Identification, Characterization, and Genomic Analysis of Novel Serratia Temperate Phages from a Gold Mine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification and Characterization of the Temperate vB_SspM_BZS1 Phage
2.2. Genomic Analysis of the vB_SspM_BZS1 Phage
2.2.1. Identification of the Genome Termini of the vB_SspM_BZS1 Phage
2.2.2. Identification of the vB_SspM_BZS1 Attachment Site
2.2.3. Module Analysis of the vB_SspM_BZS1 Genome
2.3. Genomic Analysis of the vB_SspS_OS31
2.4. Comparative Genomic Analyses
2.4.1. vB_SspM_BZS1 vs. vB_SspS_OS31
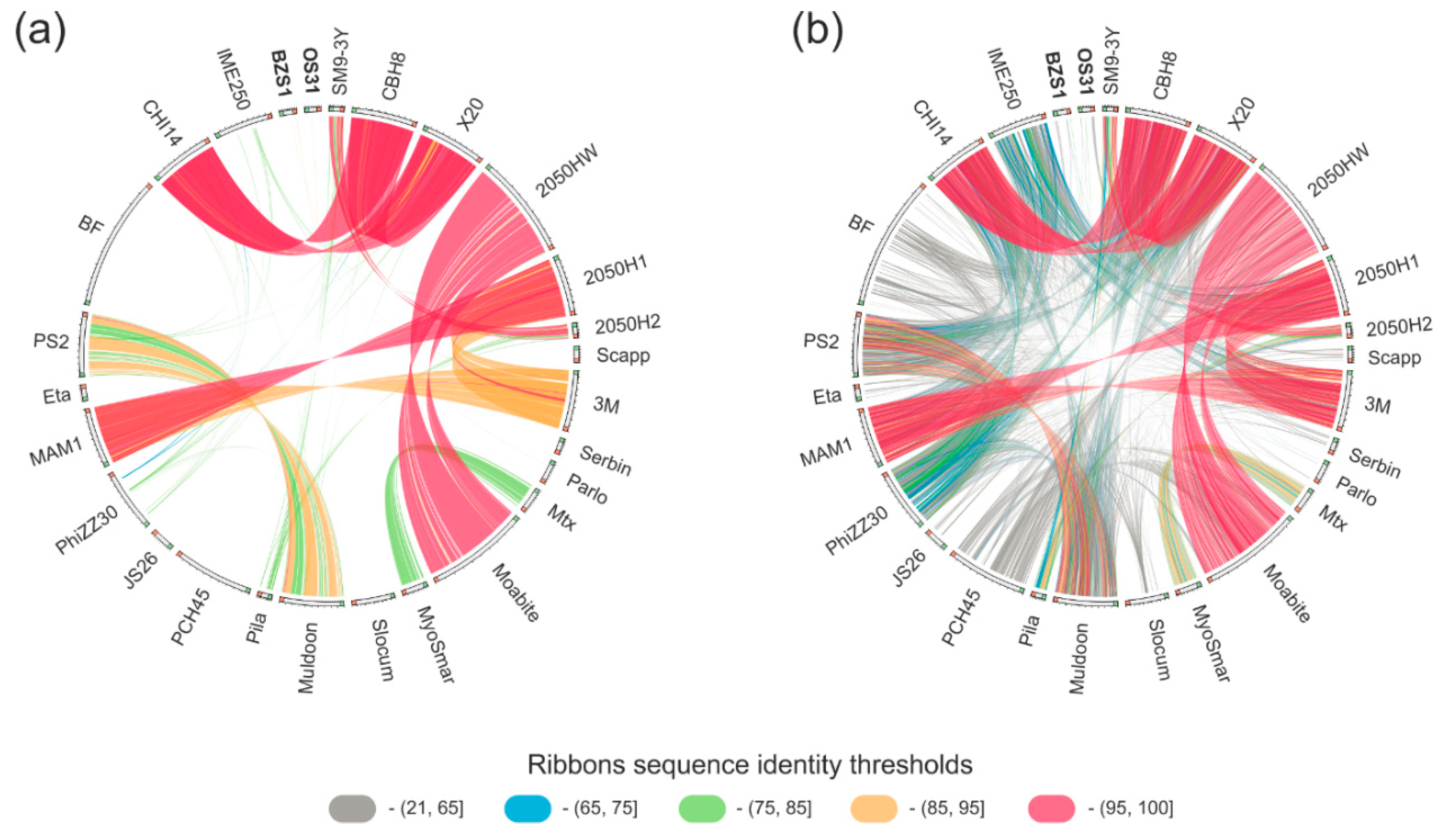
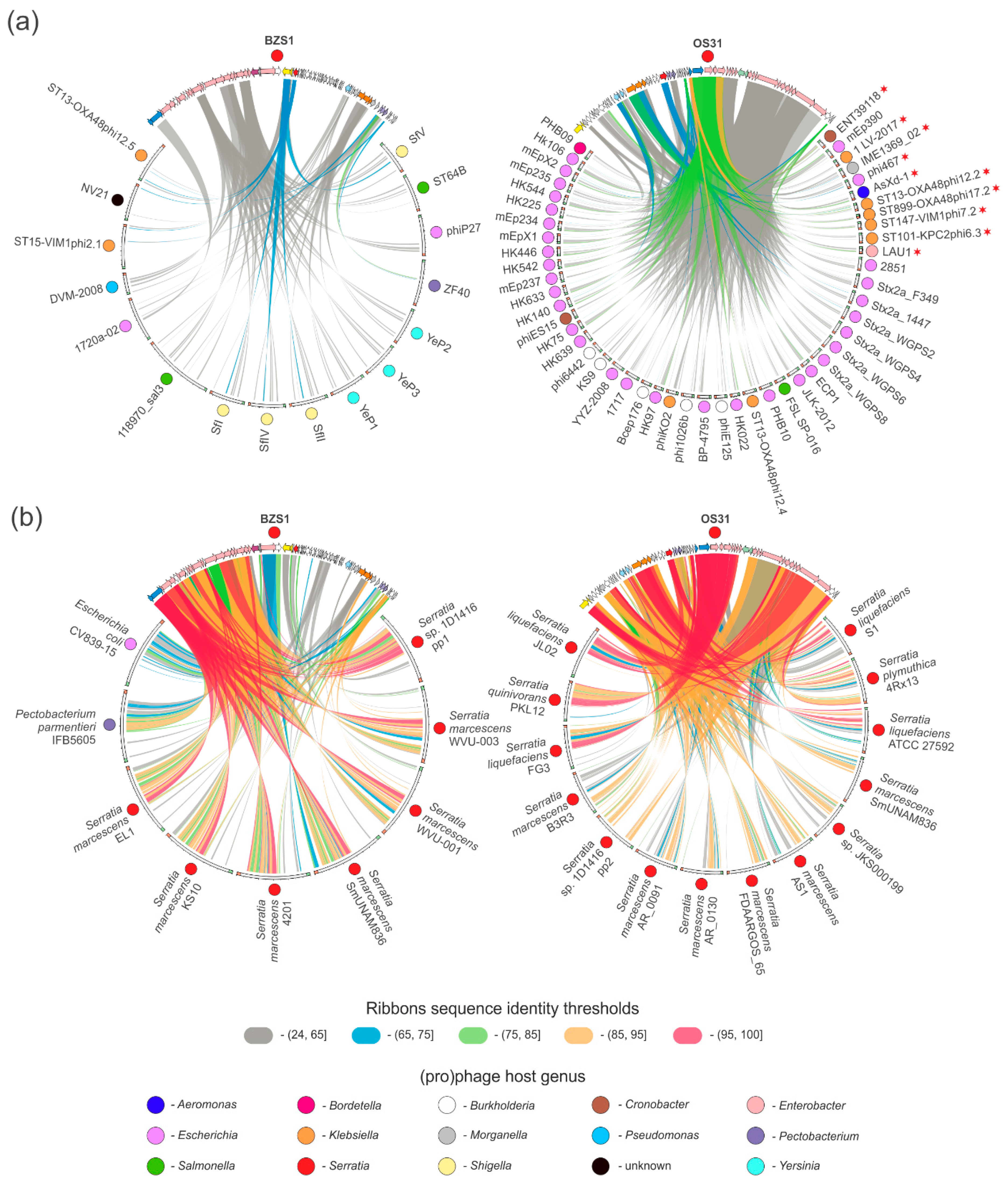
2.4.2. vB_SspM_BZS1 and vB_SspS_OS31 vs. other Serratia Phages
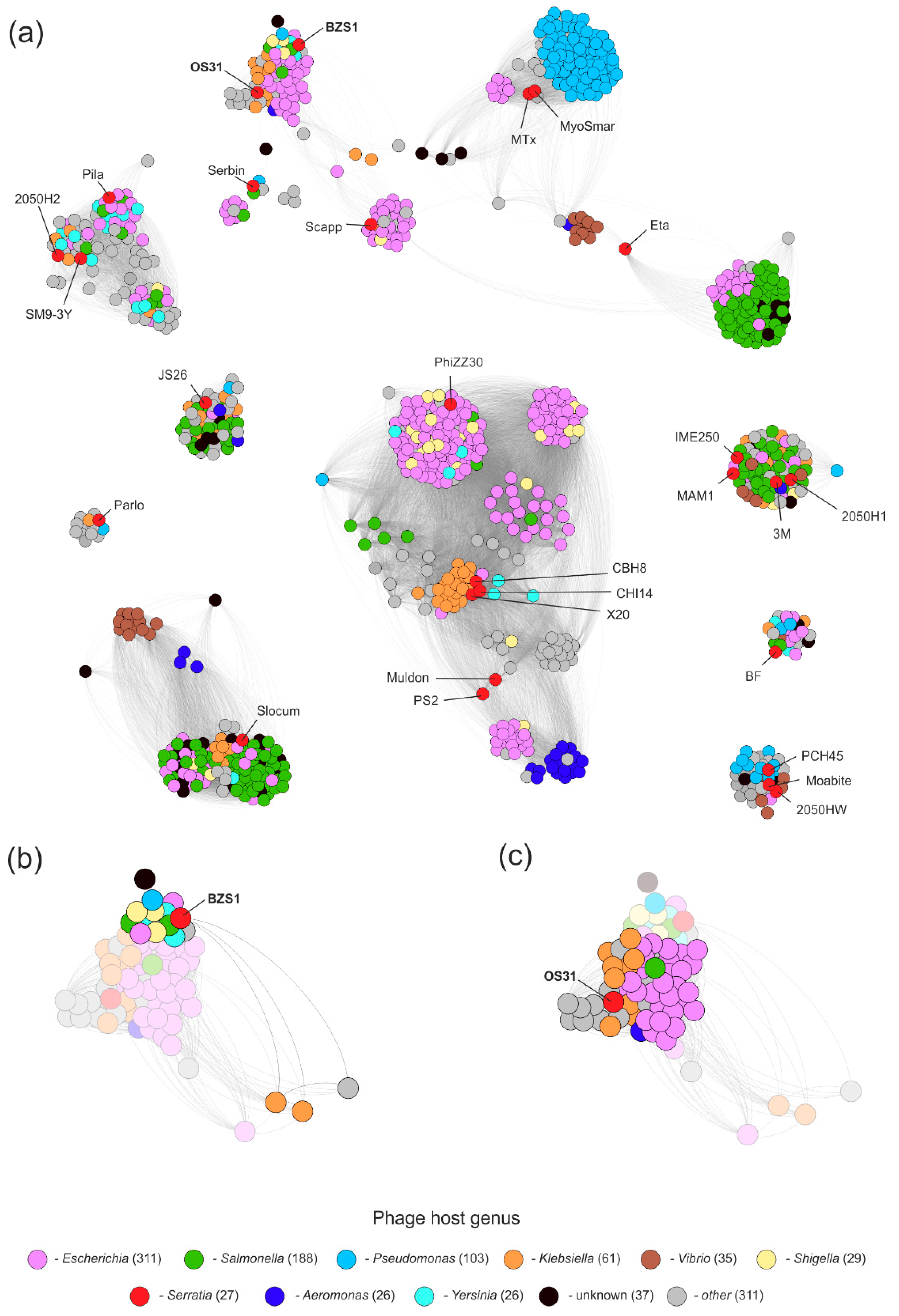
2.4.3. vB_SspM_BZS1 and vB_SspS_OS31 vs. All Known Phages
2.4.4. Prophages in Serratia Genomes Similar to vB_SspM_BZS1 and vB_SspS_OS31
2.5. Functional Characterization of the vB_SspM_BZS1 and vB_SspS_OS31 Phages
2.5.1. Host Range
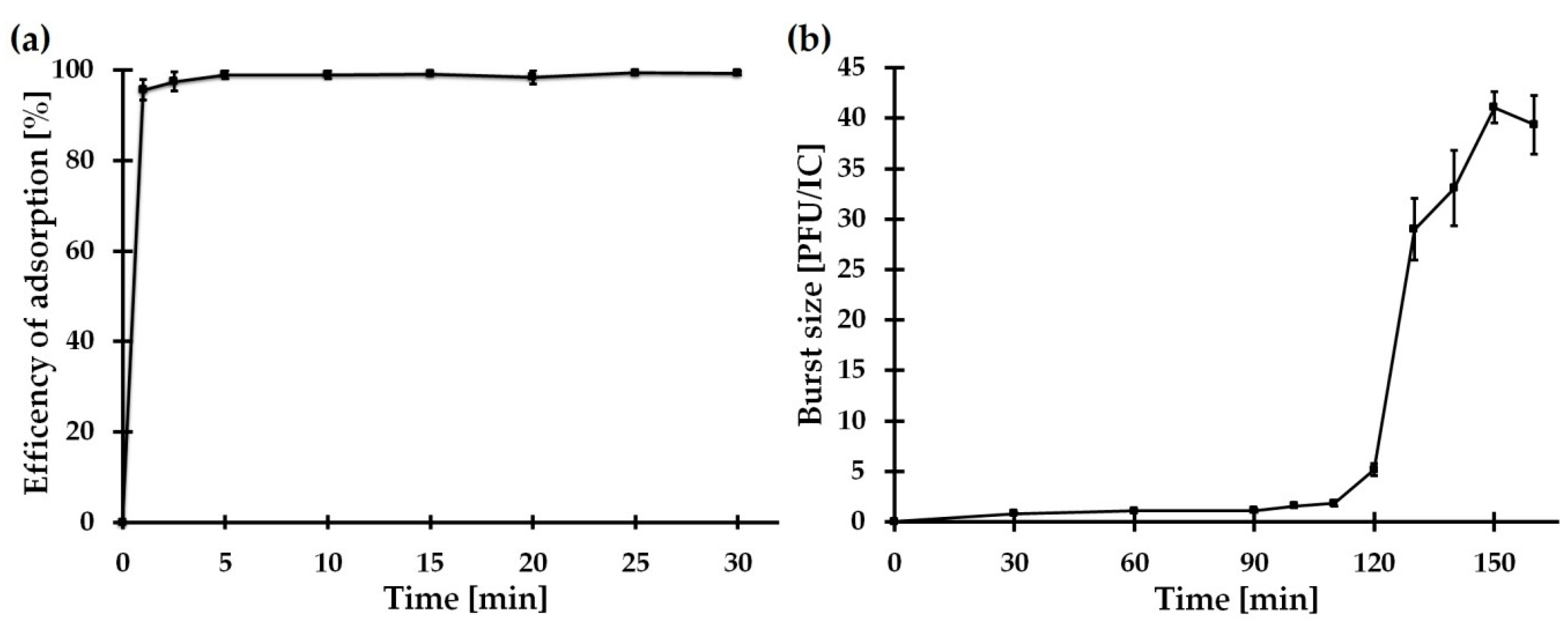
2.5.2. Adsorption Assay and One-Step Growth Curve of vB_SspM_BZS1
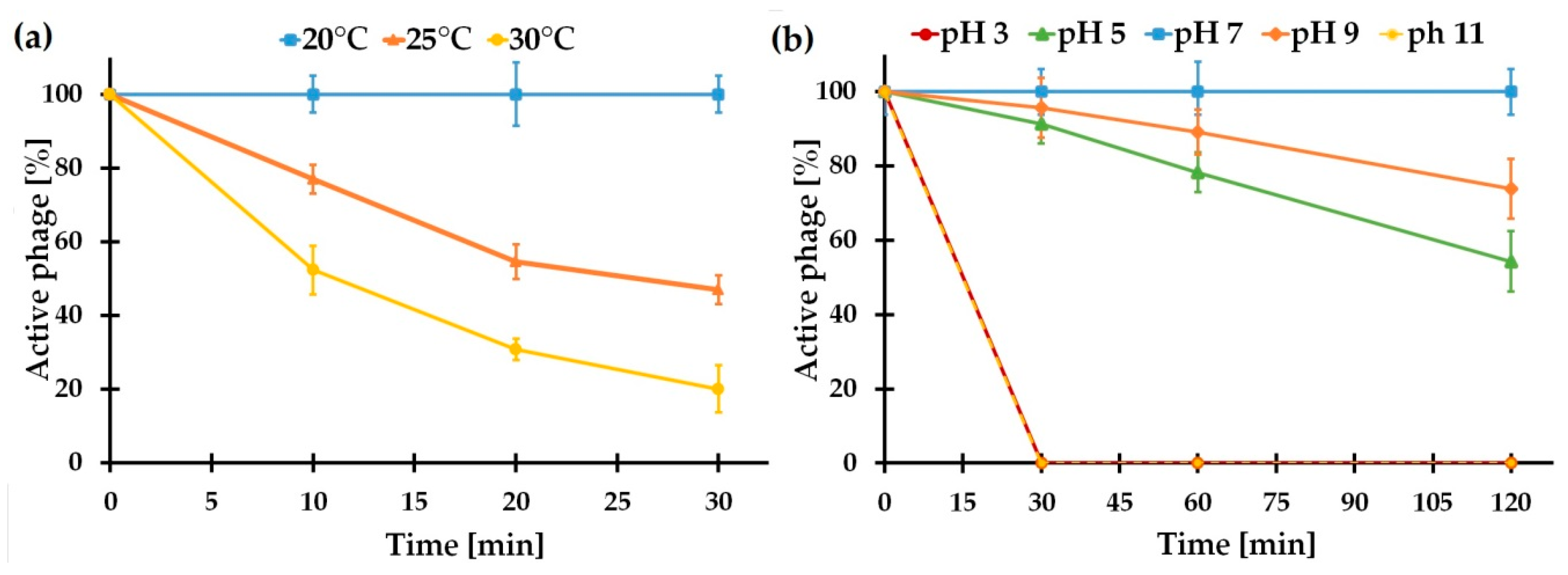
2.5.3. vB_SspM_BZS1 Stability under Various Conditions
3. Materials and Methods
3.1. Bacterial Strains, Plasmids, and Culture Conditions
3.2. Isolation of the vB_SspM_BZS1 Phage
3.3. Isolation of the Lysogenized Strain of Serratia sp. OS31
3.4. Induction of vB_SspS_OS31 Prophage
3.5. DNA Sequencing
3.6. Genome Annotation
3.7. Comparative Genomics
3.8. Verification of Attachment Sites of vB_SspM_BZS1 Phage
3.9. Cloning, Overexpression, Purification, and Testing the Activity of DNA MTases
