Tissue-Specific Delivery of CRISPR Therapeutics: Strategies and Mechanisms of Non-Viral Vectors


Abstract
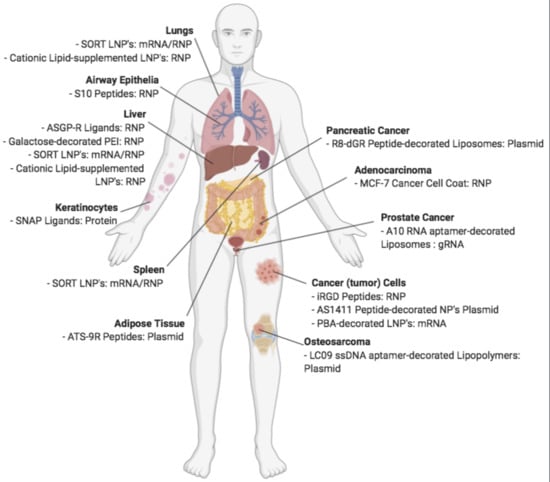
:
1. Introduction
2. Potential and Application of CRISPR Therapeutics
3. Limitations
3.1. Off-Targets
3.2. Delivery
4. Strategies for CRISPR-Cas Delivery
- 1-
- DNA
- 2-
- RNA
- Cas protein encoded in a messenger RNA (mRNA) and the gRNA as an in vitro transcribed synthetic oligonucleotide [72].
- 3-
- Protein
5. Non-Viral Targeted Delivery Strategies for the CRISPR-Cas System
5.1. Advantages of Non-Viral over Viral Vectors
- (1)
- (2)
- They are customizable and do not present a limited packaging capacity, which is compatible with the large-sized CRISPR components such as RNP or DNA.
- (3)
- (4)
- (5)
- They carry a very good scale-up potential, which greatly eases the process for clinical translation [98].
5.2. Non-Viral Vectors for Targeted Delivery of CRISPR
6. The Evolution of Peptide Delivery Systems for CRISPR-Cas Components
7. CPPs
8. Mechanism of CPP-Uptake and Release
8.1. Cellular Uptake
8.2. Endosomal Release
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 9R | Nona-arginine |
| AAV | Adeno-Associated Virus |
| ASGPr | Asialoglycoprotein receptor |
| ATS | Adipocyte Targeting Sequence |
| bp | Base pair |
| Cas | CRISPR-associated |
| CPP | Cell-penetrating peptides |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| crRNA | CRISPR RNAs |
| dCas9 | Deficient Cas9 |
| DNA | De-oxyribonucleic acid |
| DSB | Double-stranded break |
| gRNA | Guide RNA |
| HDR | Homology-Directed Repair |
| HSPG | Heparan sulfate proteoglycans |
| mRNA | Messenger RNA |
| NHEJ | Non-Homologous End Joining |
| PAM | Protospacer adjacent motif |
| RNA | Ribonucleic acid |
| RNP | Ribonucleoproteins |
| RVG | Rabies Virus Glycoprotein |
| sgRNA | Single guide RNA |
| SORT | Selective ORgan Targeting |
| SPIRIT | Superior Peptide InseRtions for Improved Targeting |
| TAT | Trans-activator of transcription |
| tracrRNA | Trans-activating RNA |
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Karginov, F.V.; Hannon, G.J. The CRISPR System: Small RNA-Guided Defense in Bacteria and Archaea. Mol. Cell 2010, 37, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; Van Der Oost, J. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J.A. RNA-programmed genome editing in human cells. eLife 2013, 2. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Ren, M.; Wang, Z.; Zhang, B.; Rong, Y.S.; Jiao, R.; Gao, G. Highly Efficient Genome Modifications Mediated by CRISPR/Cas9 in Drosophila. Genet. 2013, 195, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Xing, H.-L.; Dong, L.; Wang, Z.; Zhang, H.-Y.; Han, C.-Y.; Liu, B.; Wang, X.-C.; Chen, Q.-J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, D.; Ward, J.D.; Reiner, D.; Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 2013, 10, 1028–1034. [Google Scholar] [CrossRef] [Green Version]
- Mashiko, D.; Fujihara, Y.; Satouh, Y.; Miyata, H.; Isotani, A.; Ikawa, M. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 2013, 3, 3355. [Google Scholar] [CrossRef]
- Chylinski, K.; Makarova, K.S.; Charpentier, E.; Koonin, E.V. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014, 42, 6091–6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, P.; Goedecke, W.; Obe, G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 2000, 15, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Genet. 2019, 18, 67–83. [Google Scholar] [CrossRef]
- Swarts, D.C.; Jinek, M. Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing. Wiley Interdiscip. Rev. RNA 2018, 9, e1481. [Google Scholar] [CrossRef]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; A Scott, D.; A Weinstein, J.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Huck, S.; Bertrand, P.; Pirzio, L.; Desmaze, C.; Sabatier, L.; Lopez, B.S. Impact of the KU80 Pathway on NHEJ-Induced Genome Rearrangements in Mammalian Cells. Mol. Cell 2004, 14, 611–623. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Song, M.; Lee, J.; Menon, V.; Jung, S.; Kang, Y.-M.; Choi, J.; Woo, E.-J.; Koh, H.C.; Nam, J.-W.; et al. In vivo high-throughput profiling of CRISPR–Cpf1 activity. Nat. Methods 2016, 14, 153–159. [Google Scholar] [CrossRef]
