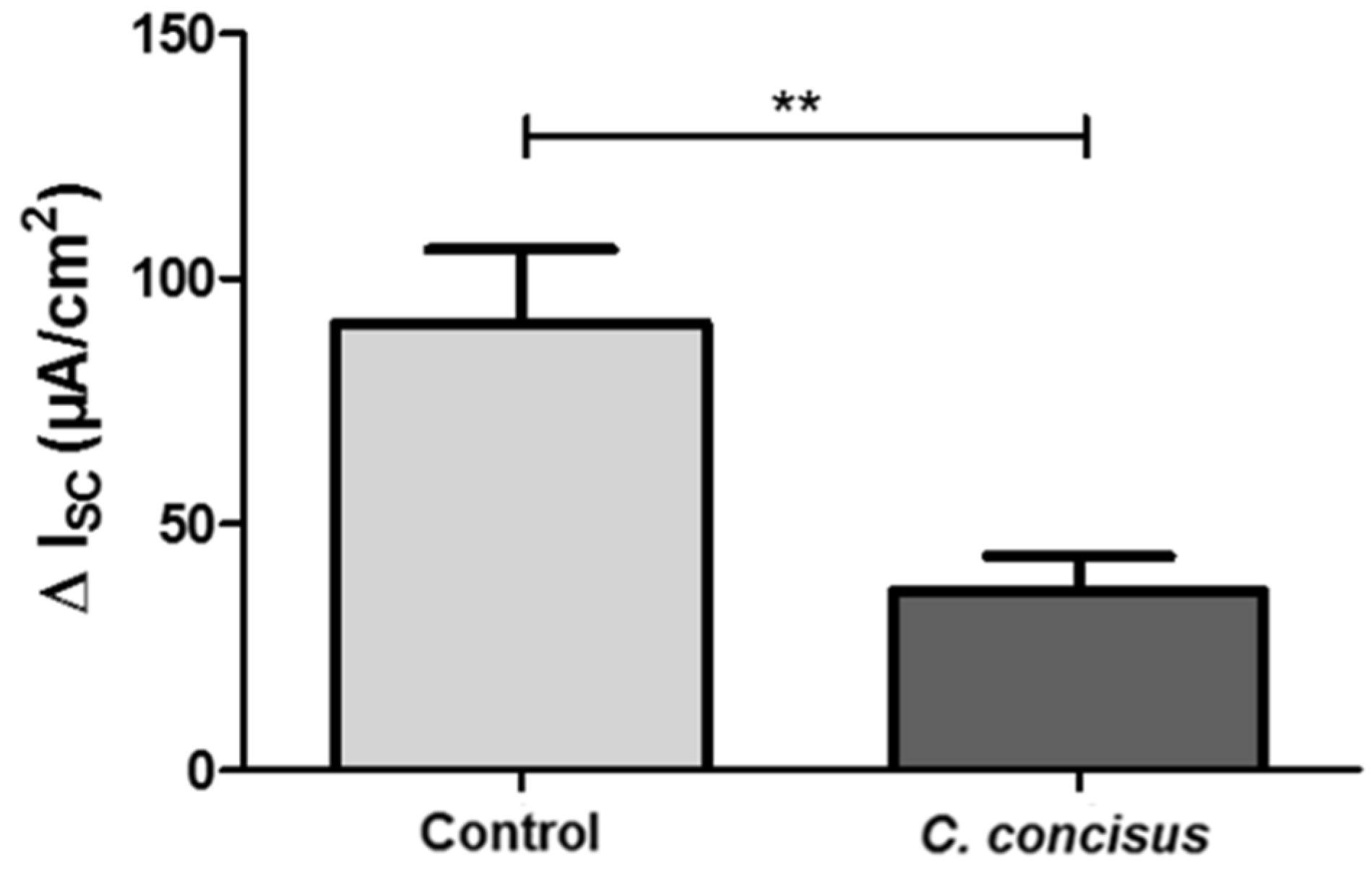
2.1. Campylobacter concisus Impairs Sodium Absorption via ENaC Dysfunction In Vitro
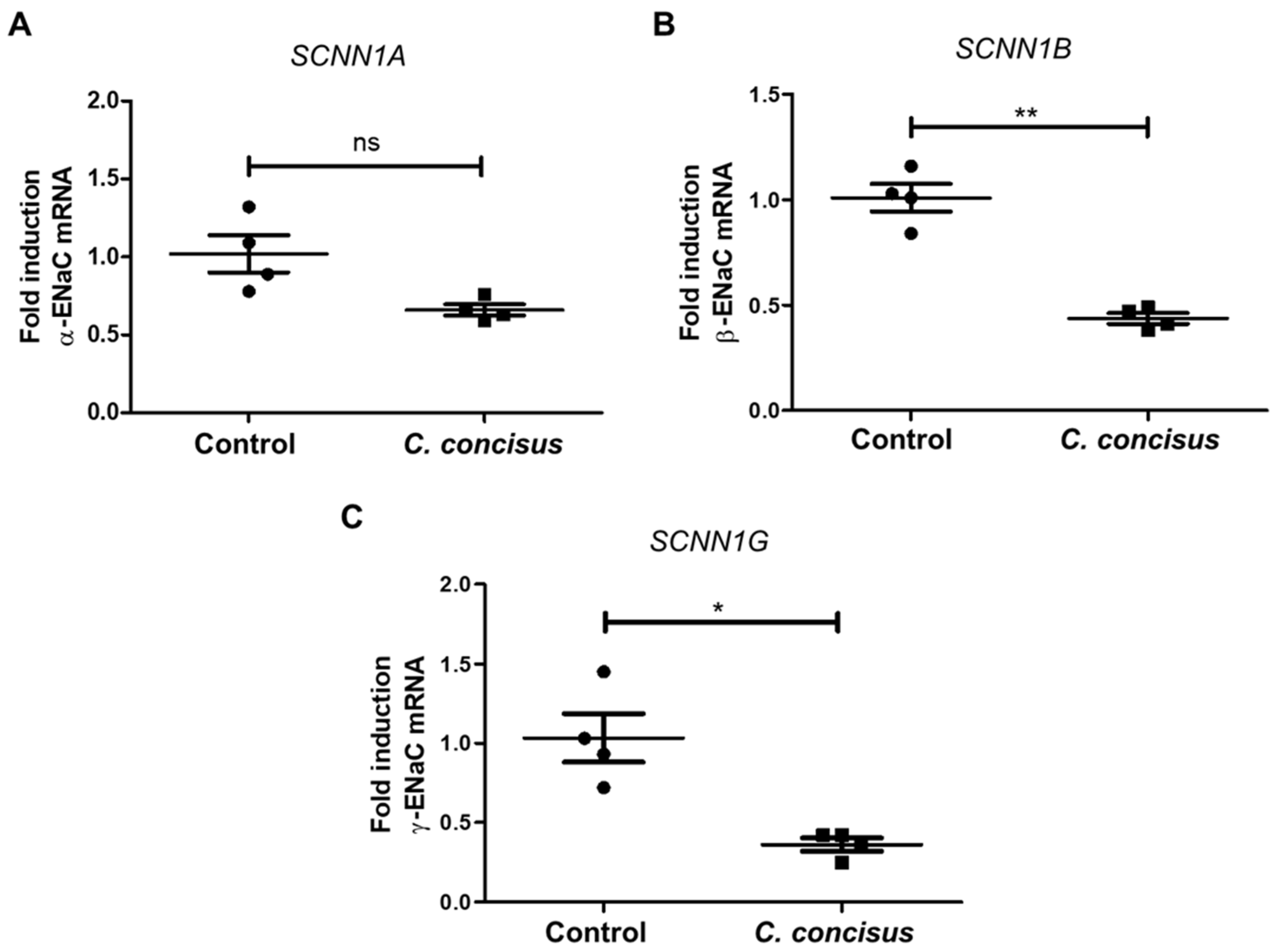
2.2. Campylobacter concisus Down-Regulates the mRNA Expression of β- and γ-ENaC Subunits
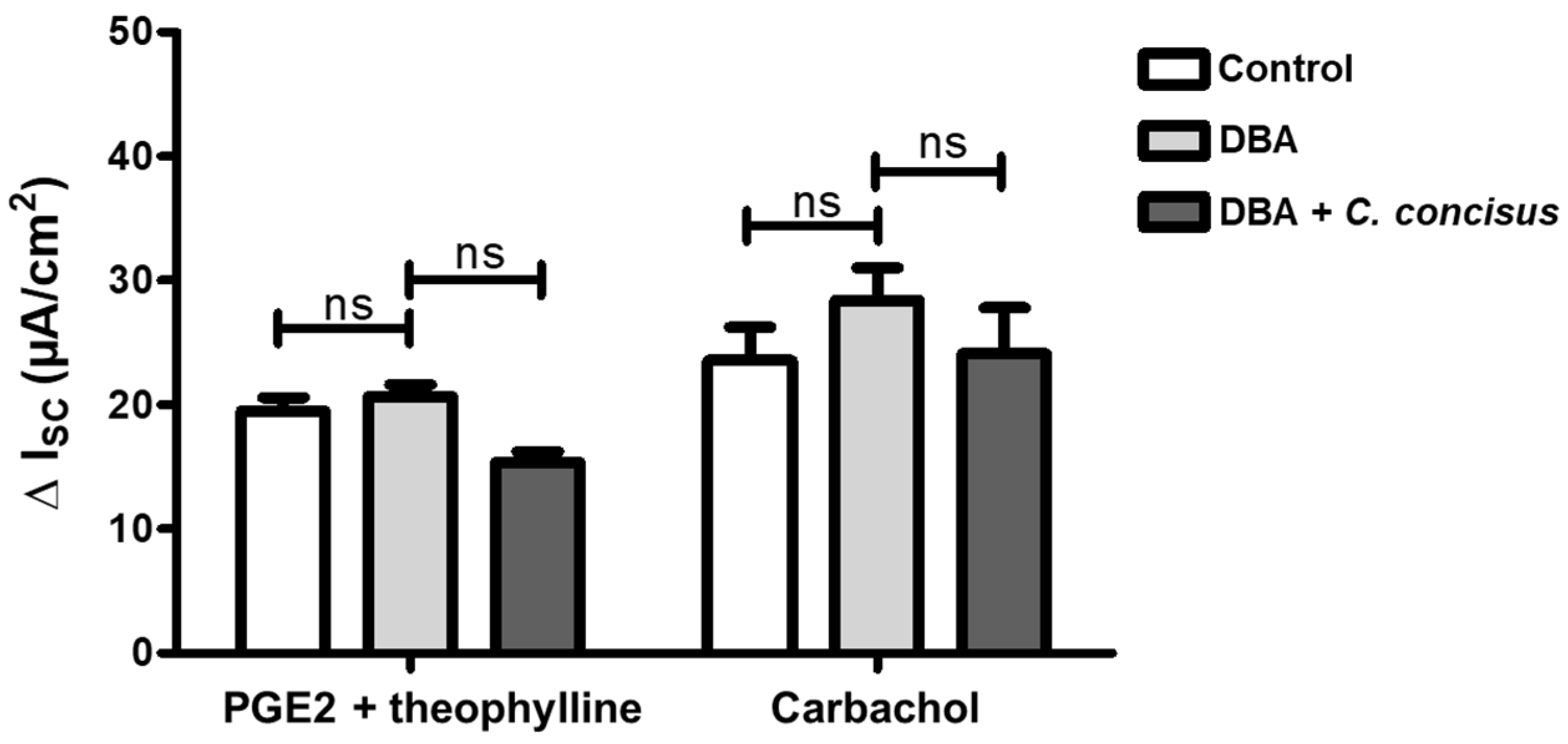
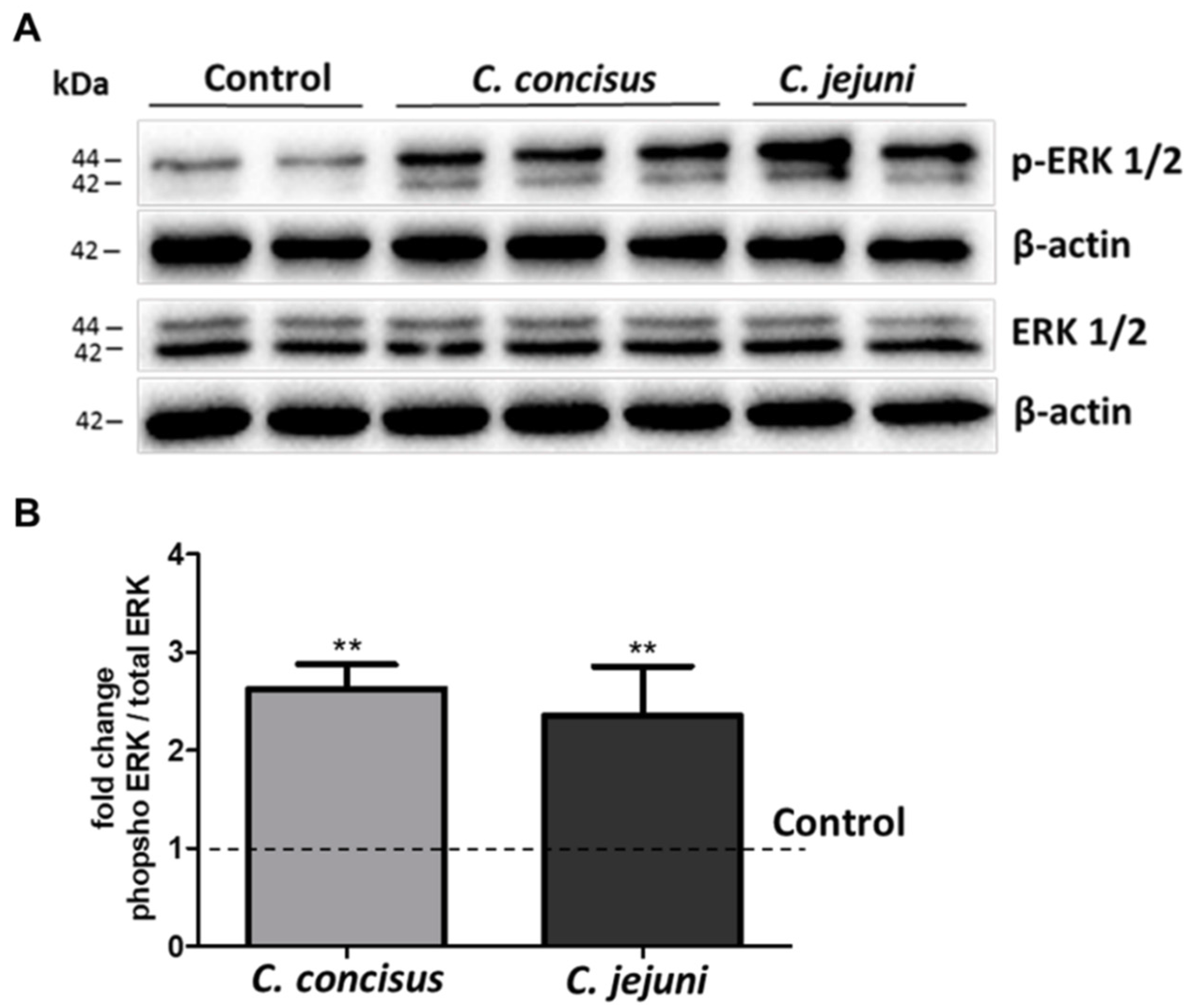
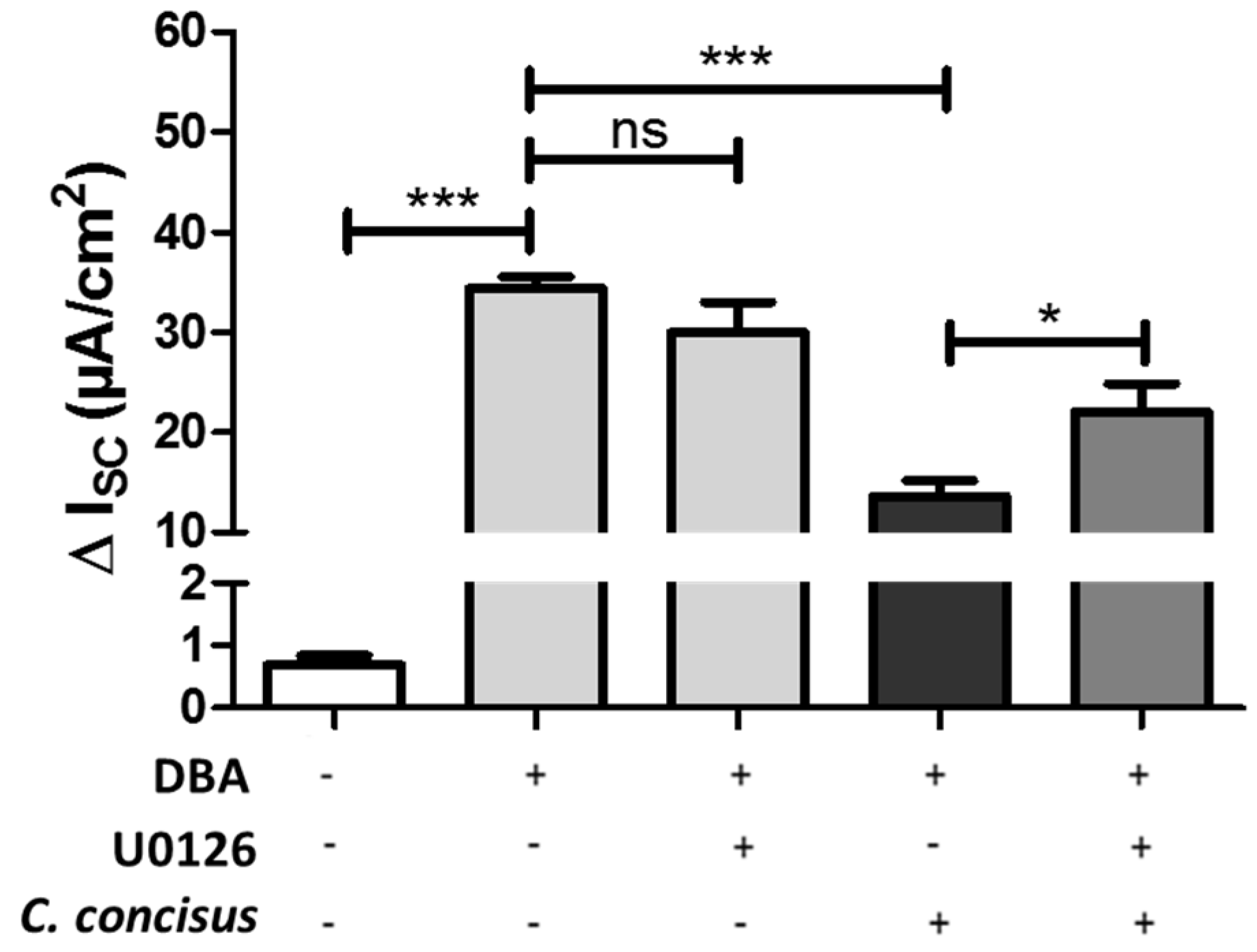
2.3. C. jejuni and C. concisus Dysregulate ENaC Function via ERK Activation
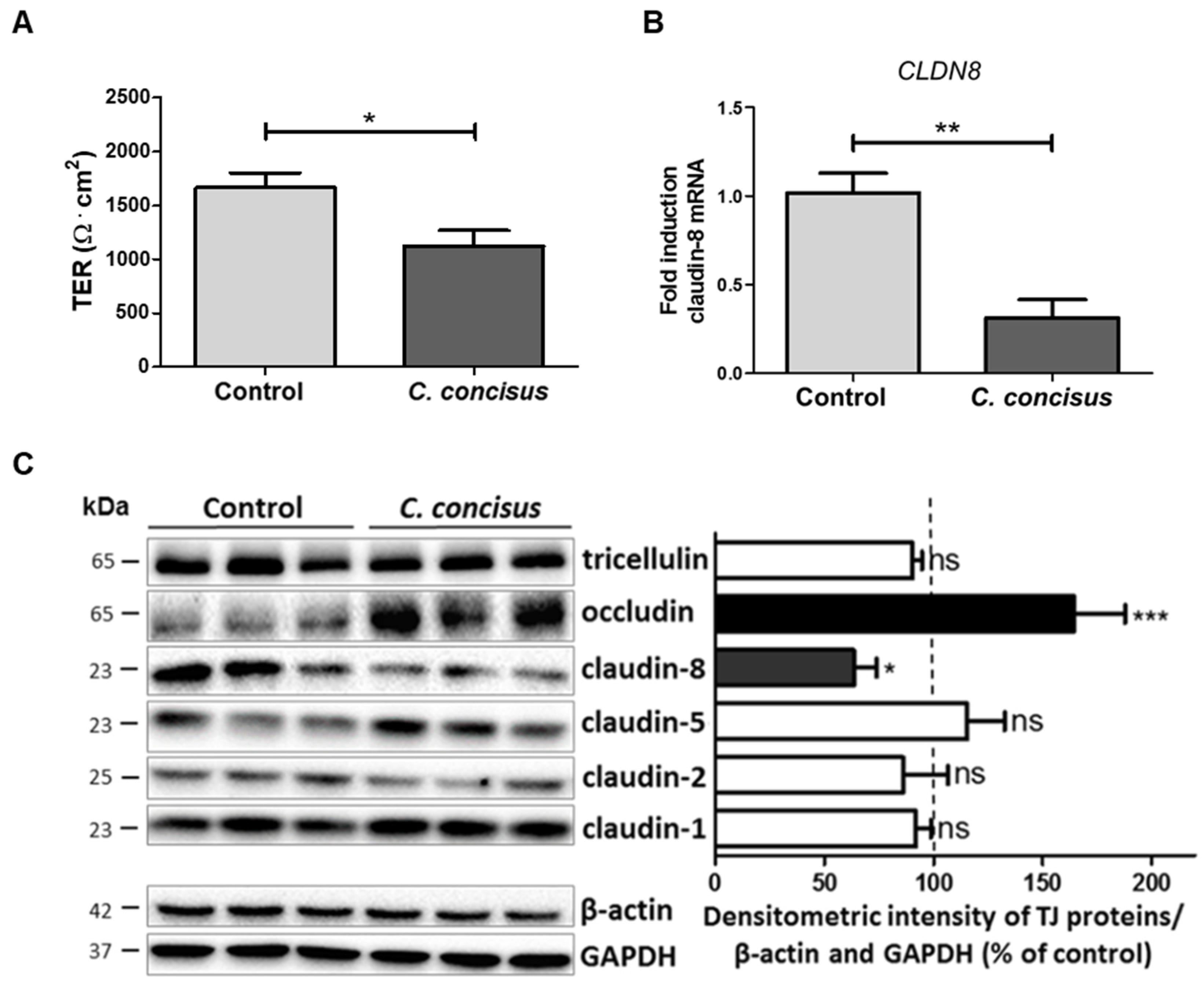
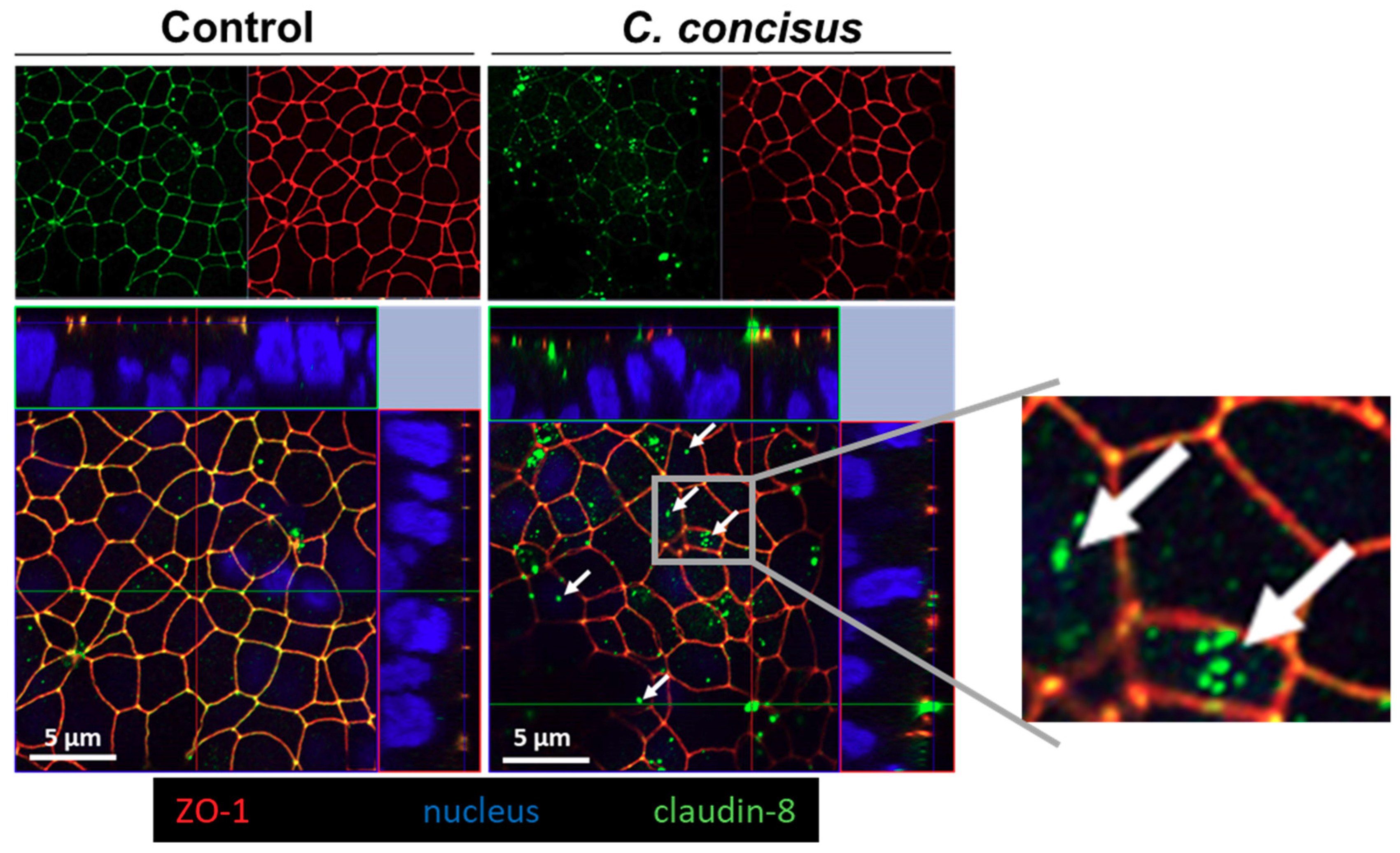
2.4. Campylobacter concisus Decreases Transepithelial Electrical Resistance and Promotes Changes in Tight Junction Protein Expression
2.5. Campylobacter concisus Impairs Sodium Absorption via ENaC in the Colon of IL-10−/− Mouse
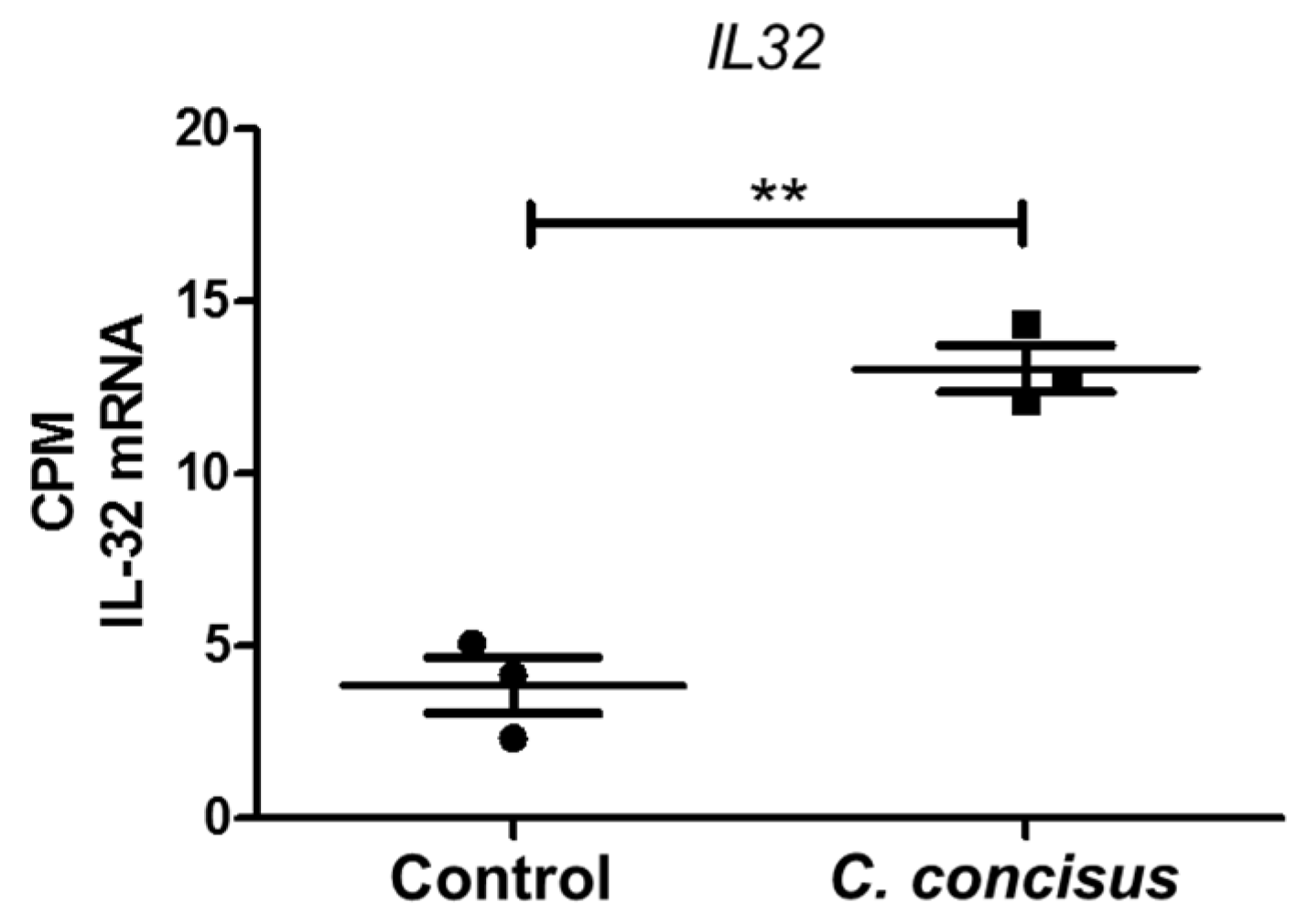
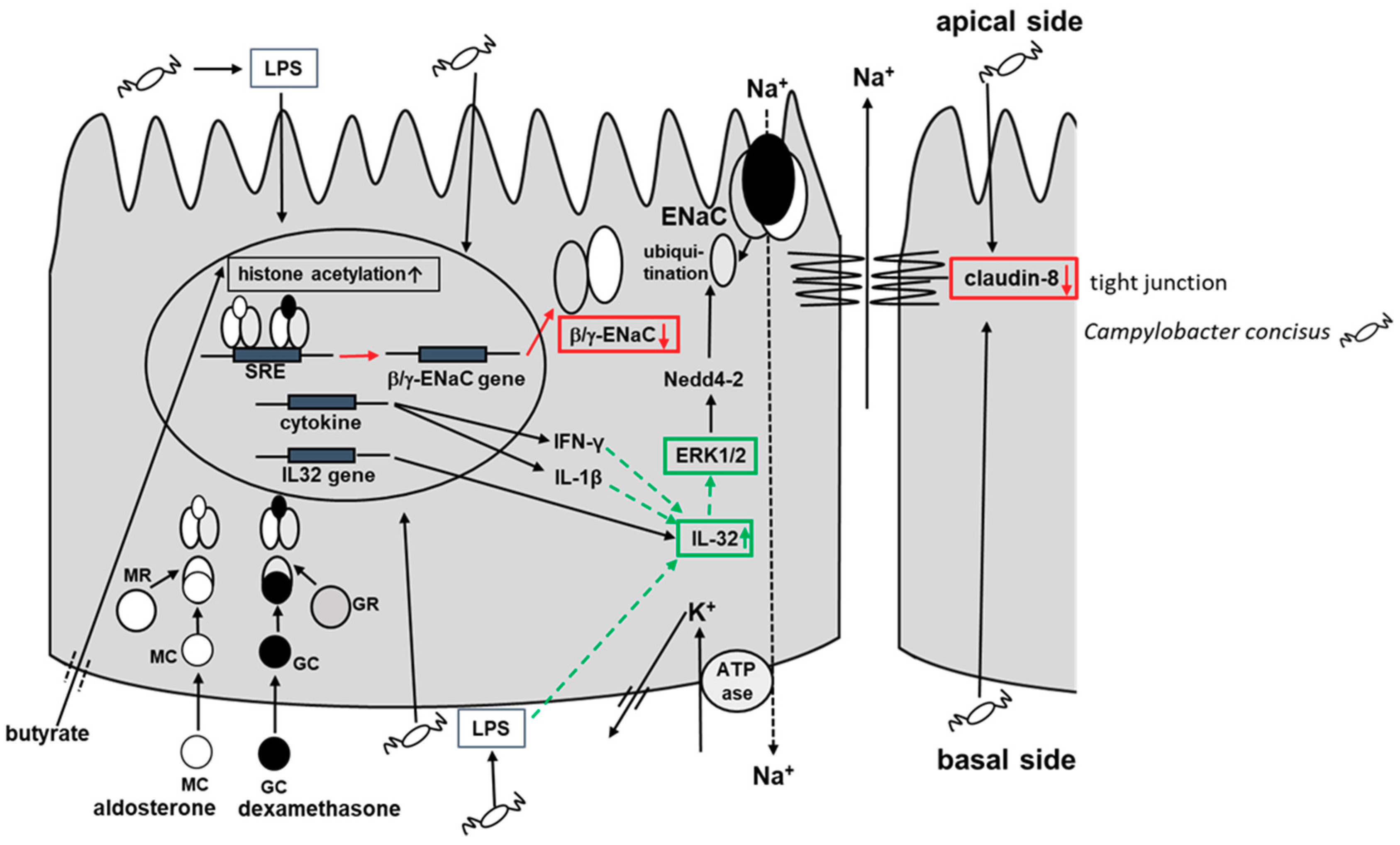
3. Discussion
4. Materials and Methods
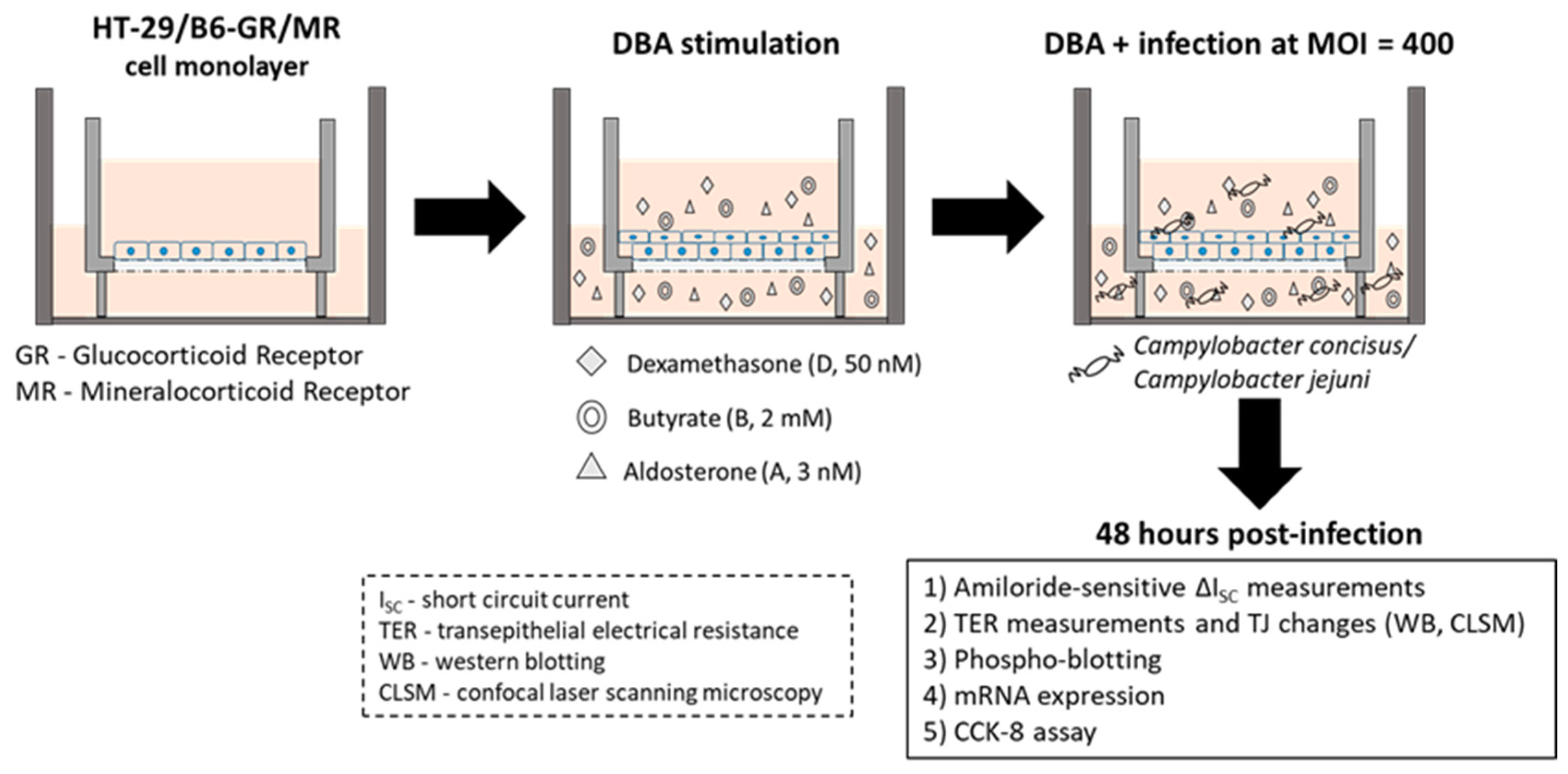
4.1. Cell Culture and Campylobacter Infection
4.2. Electrophysiological Determination of ENaC Function In Vitro
4.3. Western Blot Assessment of Tight Junction Protein Expression
4.4. Western Blot Assessment of ERK Phosphorylation
4.5. Functional Blockade of ERK to Determine the Changes in ENaC Function In Vitro
4.6. ENaC Regulatory β- and γ-Subunit and Claudin-8 mRNA Expression Analyzed by RT-qPCR
4.7. Tight Junction Protein Localization Evaluated by Immunofluorescence and Confocal Laser Scanning Microscopy
4.8. Electrophysiological Determination of ENaC Function in an In Vivo Model of C. concisus Infection
4.9. Ethics Statement
4.10. RNA-Seq Expression Analysis
4.11. Cell Proliferation and Cytotoxicity Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cAMP | Cyclic adenosine monophosphate |
| cDNA | Complementary DNA |
| CFU | Colony forming unit |
| CLSM | Confocal laser scanning microscopy |
| DAPI | 4′-6-diamidino-2-phenylindole dihydrochloride |
| DBA | Dexamethasone, butyrate and aldosterone |
| EDTA | Ethylenediaminetetraacetic acid |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| ENaC | Epithelial sodium channel |
| ERK | Extracellular signal-regulated kinase |
| FCS | Fetal calf serum |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GC | Glucocorticoids |
| GR | Glucocorticoid receptor |
| H2PO4− | Dihydrogen phosphate |
| HCO3− | Hydrogen carbonate |
| h-f FCS | Hormone-free FCS |
| HPO42− | Hydrogen phosphate |
| IFN | Interferon |
| IL | Interleukin |
| ISC | Short circuit current |
| JAM | Junctiunal adhesion molecule |
| JNK | c-Jun N-terminal kinase |
| LaGeSo | Landesamt für Gesundheit und Soziales |