3.10. Transmission Electron Microscopy
3.11. SDS-PAGE and Mass Spectrometry Protein Analysis
3.12. Adsorption Kinetics
3.13. One-Step Growth Curve
3.14. Thermal and pH Stability
3.15. Determination of the Phage Host Range by Spot Testing
3.16. Nucleotide Sequence Accession Numbers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [PubMed]
- Hejazi, A.; Falkiner, F.R. Serratia marcescens. J. Med. Microbiol. 1997, 46, 903–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlen, S.D. Serratia infections: From military experiments to current practice. Clin. Microbiol. Rev. 2011, 24, 755–791. [Google Scholar] [CrossRef] [Green Version]
- Samonis, G.; Vouloumanou, E.K.; Christofaki, M.; Dimopoulou, D.; Maraki s Triantafyllou, E.; Kofteridis, D.P.; Falagas, M.E. Serratia infections in a general hospital: Characteristics and outcomes. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 653–660. [Google Scholar] [CrossRef]
- Parmar, K.M.; Dafale, N.A.; Tikariha, H.; Purohit, H.J. Genomic characterization of key bacteriophages to formulate the potential biocontrol agent to combat enteric pathogenic bacteria. Arch. Microbiol. 2018, 200, 611–622. [Google Scholar] [CrossRef]
- Denyes, J.M.; Krell, P.J.; Manderville, R.A.; Ackermann, H.W.; She, Y.M.; Kropinski, A.M. The genome and proteome of Serratia bacteriophage η which forms unstable lysogens. Virol. J. 2014, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Bockoven, R.; Gutierrez, J.; Newkirk, H.; Liu, M.; Cahill, J.; Ramsey, J. Complete Genome Sequence of Serratia marcescens Podophage Parlo. Microbiol. Resour. Announc. 2019, 8, e00569-19. [Google Scholar] [CrossRef] [Green Version]