- Gao, P.; Yang, H.; Rajashankar, K.R.; Huang, Z.; Patel, D.J. Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Res. 2016, 26, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.-I.; Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2015, 11, 118–133. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [Green Version]
- Hess, G.T.; Tycko, J.; Yao, D.; Bassik, M.C. Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol. Cell 2017, 68, 26–43. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; A Gilbert, L.; Wang, X.; A Lim, W.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; A Abulfaraj, A.; Alshareef, S.; Aouida, M.; Mahfouz, M.M. RNA-guided transcriptional regulationin plantavia synthetic dCas9-based transcription factors. Plant Biotechnol. J. 2014, 13, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Piatek, A.; Mahfouz, M.M. Targeted genome regulation via synthetic programmable transcriptional regulators. Crit. Rev. Biotechnol. 2016, 37, 429–440. [Google Scholar] [CrossRef]
- Enríquez, P. CRISPR-Mediated Epigenome Editing. Yale J. Biol. Med. 2016, 89, 471–486. [Google Scholar]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- WHO, Index 2. 2016. Available online: http://www.who.int/genomics/public/geneticdiseases/en/index2.html#HD (accessed on 25 September 2020).
- OMIM, geneMap. 2020. Available online: https://www.omim.org/statistics/geneMap (accessed on 25 September 2020).
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479. [Google Scholar] [CrossRef]
- First CRISPR therapy dosed. Nat. Biotechnol. 2020, 38, 382. [CrossRef] [Green Version]
- Allife Medical Science and Technology Co. “iHSCs With the Gene Correction of HBB Intervent Subjests With β-thalassemia Mutations” Clinical Trial, Identifier: NCT03728322. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03728322. (accessed on 25 September 2020).
- CRISPR Therapeutics and Vertex Pharmaceuticals Incorporated, “CLIMB-121 Trial in Severe Sickle Cell Disease Updated Results Data Presented Today at EHA Reflect Longer-Duration Follow-up Data for the First Patient with SCD Treated with CTX001. CRISPR Therapeutics and Vertex Announced Initial Data for This First SCD”. 2020. Available online: https://crisprtx.gcs-web.com/news-releases/news-release-details/crispr-therapeutics-and-vertex-announce-new-clinical-data (accessed on 25 September 2020).
- Editas Medicine, Allergan. “Single Ascending Dose Study in Participants With LCA10” Clinical Trial, Identifier: NCT03872479. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03872479 (accessed on 25 September 2020).
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Song, C.-Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016, 34, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-Q.; Jiang, T.; Richter, M.; Rhym, L.H.; Koblan, L.W.; Zafra, M.P.; Schatoff, E.M.; Doman, J.L.; Cao, Y.; Dow, L.E.; et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2019, 4, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankowicz, F.P.; Barzi, M.; Legras, X.; Hubert, L.; Mi, T.; Tomolonis, J.; Ravishankar, M.; Sun, Q.; Yang, D.; Borowiak, M.; et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun. 2016, 7, 12642. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Hirai, H.; Yang, D.; Ma, L.; Hou, X.; Jiang, H.; Wei, H.; Rajagopalan, C.; Mou, H.; Wang, G.; et al. Efficient Gene Editing at Major CFTR Mutation Loci. Mol. Ther. Nucleic Acids 2019, 16, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [Green Version]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876.e12. [Google Scholar] [CrossRef]
- Sun, J.; Carlson-Stevermer, J.; Das, U.; Shen, M.; Delenclos, M.; Snead, A.M.; Koo, S.Y.; Wang, L.; Qiao, D.; Loi, J.; et al. CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat. Commun. 2019, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 2020, 181, 590–603.e16. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2013, 24, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Ge, X.; Yang, F.; Wang, B.; Li, S.; Duan, J.; Lv, X.; Cheng, C.; Song, Z.; Liu, C.; et al. High-fidelity SaCas9 identified by directional screening in human cells. PLoS Biol. 2020, 18, e3000747. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aouida, M.; Eid, A.; Ali, Z.; Cradick, T.J.; Lee, C.M.; Deshmukh, H.; Atef, A.; Abusamra, D.; Gadhoum, S.Z.; Merzaban, J.S.; et al. Efficient fdCas9 Synthetic Endonuclease with Improved Specificity for Precise Genome Engineering. PLoS ONE 2015, 10, e0133373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.R.; Aryee, M.J.; Joung, J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Javadzadeh, Y.; Bahari, L.A. Therapeutic Nanostructures for Dermal and Transdermal Drug Delivery. In Nano- and Microscale Drug Delivery Systems; Grumezescu, A.M., Ed.; Elsevier: Oxford, UK, 2017; pp. 131–146. [Google Scholar]