| MAPK | Mitogen-activated protein kinase |
| MC | Mineralocorticoids |
| MEK | Mitogen-activated protein kinase kinase |
| miRNA | MicroRNA |
| MOI | Multiplicity of infection |
| MR | Mineralocorticoid receptor |
| mRNA | Messenger RNA |
| PBS | Phosphate-buffered saline |
| PGE2 | Prostaglandin E2 |
| PMSF | Phenylmethylsulfonyl fluoride |
| PVDF | Polyvinylidene fluoride |
| PVP | Polyvinylpyrrolidone |
| STAT-6 | Signal transducer and activator of transcription 6 |
| TER | Transepithelial electrical resistance |
| TJ | Tight junction |
| TNF | Tumor necrosis factor |
References
- Tanner, A.C.; Badger, S.; Lai, C.-H.; Listgarten, M.A.; Visconti, R.A.; Socransky, S.S. Wolinella gen. nov., Wolinella succinogenes (Vibrio succinogenes Wolin et al.) comb. nov., and Description of Bacteroides gracilis sp. nov., Wolinella recta sp. nov., Campylobacter concisus sp. nov., and Eikenella corrodens from Humans with Periodontal Disease. Int. J. Syst. Bacteriol. 1981, 31, 432–435. [Google Scholar]
- Tanner, A.C.; Dzink, J.L.; Ebersole, J.L.; Socransky, S.S. Wolinella recta, Campylobacter concisus, Bacteroides gracilis, and Eikenella corrodens from periodontal lesions. J. Periodontal Res. 1987, 22, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Macuch, P.J.; Tanner, A.C. Campylobacter species in health, gingivitis, and periodontitis. J. Dent. Res. 2000, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, G.B.; Sjogren, E.; Hansson-Westerberg, J.; Kaijser, B. Campylobacter upsaliensis, C. Sputorum sputorum and C. Concisus as common causes of diarrhoea in Swedish children. Scand. J. Infect. Dis. 1995, 27, 187–188. [Google Scholar] [CrossRef]
- Aabenhus, R.; Permin, H.; On, S.L.; Andersen, L.P. Prevalence of Campylobacter concisus in diarrhoea of immunocompromised patients. Scand. J. Infect. Dis. 2002, 34, 248–252. [Google Scholar] [CrossRef] [Green Version]
- Van Etterijck, R.; Breynaert, J.; Revets, H.; Devreker, T.; Vandenplas, Y.; Vandamme, P.; Lauwers, S. Isolation of Campylobacter concisus from feces of children with and without Diarrhea. J. Clin. Microbiol. 1996, 34, 2304–2306. [Google Scholar] [CrossRef] [Green Version]