- Drewniak, L.; Maryan, N.; Lewandowski, W.; Kaczanowski, S.; Sklodowska, A. The contribution of microbial mats to the arsenic geochemistry of an ancient gold mine. Environ. Pollut. 2012, 162, 190–201. [Google Scholar] [CrossRef]
- Drewniak, L.; Krawczyk, P.S.; Mielnicki, S.; Adamska, D.; Sobczak, A.; Lipinski, L.; Burec-Drewniak, W.; Sklodowska, A. Physiological and Metagenomic Analyses of Microbial Mats Involved in Self-Purification of Mine Waters Contaminated with Heavy Metals. Front. Microbiol. 2016, 7, 1252. [Google Scholar] [CrossRef]
- Tomczyk-Żak, K.; Kaczanowski, S.; Drewniak, Ł.; Dmoch, Ł.; Sklodowska, A.; Zielenkiewicz, U. Bacteria diversity and arsenic mobilization in rock biofilm from an ancient gold and arsenic mine. Sci. Total Environ. 2013, 461, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Uhrynowski, W.; Decewicz, P.; Dziewit, L.; Radlinska, M.; Krawczyk, P.S.; Lipinski, L. Dorota Adamska, Lukasz Drewniak. Analysis of the Genome and Mobilome of a Dissimilatory Arsenate Reducing Aeromonas sp. O23A Reveals Multiple Mechanisms for Heavy Metal Resistance and Metabolism. Front. Microbiol. 2017, 8, 936. [Google Scholar] [CrossRef]
- Uhrynowski, W.; Radlinska, M.; Drewniak, L. Genomic Analysis of Shewanella sp. O23S-The Natural Host of the pSheB Plasmid Carrying Genes for Arsenic Resistance and Dissimilatory Reduction. Int. J. Mol. Sci. 2019, 20, 1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, E.; Fitzgerald, B.; Mahony, J.; Lugli, G.A.; Ventura, M.; van Sinderen, D. Genome Sequence of Serratia marcescens Phage BF. Genome Announc. 