- Wasels, F.; Jean-Marie, J.; Collas, F.; López-Contreras, A.M.; Ferreira, N.L. A two-plasmid inducible CRISPR/Cas9 genome editing tool for Clostridium acetobutylicum. J. Microbiol. Methods 2017, 140, 5–11. [Google Scholar] [CrossRef]
- Lauritsen, I.; Porse, A.; Sommer, M.O.A.O.A.; Nørholm, M.H.H. A versatile one-step CRISPR-Cas9 based approach to plasmid-curing. Microb. Cell Factories 2017, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [Green Version]
- Suresh, B.; Ramakrishna, S.; Kim, H. Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA for Genome Editing. Methods. Mol. Biol. 2016, 1507, 81–94. [Google Scholar]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 2579–2586. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.H.; Cho, H.-J.; Shim, G.; Lee, S.; Kim, S.-H.; Choi, H.-G.; Kim, C.-W.; Oh, Y.-K. Cationic Liposomal Co-delivery of Small Interfering RNA and a MEK Inhibitor for Enhanced Anticancer Efficacy. Pharm. Res. 2011, 28, 3069–3078. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Chakraborty, S. Sequencing data from Massachusetts General Hospital shows Cas9 integration into the genome, highlighting a serious hazard in gene-editing therapeutics. F1000Research 2019, 8, 1846. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2012, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mout, R.; Ray, M.; Lee, Y.-W.; Scaletti, F.; Rotello, V. In Vivo Delivery of CRISPR/Cas9 for Therapeutic Gene Editing: Progress and Challenges. Bioconjugate Chem. 2017, 28, 880–884. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Alphonse, M.; Liu, Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12. [Google Scholar] [CrossRef]
- DiCarlo, J.E.; Mahajan, V.B.; Tsang, S.H. Gene therapy and genome surgery in the retina. J. Clin. Investig. 2018, 128, 2177–2188. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; A Glass, Z.; Xu, Q. Non-viral delivery of genome-editing nucleases for gene therapy. Gene Ther. 2016, 24, 144–150. [Google Scholar] [CrossRef]
- Jo, Y.-I.; Suresh, B.; Kim, H.; Ramakrishna, S. CRISPR/Cas9 system as an innovative genetic engineering tool: Enhancements in sequence specificity and delivery methods. Biochim. Biophys. Acta 2015, 1856, 234–243. [Google Scholar] [CrossRef]
- Börner, K.; Kienle, E.; Huang, L.-Y.; Weinmann, J.; Sacher, A.; Bayer, P.; Stüllein, C.; Fakhiri, J.; Zimmermann, L.; Westhaus, A.; et al. Pre-arrayed Pan-AAV Peptide Display Libraries for Rapid Single-Round Screening. Mol. Ther. 2020, 28, 1016–1032. [Google Scholar] [CrossRef]
- Horii, T.; Arai, Y.; Yamazaki, M.; Morita, S.; Kimura, M.; Itoh, M.; Abe, Y.; Hatada, I. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Qin, W.; Dion, S.L.; Kutny, P.M.; Zhang, Y.; Cheng, A.W.; Jillette, N.L.; Malhotra, A.; Geurts, A.M.; Chen, Y.-G.; Wang, H. Efficient CRISPR/Cas9-Mediated Genome Editing in Mice by Zygote Electroporation of Nuclease. Genetics 2015, 200, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2014, 33, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.M.; Kornepati, A.V.R.; Goldstein, M.; Bogerd, H.P.; Poling, B.C.; Whisnant, A.W.; Kastan, M.B.; Cullen, B.R. Inactivation of the Human Papillomavirus E6 or E7 Gene in Cervical Carcinoma Cells by Using a Bacterial CRISPR/Cas RNA-Guided Endonuclease. J. Virol. 2014, 88, 11965–11972. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, S.; Dad, A.-B.K.; Beloor, J.; Gopalappa, R.; Lee, S.-K.; Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Ji, W.; Hall, J.M.; Hu, Q.; Wang, C.; Beisel, C.L.; Gu, Z. Self-Assembled DNA Nanoclews for the Efficient Delivery of CRISPR-Cas9 for Genome Editing. Angew. Chem. Int. Ed. 2015, 54, 12029–12033. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Yang, Z.; Zhang, R.; Li, L.; Fan, Y.; Kuang, Y.; Gao, Y.; Wang, T.; Lu, W.W.; Xu, B. Supramolecular Hydrogel of ad-Amino Acid Dipeptide for Controlled Drug Release in Vivo. Langmuir 2009, 25, 8419–8422. [Google Scholar] [CrossRef]