- Maher, M.; Finnegan, C.; Collins, E.; Ward, B.; Carroll, C.; Cormican, M. Evaluation of culture methods and a DNA probe-based PCR assay for detection of campylobacter species in clinical specimens of feces. J. Clin. Microbiol. 2003, 41, 2980–2986. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.L.; Engberg, J.; Ejlertsen, T.; Bucker, R.; Nielsen, H. Short-term and medium-term clinical outcomes of Campylobacter concisus infection. Clin. Microbiol. Infect. 2012, 18, E459–E465. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.L.; Nielsen, H.; Ejlertsen, T.; Engberg, J.; Gunzel, D.; Zeitz, M.; Hering, N.A.; Fromm, M.; Schulzke, J.D.; Bucker, R. Oral and fecal Campylobacter concisus strains perturb barrier function by apoptosis induction in HT-29/B6 intestinal epithelial cells. PLoS ONE 2011, 6, e23858. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, P.J.; Clarke, L.L.; Meneton, P.; Miller, M.L.; Soleimani, M.; Gawenis, L.R.; Riddle, T.M.; Duffy, J.J.; Doetschman, T.; Wang, T.; et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 1998, 19, 282–285. [Google Scholar] [CrossRef]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.D.; Rossier, B.C. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994, 367, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Renard, S.; Voilley, N.; Bassilana, F.; Lazdunski, M.; Barbry, P. Localization and regulation by steroids of the alpha, beta and gamma subunits of the amiloride-sensitive Na+ channel in colon, lung and kidney. Pflüg. Arch. 1995, 430, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Epple, H.J.; Amasheh, S.; Mankertz, J.; Goltz, M.; Schulzke, J.D.; Fromm, M. Early aldosterone effect in distal colon by transcriptional regulation of ENaC subunits. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G718–G724. [Google Scholar] [CrossRef] [PubMed]
- Greig, E.R.; Baker, E.H.; Mathialahan, T.; Boot-Handford, R.P.; Sandle, G.I. Segmental variability of ENaC subunit expression in rat colon during dietary sodium depletion. Pflüg. Arch. 2002, 444, 476–483. [Google Scholar]
- Bergann, T.; Fromm, A.; Borden, S.A.; Fromm, M.; Schulzke, J.D. Glucocorticoid receptor is indispensable for physiological responses to aldosterone in epithelial Na+ channel induction via the mineralocorticoid receptor in a human colonic cell line. Eur. J. Cell Biol. 2011, 90, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Kuntzsch, D.; Bergann, T.; Dames, P.; Fromm, A.; Fromm, M.; Davis, R.A.; Melzig, M.F.; Schulzke, J.D. The plant-derived glucocorticoid receptor agonist Endiandrin A acts as co-stimulator of colonic epithelial sodium channels (ENaC) via SGK-1 and MAPKs. PLoS ONE 2012, 7, e49426. [Google Scholar] [CrossRef] [Green Version]
- Dames, P.; Bergann, T.; Fromm, A.; Bucker, R.; Barmeyer, C.; Krug, S.M.; Fromm, M.; Schulzke, J.D. Interleukin-13 affects the epithelial sodium channel in the intestine by coordinated modulation of stat6 and p38 MAPK activity. J. Physiol. 2015, 593, 5269–5282. [Google Scholar] [CrossRef] [Green Version]