2017, 5, e00211–e00217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, K.; Freeman, M.; Newkirk, H.; Liu, M.; Cahill, J.; Ramsey, J. Complete Genome Sequence of Serratia marcescens Phage MTx. Microbiol. Resour. Announc. 2019, 8, e00573-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.; Nguyen, Q.; Newkirk, H.; Liu, M.; Cahill, J.; Ramsey, J. Complete Genome Sequence of Serratia marcescens Myophage MyoSmar. Microbiol. Resour. Announc. 2019, 8, e01046-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Catalano, C.E.; Cue, D.; Feiss, M. Virus DNA packaging: The strategy used by phage lambda. Mol. Microbiol. 1995, 16, 1075–1086. [Google Scholar] [CrossRef]
- Reiter, W.D.; Palm, P.; Yeats, S. Transfer RNA genes frequently serve as integration sites for prokaryotic genetic elements. Nucleic Acids Res. 1989, 17, 1907–1914. [Google Scholar] [CrossRef] [Green Version]
- Grainge, I.; Jayaram, M. The integrase family of recombinase: Organization and function of the active site. Mol. Microbiol. 1999, 33, 449–456. [Google Scholar] [CrossRef]
- Vander Byl, C.; Kropinski, A.M. Sequence of the genome of Salmonella bacteriophage P22. J. Bacteriol. 2000, 182, 6472–6481. [Google Scholar] [PubMed] [Green Version]
- Bujnicki, J.M.; Feder, M.; Radlinska, M.; Blumenthal, R.M. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J. Mol. Evol. 2002, 55, 431–444. [Google Scholar] [CrossRef]