- Tugyi, R.; Mezö, G.; Fellinger, E.; Andreu, D.; Hudecz, F. The effect of cyclization on the enzymatic degradation of herpes simplex virus glycoprotein D derived epitope peptide. J. Pept. Sci. 2005, 11, 642–649. [Google Scholar] [CrossRef]
- Wang, X.; Yun, W.; Jiang, W.; Wang, D.; Zhang, L.; Tang, J. An amphiphilic non-viral gene vector prepared by a combination of enzymatic atom transfer radical polymerization and enzymatic ring-opening polymerization. RSC Adv. 2017, 7, 9926–9932. [Google Scholar] [CrossRef] [Green Version]
- Aldayel, A.M.; Naguib, Y.W.; O’Mary, H.L.; Li, X.; Niu, M.; Ruwona, T.B.; Cui, Z. Acid-Sensitive Sheddable PEGylated PLGA Nanoparticles Increase the Delivery of TNF-α siRNA in Chronic Inflammation Sites. Mol. Ther. 2016, 5, e340. [Google Scholar] [CrossRef]
- Chung, J.Y.; Ain, Q.U.; Song, Y.; Yong, S.-B.; Kim, Y.-H. Targeted delivery of CRISPR interference system against Fabp4 to white adipocytes ameliorates obesity, inflammation, hepatic steatosis, and insulin resistance. Genome Res. 2019, 29, 1442–1452. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.K. Nanotechnology-based Drug Delivery for Cancer. Technol. Cancer Res. Treat. 2005, 4, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.R.; Paliwal, R.; Vyas, S.P. A review of mechanistic insight and application of pH-sensitive liposomes in drug delivery. Drug Deliv. 2014, 22, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; DeWitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouet, R.; Thuma, B.A.; Roy, M.D.; Lintner, N.G.; Rubitski, D.M.; Finley, J.E.; Wisniewska, H.M.; Mendonsa, R.; Hirsh, A.; De Oñate, L.; et al. Receptor-Mediated Delivery of CRISPR-Cas9 Endonuclease for Cell-Type-Specific Gene Editing. J. Am. Chem. Soc. 2018, 140, 6596–6603. [Google Scholar] [CrossRef]
- Sun, W.; Wang, J.; Hu, Q.; Zhou, X.; Khademhosseini, A.; Gu, Z. CRISPR-Cas12a delivery by DNA-mediated bioresponsive editing for cholesterol regulation. Sci. Adv. 2020, 6, eaba2983. [Google Scholar] [CrossRef]
- Qi, Y.; Liu, Y.; Yu, B.; Hu, Y.; Zhang, N.; Zheng, Y.; Yang, M.; Liu, F. A Lactose-Derived CRISPR/Cas9 Delivery System for Efficient Genome Editing In Vivo to Treat Orthotopic Hepatocellular Carcinoma. Adv. Sci. 2020, 7, 2001424. [Google Scholar] [CrossRef]
- Chaverra-Rodríguez, D.; Macias, V.M.; Hughes, G.L.; Pujhari, S.; Suzuki, Y.; Peterson, D.R.; Kim, D.; McKeand, S.; Rasgon, J.L. Targeted delivery of CRISPR-Cas9 ribonucleoprotein into arthropod ovaries for heritable germline gene editing. Nat. Commun. 2018, 9, 3008. [Google Scholar] [CrossRef] [Green Version]
- Maffei, M.; Morelli, C.; Graham, E.; Patriarca, S.; Donzelli, L.; Doleschall, B.; Reis, F.D.C.; Nocchi, L.; Chadick, C.H.; Reymond, L.; et al. A ligand-based system for receptor-specific delivery of proteins. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, J.; Jiang, Y.; Zhang, M.; Mao, L.; Wang, M. Cell-Selective Messenger RNA Delivery and CRISPR/Cas9 Genome Editing by Modulating the Interface of Phenylboronic Acid-Derived Lipid Nanoparticles and Cellular Surface Sialic Acid. ACS Appl. Mater. Interfaces 2019, 11, 46585–46590. [Google Scholar] [CrossRef]
- Zhuang, J.; Tan, J.; Wu, C.; Zhang, J.; Liu, T.; Fan, C.; Li, J.; Zhang, Y. Extracellular vesicles engineered with valency-controlled DNA nanostructures deliver CRISPR/Cas9 system for gene therapy. Nucleic Acids Res. 2020, 48, 8870–8882. [Google Scholar] [CrossRef]
- He, X.; Ren, X.; Peng, Y.; Zhang, J.; Ai, S.; Liu, B.; Xu, C.; Cheng, S. Aptamer/Peptide-Functionalized Genome-Editing System for Effective Immune Restoration through Reversal of PD-L1-Mediated Cancer Immunosuppression. Adv. Mater. 2020, 32, e2000208. [Google Scholar] [CrossRef]