- Amasheh, S.; Barmeyer, C.; Koch, C.S.; Tavalali, S.; Mankertz, J.; Epple, H.J.; Gehring, M.M.; Florian, P.; Kroesen, A.J.; Zeitz, M.; et al. Cytokine-dependent transcriptional down-regulation of epithelial sodium channel in ulcerative colitis. Gastroenterology 2004, 126, 1711–1720. [Google Scholar] [CrossRef]
- Zeissig, S.; Bergann, T.; Fromm, A.; Bojarski, C.; Heller, F.; Guenther, U.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Altered ENaC expression leads to impaired sodium absorption in the noninflamed intestine in Crohn’s disease. Gastroenterology 2008, 134, 1436–1447. [Google Scholar] [CrossRef]
- Barmeyer, C.; Erko, I.; Fromm, A.; Bojarski, C.; Loddenkemper, C.; Dames, P.; Kerick, M.; Siegmund, B.; Fromm, M.; Schweiger, M.R.; et al. ENaC dysregulation through activation of MEK1/2 contributes to impaired Na+ absorption in lymphocytic colitis. Inflamm. Bowel Dis. 2016, 22, 539–547. [Google Scholar] [CrossRef]
- Chiba, H.; Osanai, M.; Murata, M.; Kojima, T.; Sawada, N. Transmembrane proteins of tight junctions. Biochim. Biophys. Acta 2008, 1778, 588–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.I. Structure and regulation of intestinal epithelial tight junctions: Current concepts and unanswered questions. Adv. Exp. Med. Biol. 2012, 763, 132–148. [Google Scholar] [PubMed]
- Krug, S.M.; Gunzel, D.; Conrad, M.P.; Lee, I.F.; Amasheh, S.; Fromm, M.; Yu, A.S. Charge-selective claudin channels. Ann. N. Y. Acad. Sci. 2012, 1257, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Hayashi, H.; Imasato, M.; Yamazaki, Y.; Hagiwara, A.; Wada, M.; Noda, T.; Watanabe, M.; Suzuki, Y.; Tsukita, S. Loss of claudin-15, but not claudin-2, causes Na+ deficiency and glucose malabsorption in mouse small intestine. Gastroenterology 2011, 140, 913–923. [Google Scholar] [CrossRef]
- Amasheh, S.; Milatz, S.; Krug, S.M.; Bergs, M.; Amasheh, M.; Schulzke, J.D.; Fromm, M. Na+ absorption defends from paracellular back-leakage by claudin-8 upregulation. Biochem. Biophys. Res. Commun. 2009, 378, 45–50. [Google Scholar] [CrossRef]
- Bucker, R.; Krug, S.M.; Moos, V.; Bojarski, C.; Schweiger, M.R.; Kerick, M.; Fromm, A.; Janssen, S.; Fromm, M.; Hering, N.A.; et al. Campylobacter jejuni impairs sodium transport and epithelial barrier function via cytokine release in human colon. Mucosal Immunol. 