- Radlińska, M.; Piekarowicz, A.; Galimand, M.; Bujnicki, J.M. Cloning and preliminary characterization of a GATC-specific beta2-class DNA:m6A methyltransferase encoded by transposon Tn1549 from Enterococcus spp. Pol. J. Microbiol. 2005, 54, 249–252. [Google Scholar]
- Casadesús, J.; Low, D. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, S.; Brown, P.D. Distribution of virulence determinants among antimicrobial-resistant and antimicrobial-susceptible Escherichia coli implicated in urinary tract infections. Indian J. Med. Microbiol. 2016, 34, 448–456. [Google Scholar] [PubMed]
- Bujnicki, J.M.; Radlinska, M.; Zaleski, P.; Piekarowicz, A. Cloning of the Haemophilus influenzae Dam methyltransferase and analysis of its relationship to the Dam methyltransferase encoded by the HP1 phage. Acta Biochim. Pol. 2001, 48, 969–983. [Google Scholar] [CrossRef] [Green Version]
- Julio, S.M.; Heithoff, D.M.; Provenzano, D.; Klose, K.E.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect. Immun. 2001, 69, 7610–7615. [Google Scholar] [CrossRef] [Green Version]
- Ostendorf, T.; Cherepanov, P.; de Vries, J.; Wackernagel, W. Characterization of a dam mutant of Serratia marcescens and nucleotide sequence of the dam region. J. Bacteriol. 1999, 181, 3880–3885. [Google Scholar] [CrossRef] [Green Version]
- Coulby, J.N.; Sternberg, N.L. Characterization of the phage P1 dam gene. Gene 1988, 74, 191. [Google Scholar] [CrossRef]
- Radlinska, M.; Bujnicki, J.M. Cloning of enterohemorrhagic Escherichia coli phage VT-2 dam methyltransferase. Acta Microbiol. Pol. 2001, 50, 161–167. [Google Scholar]