- Liu, B.-Y.; He, X.-Y.; Xu, C.; Ren, X.-H.; Zhuo, R.-X.; Cheng, S. Peptide and Aptamer Decorated Delivery System for Targeting Delivery of Cas9/sgRNA Plasmid To Mediate Antitumor Genome Editing. ACS Appl. Mater. Interfaces 2019, 11, 23870–23879. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Li, F.; Wang, L.; Zhang, Z.-K.; Wang, C.; He, B.; Li, J.; Chen, Z.; Shaikh, A.B.; Liu, J.; et al. Tumor cell-targeted delivery of CRISPR/Cas9 by aptamer-functionalized lipopolymer for therapeutic genome editing of VEGFA in osteosarcoma. Biomaterials 2017, 147, 68–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Xie, H.; Liu, Y.; Xia, C.; Cun, X.; Long, Y.; Chen, X.; Deng, M.; Guo, R.; Zhang, Z.-R.; et al. Knockdown of hypoxia-inducible factor-1 alpha by tumor targeted delivery of CRISPR/Cas9 system suppressed the metastasis of pancreatic cancer. J. Control. Release 2019, 304, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Takahashi, Y.; Narita, S.; Yang, Y.-C.; Li, X. Targeted delivery of CRISPR/Cas9 to prostate cancer by modified gRNA using a flexible aptamer-cationic liposome. Oncotarget 2016, 8, 9375–9387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Liu, F.; Chen, Y.; Liu, J.; Wang, X.; Chen, A.T.; Deng, G.; Zhang, H.; Liu, J.; Hong, Z.; et al. Targeted Delivery of CRISPR/Cas9-Mediated Cancer Gene Therapy via Liposome-Templated Hydrogel Nanoparticles. Adv. Funct. Mater. 2017, 27, 1703036. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Wei, T.; Cheng, Q.; Min, Y.-L.; Olson, E.N.; Siegwart, D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Alyami, M.Z.; Alsaiari, S.K.; Li, Y.; Qutub, S.S.; Aleisa, F.A.; Sougrat, R.; Merzaban, J.S.; Khashab, N. Cell-Type-Specific CRISPR/Cas9 Delivery by Biomimetic Metal Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 1715–1720. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Cho, S.W.; Kim, S.; Song, M.; Gopalappa, R.; Kim, J.-S.; Kim, H. Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. Nat. Commun. 2014, 5, 3378. [Google Scholar] [CrossRef]
- Lostalé-Seijo, I.; Louzao, I.; Juanes, M.; Montenegro, J. Peptide/Cas9 nanostructures for ribonucleoprotein cell membrane transport and gene edition† †Electronic supplementary information (ESI) available. See doi:10.1039/c7sc03918b. Chem. Sci. 2017, 8, 7923–7931. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, M.; De Groot, A.M.; Berthelsen, J.; Franzyk, H.; Sijts, A.J.A.M.; Nielsen, H.M. Conjugation of Cell-Penetrating Peptides to Parathyroid Hormone Affects Its Structure, Potency, and Transepithelial Permeation. Bioconjugate Chem. 2015, 26, 477–488. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Yusufaly, T.I.; Li, Y.; Singh, G.; Olson, W.K. Arginine-phosphate salt bridges between histones and DNA: Intermolecular actuators that control nucleosome architecture. J. Chem. Phys. 2014, 141, 165102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehto, T.; Kurrikoff, K.; Langel, Ü. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836. [Google Scholar] [CrossRef]
- Lehto, T.; Ezzat, K.; Wood, M.J.; El Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef]
- Svensen, N.; Walton, J.G.; Bradley, M. Peptides for cell-selective drug delivery. Trends Pharmacol. Sci. 2012, 33, 186–192. [Google Scholar] [CrossRef]
- Jain, P.K.; Lo, J.H.-J.; Rananaware, S.; Downing, M.; Panda, A.; Tai, M.; Raghavan, S.; Fleming, H.E.; Bhatia, S.N. Non-viral delivery of CRISPR/Cas9 complex using CRISPR-GPS nanocomplexes. Nanoscale 2019, 11, 21317–21323. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.; Wohlford-Lenane, C.; Kandimalla, S.; Sartre, G.; Meyerholz, D.K.; Théberge, V.; Hallée, S.; Duperré, A.-M.; Guidice, T.D.; Lepetit-Stoffaes, J.-P.; et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019, 10, 4906–4912. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.; Frankel, A. Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J. 1991, 10, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- DeRossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Levchenko, T.; Torchilin, V. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef]
- Lax, R. The Future of Peptide Development in the Pharmaceutical Industry. PharManufacturing 2010, 10–15. [Google Scholar]
- Jani, P.; Manseta, P.; Patel, S. Pharmaceutical approaches related to systemic delivery of protein and peptide drugs: An overview. Int. J. Pharm. Sci. Rev. Res. 2012, 12, 42–52. [Google Scholar]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [Green Version]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Park, S.E.; Sajid, M.I.; Parang, K.; Tiwari, R. Cyclic Cell-Penetrating Peptides as Efficient Intracellular Drug Delivery Tools. Mol. Pharm. 2019, 16, 3727–3743. [Google Scholar] [CrossRef]