2018, 11, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Haag, L.M.; Fischer, A.; Otto, B.; Plickert, R.; Kuhl, A.A.; Gobel, U.B.; Bereswill, S.; Heimesaat, M.M. Campylobacter jejuni induces acute enterocolitis in gnotobiotic il-10-/- mice via toll-like-receptor-2 and -4 signaling. PLoS ONE 2012, 7, e40761. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Lugert, R.; Fischer, A.; Alutis, M.; Kuhl, A.A.; Zautner, A.E.; Tareen, A.M.; Gobel, U.B.; Bereswill, S. Impact of Campylobacter jejuni cj0268c knockout mutation on intestinal colonization, translocation, and induction of immunopathology in gnotobiotic IL-10 deficient mice. PLoS ONE 2014, 9, e90148. [Google Scholar] [CrossRef]
- Zeissig, S.; Fromm, A.; Mankertz, J.; Weiske, J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Butyrate induces intestinal sodium absorption via sp3-mediated transcriptional up-regulation of epithelial sodium channels. Gastroenterology 2007, 132, 236–248. [Google Scholar] [CrossRef]
- Barmeyer, C.; Amasheh, S.; Tavalali, S.; Mankertz, J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. IL-1β and TNFα regulate sodium absorption in rat distal colon. Biochem. Biophys. Res. Commun. 2004, 317, 500–507. [Google Scholar] [CrossRef]
- Man, S.M.; Kaakoush, N.O.; Leach, S.T.; Nahidi, L.; Lu, H.K.; Norman, J.; Day, A.S.; Zhang, L.; Mitchell, H.M. Host attachment, invasion, and stimulation of proinflammatory cytokines by Campylobacter concisus and other non-Campylobacter jejuni campylobacter species. J. Infect. Dis. 2010, 202, 1855–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Lee, C.K.; Kim, S.H.; Cho, W.S.; Mun, S.H.; Yoo, B. Interleukin-32γ Enhances the Production of IL-6 and IL-8 in Fibroblast-Like Synoviocytes Via Erk1/2 Activation. J. Clin. Immunol. 2010, 30, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Chiu, Y.M.; Lee, Y.J.; Hsieh, C.T.; Shieh, D.C.; Tsay, G.J.; Bau, D.T.; Wu, Y.Y. Interleukin-32 plays an essential role in human calcified aortic valve cells. Eur. Cytokine Netw. 2018, 29, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Hering, N.A.; Richter, J.F.; Krug, S.M.; Gunzel, D.; Fromm, A.; Bohn, E.; Rosenthal, R.; Bucker, R.; Fromm, M.; Troeger, H.; et al. Yersinia enterocolitica induces epithelial barrier dysfunction through regional tight junction changes in colonic HT-29/B6 cell monolayers. Lab. Investig. 2011, 91, 310–324. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Rong, A.; Zhou, Q.; Li, W. Cldn8 promotes colorectal cancer cell proliferation, migration, and invasion by activating MAPK/Erk signaling. Cancer Manag. Res. 2019, 11, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- Barmeyer, C.; Erko, I.; Awad, K.; Fromm, A.; Bojarski, C.; Meissner, S.; Loddenkemper, C.; Kerick, M.; Siegmund, B.; Fromm, M.; et al. Epithelial barrier dysfunction in lymphocytic colitis through cytokine-dependent internalization of claudin-5 and -8. J. Gastroenterol. 2017, 52, 1090–1100. [Google Scholar] [CrossRef]