- Hattman, S.; Malygin, E.G. Bacteriophage T2Dam and T4Dam DNA-[N6-adenine]-methyltransferases. Prog. Nucleic Acid Res. Mol. Biol. 2004, 77, 67–126. [Google Scholar] [PubMed]
- Piekarowicz, A.; Bujnicki, J. Cloning of the Dam methyltransferase gene from Haemophilus influenzae bacteriophage HP1. Acta. Microbiol Pol. 1999, 48, 123–129. [Google Scholar] [PubMed]
- Dziewit, L.; Radlinska, M. Two novel temperate bacteriophages co-existing in Aeromonas sp. ARM81—Characterization of their genomes, proteomes and DNA methyltransferases. J. Gen. Virol. 2016, 97, 2008–2022. [Google Scholar] [CrossRef] [PubMed]
- Bheemanaik, S.; Reddy, Y.V.; Rao, D.N. Structure, function and mechanism of exocyclic DNA methyltransferases. Biochem. J. 2006, 399, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokney, A.; Kobiler, O.; Amir, A.; Court, D.L.; Stavans, J.; Adhya, S.; Oppenheim, A.B. Host responses influence on the induction of lambda prophage. Mol. Microbiol. 2008, 68, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Little, J.W. Autodigestion of lexA and phage lambda repressors. Proc. Natl. Acad. Sci. USA 1984, 81, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Sharples, G.J.; Ingleston, S.M.; Lloyd, R.G. Holliday junction processing in bacteria: Insights from the evolutionary conservation of RuvABC, RecG, and RusA. J. Bacteriol. 1999, 181, 5543–5550. [Google Scholar] [CrossRef] [Green Version]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brüssow, H. Prophage genomics Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef] [Green Version]