- Schaeffer, L. The Role of Functional Groups in Drug-Receptor Interactions. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth, C.G., Aldous, D., Raboisson, P., Rognan, D., Eds.; Academic Press: London, UK, 2008. [Google Scholar]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef]
- Nakase, I.; Tadokoro, A.; Kawabata, N.; Takeuchi, T.; Katoh, H.; Hiramoto, K.; Negishi, M.; Nomizu, M.; Sugiura, Y.; Futaki, S. Interaction of Arginine-Rich Peptides with Membrane-Associated Proteoglycans Is Crucial for Induction of Actin Organization and Macropinocytosis. Biochemistry 2007, 46, 492–501. [Google Scholar] [CrossRef]
- Mager, I.; Eiríksdóttir, E.; Langel, K.; El Andaloussi, S.; Langel, Ü. Assessing the uptake kinetics and internalization mechanisms of cell-penetrating peptides using a quenched fluorescence assay. Biochim. Biophys. Acta 2010, 1798, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Drin, G.; Cottin, S.; Blanc, E.; Rees, A.R.; Temsamani, J. Studies on the Internalization Mechanism of Cationic Cell-penetrating Peptides. J. Biol. Chem. 2003, 278, 31192–31201. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; Pellegrini, V.; Arcangeli, C.; Fittipaldi, A.; Giacca, M.; Beltram, F. Caveolae-Mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol. Ther. 2003, 8, 284–294. [Google Scholar] [CrossRef]
- Fuchs, S.M.; Raines, R.T. Pathway for Polyarginine Entry into Mammalian Cells. Biochemistry 2004, 43, 2438–2444. [Google Scholar] [CrossRef] [Green Version]
- Belting, M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem. Sci. 2003, 28, 145–151. [Google Scholar] [CrossRef]
- Hakansson, S.; Caffrey, M. Structural and Dynamic Properties of the HIV-1 Tat Transduction Domain in the Free and Heparin-Bound States. Biochemistry 2003, 42, 8999–9006. [Google Scholar] [CrossRef] [PubMed]
- Christianson, H.C.; Belting, M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.-Q.; LaRochelle, J.R.; Jiang, B.; Lian, W.; Hard, R.L.; Selner, N.G.; Luechapanichkul, R.; Barrios, A.M.; Pei, D. Early Endosomal Escape of a Cyclic Cell-Penetrating Peptide Allows Effective Cytosolic Cargo Delivery. Biochemistry 2014, 53, 4034–4046. [Google Scholar] [CrossRef] [PubMed]
- Witte, K.; Olausson, B.E.; Walrant, A.; Alves, I.; Vogel, A. Structure and dynamics of the two amphipathic arginine-rich peptides RW9 and RL9 in a lipid environment investigated by solid-state NMR and MD simulations. Biochim. Biophys. Acta 2013, 1828, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Pallerla, M.; Zaltsman, Y.; Burlina, F.; Alves, I.; Lequin, O.; Sagan, S. Tryptophan within basic peptide sequences triggers glycosaminoglycan-dependent endocytosis. FASEB J. 2012, 27, 738–749. [Google Scholar] [CrossRef]
- Lentz, T.L. Rabies virus binding to an acetylcholine receptor α-subunit peptide. J. Mol. Recognit. 1990, 3, 82–88. [Google Scholar] [CrossRef]
- Won, Y.-W.; Adhikary, P.; Lim, K.S.; Kim, H.J.; Kim, J.K.; Kim, Y.-H. Oligopeptide complex for targeted non-viral gene delivery to adipocytes. Nat. Mater. 2014, 13, 1157–1164. [Google Scholar] [CrossRef]
- Givens, B.E.; Naguib, Y.W.; Geary, S.M.; Devor, E.J.; Salem, A.K. Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Therapeutics. AAPS J. 2018, 20, 108. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.-Y.; Pellois, J.-P. Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef]
- Ignatovich, I.A.; Dizhe, E.B.; Pavlotskaya, A.V.; Akifiev, B.N.; Burov, S.; Orlov, S.; Perevozchikov, A. Complexes of Plasmid DNA with Basic Domain 47–57 of the HIV-1 Tat Protein Are Transferred to Mammalian Cells by Endocytosis-mediated Pathways. J. Biol. Chem. 2003, 278, 42625–42636. [Google Scholar] [CrossRef] [Green Version]
- Rinne, J.; Albarran, B.; Jylhävä, J.; Ihalainen, T.O.; Kankaanpää, P.; Hytönen, V.P.; Stayton, P.S.; Kulomaa, M.S.; Vihinen-Ranta, M. Internalization of novel non-viral vector TAT-streptavidin into human cells. BMC Biotechnol. 2007, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youngblood, D.S.; Hatlevig, S.A.; Hassinger, J.N.; Iversen, P.L.; Moulton, H.M. Stability of Cell-Penetrating Peptide−Morpholino Oligomer Conjugates in Human Serum and in Cells. Bioconjugate Chem. 2007, 18, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem. 2018, 30, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.P. The proton sponge: A trick to enter cells the viruses did not exploit. Chimia 1997, 2, 34–36. [Google Scholar]