- Nielsen, H.L.; Engberg, J.; Ejlertsen, T.; Nielsen, H. Evaluation of fecal calprotectin in Campylobacter concisus and Campylobacter jejuni/coli gastroenteritis. Scand. J. Gastroenterol. 2013, 48, 633–635. [Google Scholar] [CrossRef]
- Burgos-Portugal, J.A.; Mitchell, H.M.; Castano-Rodriguez, N.; Kaakoush, N.O. The role of autophagy in the intracellular survival of Campylobacter concisus. FEBS Open Bio 2014, 4, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Ganapathy, A.S.; Suchanec, E.; Laidler, L.; Ma, T.; Nighot, P. Intestinal epithelial tight junction barrier regulation by autophagy-related protein atg6/beclin 1. Am. J. Physiol. Cell Physiol. 2019, 316, C753–C765. [Google Scholar] [CrossRef]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [Green Version]
- Schulzke, J.D.; Gitter, A.H.; Mankertz, J.; Spiegel, S.; Seidler, U.; Amasheh, S.; Saitou, M.; Tsukita, S.; Fromm, M. Epithelial transport and barrier function in occludin-deficient mice. Biochim. Biophys. Acta 2005, 1669, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Bereswill, S.; Fischer, A.; Plickert, R.; Haag, L.M.; Otto, B.; Kühl, A.A.; Dashti, J.I.; Zautner, A.E.; Muñoz, M.; Loddenkemper, C.; et al. Novel murine Infection Models Provide Deep Insights into the ‘‘Ménage à Trois’’ of Campylobacter jejuni, Microbiota and Host Innate Immunity. PLoS ONE 2011, 6, e209523. [Google Scholar] [CrossRef]
- Masanta, W.O.; Heimesaat, M.M.; Bereswill, S.; Tareen, A.M.; Lugert, R.; Groß, U.; Zautner, A.E. Modification of Intestinal Microbiota and Its Consequences for Innate Immune Response in the Pathogenesis of Campylobacteriosis. Clin. Dev. Immunol. 2013, 526860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, S.A.; Care, A.S.; Skinner, R.J. Interleukin 10 Regulates Inflammatory Cytokine Synthesis to Protect Against Lipopolysaccharide-Induced Abortion and Fetal Growth Restriction in Mice. Biol. Reprod. 2007, 76, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, H.S.; Fitting, C.; Hiff, E.; Adib-Conquy, M.; Beasley-Topliffe, L.; Tesini, B.; Liang, X.; Valentine, C.; Hellman, J.; Hayden, D.; et al. Resilience to bacterial infection: Difference between species could be due to proteins in serum. J. Infect. Dis. 2010, 201, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Taveira da Silva, A.M.; Kaulbach, H.C.; Chuidian, F.S.; Lambert, D.R.; Suffredeni, A.F.; Danner, R.L. Brief Report: Shock and Multiple-Organ Dysfunction after Self-Administration of Salmonella Endotoxin. N. Engl. J. Med. 1993, 328, 1457–1460. [Google Scholar] [CrossRef]