- Zelphati, O.; Szoka, F.C. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. USA 1996, 93, 11493–11498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herce, H.; García, A.E.; Litt, J.; Kane, R.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-Rich Peptides Destabilize the Plasma Membrane, Consistent with a Pore Formation Translocation Mechanism of Cell-Penetrating Peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-T.; Zaitseva, E.; Chernomordik, L.V.; Melikov, K. Cell-Penetrating Peptide Induces Leaky Fusion of Liposomes Containing Late Endosome-Specific Anionic Lipid. Biophys. J. 2010, 99, 2525–2533. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.-Q.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; A Phelps, M.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Mandal, D.; Shirazi, A.N.; Parang, K. Cell-Penetrating Homochiral Cyclic Peptides as Nuclear-Targeting Molecular Transporters. Angew. Chem. Int. Ed. 2011, 50, 9633–9637. [Google Scholar] [CrossRef]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.; Hackenberger, C.P.R. Covalent Attachment of Cyclic TAT Peptides to GFP Results in Protein Delivery into Live Cells with Immediate Bioavailability. Angew. Chem. Int. Ed. 2014, 54, 1950–1953. [Google Scholar] [CrossRef]
- Lättig-Tünnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Ma, H.; Dahl, K.N.; Zhu, J.; Diamond, S.L. Adenovirus or HA-2 fusogenic peptide-assisted lipofection increases cytoplasmic levels of plasmid in nondividing endothelium with little enhancement of transgene expression. J. Gene Med. 2002, 4, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.-F.; Tian, M.-M.; Wang, T.-X.; Ren, L.; Wang, D.; Shen, L.; Shang, T. Synergistic effects of cell-penetrating peptide Tat and fusogenic peptide HA2-enhanced cellular internalization and gene transduction of organosilica nanoparticles. Nanomedicine 2012, 8, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Guidice, T.D.; Lepetit-Stoffaes, J.-P.; Bordeleau, L.-J.; Roberge, J.; Théberge, V.; Lauvaux, C.; Barbeau, X.; Trottier, J.; Davé, V.; Roy, D.-C.; et al. Membrane permeabilizing amphiphilic peptide delivers recombinant transcription factor and CRISPR-Cas9/Cpf1 ribonucleoproteins in hard-to-modify cells. PLoS ONE 2018, 13, e0195558. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Yuan, X.; Luo, L.; Lu, Y.; Qin, B.; Zhang, J.; Shi, Y.; Zhu, C.; Yang, J.; Li, X.; et al. Appropriate Delivery of the CRISPR/Cas9 System through the Nonlysosomal Route: Application for Therapeutic Gene Editing. Adv. Sci. 2020, 7, 1903381. [Google Scholar] [CrossRef]
- Salomone, F.; Cardarelli, F.; Di Luca, M.; Boccardi, C.; Nifosì, R.; Bardi, G.; Di Bari, L.; Serresi, M.; Beltram, F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 2012, 163, 293–303. [Google Scholar] [CrossRef]
- Meyer, M.; Philipp, A.; Oskuee, R.; Schmidt, C.; Wagner, E. Breathing Life into Polycations: Functionalization with pH-Responsive Endosomolytic Peptides and Polyethylene Glycol Enables siRNA Delivery. J. Am. Chem. Soc. 2008, 130, 3272–3273. [Google Scholar] [CrossRef] [Green Version]
- Zhelev, D.V.; Stoicheva, N.; Scherrer, P.; Needham, D. Interaction of Synthetic HA2 Influenza Fusion Peptide Analog with Model Membranes. Biophys. J. 2001, 81, 285–304. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Type | Nuclease | gRNA Structure | Target | Cleavage | ||||
|---|---|---|---|---|---|---|---|---|---|
| crRNA | tracrRNA | gRNA Length | Molecule | PAM | Availability in Human Genome | ||||
| 2 | 2 | Cas9 | 20nt (complementary to target) | 85nt | 105nt | dsDNA | 5′-NGG (SpCas9) | Every ~8 bp | Blunt-ended DSB |
| 5 | Cas12 | 40nt (20-24nt complementary to target) | None | 40nt | dsDNA | 5′-TTTN | Every ~23 bp | Sticky-ended DSB | |
| 6 | Cas13 | 64nt (23-30nt complementary to target) | None | 64nt | ssRNA | None | Any location | Arbitrary cleavage around target site | |
| Feature | DNA | RNA | Protein |
|---|---|---|---|
| Cost | ++ | + | +++ |
| Stability | +++ | + | ++ |
| Editing Efficiency | + | ++ | +++ |
| Rapidity | + | ++ | +++ |
| Insertional Mutagenesis | + | − | − |
| Immunogenicity | +++ | ++ | + |
| Off-targets | +++ | ++ | + |
| Duration in cells | +++ | ++ | + |
| Carrier | Molecule | Target | Model | Disease/Gene | Reference |
|---|---|---|---|---|---|
| Valency-controlled tetrahedral DNA nanostructures conjugated with DNA aptamer | RNP | Tumor cells | In vivo | Cancer: WNT10B | Zhuang et al. 2020 [100] |
| Lactose-derived branched cationic biopolymer | Plasmid | ASGPrs on Liver cells | In vivo | Hepatocellular carcinoma: survivin | Qi et al. 2020 [101] |
| Classic Lipid Nanoparticles supplemented with permanently cationic lipid formulations | RNP | Lung, Liver | In vivo | Hypercholesterolemia: PCSK9, Lung Cancer: PTEN | Wei et al. 2020 [102] |
| Galactose-functionalized polyethyleneimine-coated DNA nanoclews | RNP | ASGPrs on Liver cells | In vivo | Hypercholesterolemia: PCSK9 | Sun et al. 2020 [103] |
| Lipid Nanoparticles with SORT Supplemental molecules | mRNA/gRNA RNP | Liver, Lungs, Spleen | In vivo | Cardiovascular Disease: PCSK9 | Cheng et al. 2020 [104] |
| Functionalized carrier: - Ca2+, Protamine, CO32−, Hyaluronic acid chain, - Targeting Aptamer (AS1411), - Cell-Penetrating Peptide (TAT-NLS) | Plasmid | CD44 receptors on tumor cells | In vitro | Cancer: CTNNB1 | He et al. 2020 [105] |
| Metal Organic Frameworks (Zeolitic imidazolate) encapsulating CRISPR/Cas9, coated with MCF-7 cancer cell membrane | RNP | Antigens on adenocarcinoma cells | In vivo | EGFP | Alyami et al. 2020 [106] |
| Phenylboronic acid-functionalized Lipid Nanoparticles | mRNA | Cellular surface sialic acid on cancer cells | In vitro | Cancer: HPV18E6 | Tang et al. 2019 [107] |
| Cyclic ATS-9R Peptide: CKGGRAKD-rrrrrrrrrC | Plasmid | Prohibitin on adipocytes | In vivo | Diabetes: Fabp4 | Chung et al. 2019 [96] |
| Functionalized carrier: - Ca2+, Protamine, CO32-, Alginate chain, - Targeting Aptamer (AS1411), - Cell-Penetrating Peptide (NLS) | Plasmid | AS1411 receptors on tumor cells | In vitro | Cancer: PTK2 | Liu et al. 2019 [108] |
| Liposome functionalized with R8-dGR peptide: Cys-RRRRRRRRdGR | Plasmid | NRP-1 and integrin αvβ3 on tumor cells | In vivo | Pancreatic cancer: HIF-1α | Li et al. 2019 [109] |
| CRISPR-GPS: - Targeting Peptide: Cyclic iRGD: CRGDKGPDC - Cell-Penetrating Peptide (mTP): GWTLNSAGYLLGKINLKALAALAKKIL | RNP | αvβ3 integrins on cancer cells | In vitro | Cancer: CD71 | Jain et al. 2019 [110] |
| - IL-31 or NGF SNAP-ligands, - Cationic Protamine Peptides, - Endosomolytic Peptide (ppTG21) | Protein | IL-31 and NGF receptors on keratinocytes | In vivo | Skin disease: Atat1 | Maffei et al. 2019 [111] |
| - S10 Peptide: KWKLARAFARAIKKLGGSGGGSYARALRRQARTG | RNP | Airway Epithelia | In vivo | Airway diseases: CFTR, HPRT1 | Krishnamurthy et al. 2019 [112] |
| - Asialoglycoprotein receptor ligands, - Endosomolytic Peptide (ppTG21): GLFHALLHLLHSLWHLLLHA | RNP | ASGPrs on liver cells | In vitro | EMX1 | Rouet et al. 2018 [113] |
| Recombinant Cas9 protein fused to Targeting Peptide: P2C on C-terminus | RNP | Yolk Protein receptors on mosquito oocytes | In vivo | kmo | Chaverra-Rodriguez et al. 2018 [114] |
| ssDNA LC09-functionalized PPC lipopolymer | Plasmids | Osteosarcoma | In vitro | Osteosarcoma: VEGFA | Liang et al. 2017 [115] |
| RNA aptamer A10-functionalized liposome | gRNA | Prostate-specific membrane antigen on cancer cells | In vivo | Prostate Cancer: PLK1 | Zhen et al. 2017 [116] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalaby, K.; Aouida, M.; El-Agnaf, O. Tissue-Specific Delivery of CRISPR Therapeutics: Strategies and Mechanisms of Non-Viral Vectors. Int. J. Mol. Sci. 2020, 21, 7353. https://doi.org/10.3390/ijms21197353
Shalaby K, Aouida M, El-Agnaf O. Tissue-Specific Delivery of CRISPR Therapeutics: Strategies and Mechanisms of Non-Viral Vectors. International Journal of Molecular Sciences. 2020; 21(19):7353. https://doi.org/10.3390/ijms21197353
Chicago/Turabian StyleShalaby, Karim, Mustapha Aouida, and Omar El-Agnaf. 2020. "Tissue-Specific Delivery of CRISPR Therapeutics: Strategies and Mechanisms of Non-Viral Vectors" International Journal of Molecular Sciences 21, no. 19: 7353. https://doi.org/10.3390/ijms21197353