- Stahl, M.; Ries, J.; Vermeulen, J.; Yang, H.; Sham, H.P.; Crowley, S.M.; Badayeva, Y.; Turvey, S.E.; Gaynor, E.C.; Li, X.; et al. A Novel Mouse Model of Campylobacter jejuni Gastroenteritis Reveals Key Pro-inflammatory and Tissue Protective Roles for Toll-like Receptor Signaling during Infection. PLoS Pathog. 2014, 10, e1004624. [Google Scholar] [CrossRef] [Green Version]
- Haag, L.M.; Fischer, A.; Otto, B.; Plickert, R.; Kühl, A.A.; Göbel, U.B.; Bereseswill, S.; Heimesaat, M.M. Intestinal Microbiota Shift towards Elevated Commensal Escherichia coli Loads Abrogate Colinization Resistance against Campylobacter jejuni in Mice. PLoS ONE 2012, 7, e35988. [Google Scholar] [CrossRef]
- Alutis, M.E.; Grundmann, U.; Fischer, A.; Kühl, A.A.; Bereswill, S.; Heimesaat, M.M. Selective Gelatinase Inhibition reduces Apoptosis and Pro-inflammatory Immune Cell Responses in Campylobacter jejuni-infected Gnotobiotic IL-10 Deficient Mice. Eur. J. Microbiol. Immunol. 2014, 4, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Grundmann, U.; Alutis, M.E.; Fischer, A.; Bereswill, S. Microbiota Composition and Immune Responses during Campylobacter jejuni Infection in Conventionally Colonized IL-10−/− Mice Lacking Nucleotide Oligomerization Domain 2. Eur. J. Microbiol. Immunol. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bereswill, S.; Grundmann, U.; Alutis, M.E.; Fischer, A.; Heimesaat, M.M. Immune responses upon Campylobacter jejuni infection of secondary abiotic mice lacking nucleotide-oligomerization-domain-2. Gut Pathog. 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo de Sa, F.D.; Butkevych, E.; Nattramilarasu, P.K.; Fromm, A.; Mousavi, S.; Moos, V.; Golz, J.C.; Stingl, K.; Kittler, S.; Seinige, D.; et al. Curcumin mitigates immune-induced epithelial barrier dysfunction by Campylobacter jejuni. Int. J. Mol. Sci. 2019, 20, 4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, S.; Lobo de Sa, F.D.; Schulzke, J.D.; Bucker, R.; Bereswill, S.; Heimesaat, M.M. Vitamin d in acute campylobacteriosis-results from an intervention study applying a clinical Campylobacter jejuni induced enterocolitis model. Front. Immunol. 2019, 10, 2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aabenhus, R.; Stenram, U.; Andersen, L.P.; Permin, H.; Ljungh, A. First attempt to produce experimental Campylobacter concisus infection in mice. World J. Gastroenterol. 2008, 14, 6954–6959. [Google Scholar] [CrossRef]
- Kirk, K.F.; Meric, G.; Nielsen, H.L.; Pascoe, B.; Sheppard, S.K.; Thorlacius-Ussing, O.; Nielsen, H. Molecular epidemiology and comparative genomics of Campylobacter concisus strains from saliva, faeces and gut mucosal biopsies in inflammatory bowel disease. Sci. Rep. 2018, 8, 1902. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schulzke, J.D.; Fromm, M.; Hegel, U. Epithelial and subepithelial resistance of rat large intestine: Segmental differences, effect of stripping, time course, and action of aldosterone. Pflüg. Arch. 1986, 407, 632–637. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, M.; Miyazono, Y.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A highly water-soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta 1997, 44, 1299–1305. [Google Scholar] [CrossRef]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abiotic IL-10−/− Mice | ∆ISC (µA/cm2) after PGE2 + Theophylline | ∆ISC (µA/cm2) after Bumetanide | Resistance | ||
|---|---|---|---|---|---|
| Repi | Rsub | Rtotal | |||
| Control (n = 4–8) | 27 ± 7.99 | −15 ± 4.08 | 28.4 ± 3.62 | 24.8 ± 2.15 | 52.4 ± 3.55 |
| C. concisus-infection (n = 5–8) | 25 ± 8.12 | −16 ± 5.57 | 33.2 ± 3.10 | 28.5 ± 2.57 | 61.7 ± 3.90 |
| Significance | ns | ns | ns | ns | ns |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nattramilarasu, P.K.; Bücker, R.; Lobo de Sá, F.D.; Fromm, A.; Nagel, O.; Lee, I.-F.M.; Butkevych, E.; Mousavi, S.; Genger, C.; Kløve, S.; et al. Campylobacter concisus Impairs Sodium Absorption in Colonic Epithelium via ENaC Dysfunction and Claudin-8 Disruption. Int. J. Mol. Sci. 2020, 21, 373. https://doi.org/10.3390/ijms21020373
Nattramilarasu PK, Bücker R, Lobo de Sá FD, Fromm A, Nagel O, Lee I-FM, Butkevych E, Mousavi S, Genger C, Kløve S, et al. Campylobacter concisus Impairs Sodium Absorption in Colonic Epithelium via ENaC Dysfunction and Claudin-8 Disruption. International Journal of Molecular Sciences. 2020; 21(2):373. https://doi.org/10.3390/ijms21020373
Chicago/Turabian StyleNattramilarasu, Praveen Kumar, Roland Bücker, Fábia Daniela Lobo de Sá, Anja Fromm, Oliver Nagel, In-Fah Maria Lee, Eduard Butkevych, Soraya Mousavi, Claudia Genger, Sigri Kløve, and et al. 2020. "Campylobacter concisus Impairs Sodium Absorption in Colonic Epithelium via ENaC Dysfunction and Claudin-8 Disruption" International Journal of Molecular Sciences 21, no. 2: 373. https://doi.org/10.3390/ijms21020373