- Berry, J.; Rajaure, M.; Pang, T.; Young, R. The spanin complex is essential for lambda lysis. J. Bacteriol. 2012, 194, 5667–5674. [Google Scholar] [CrossRef] [Green Version]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, Y. Phage spanins: Diversity, topological dynamics and gene convergence. BMC Bioinformatics. 2018, 19, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertwig, S.; Klein, I.; Lurz, R.; Lanka, E.; Appel, B. PY54, a linear plasmid prophage of Yersinia enterocolitica with covalently closed ends. Mol Microbiol. 2003, 48, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Gervais, F.G.; Drapeau, G.R. Identification, cloning, and characterization of rcsF, a new regulator gene for exopolysaccharide synthesis that suppresses the division mutation ftsZ84 in Escherichia coli K-12. J. Bacteriol. 1992, 174, 8016–8022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanié-Cornet, M.P.; Cam, K.; Jacq, A. RcsF is an outer membrane lipoprotein involved in the RcsCDB phosphorelay signaling pathway in Escherichia coli. J. Bacteriol. 2006, 188, 4264–4270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, D.J. The Rcs phosphorelay: More than just a two-component pathway. Future. Microbiol. 2010, 5, 1173–1184. [Google Scholar] [CrossRef]
- Feiss, M.; Rao, V.B. The bacteriophage DNA packaging machine. Adv. Exp. Med. Biol. 2012, 726, 489–509. [Google Scholar] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Kimes, N.E.; Ghai, R. Expanding the marine virosphere using metagenomics. PLoS Genet. 2013, 9, e1003987. [Google Scholar] [CrossRef] [PubMed]
- Pickard, D.; Toribio, A.L.; Petty, N.K.; van Tonder, A.; Yu, L.; Goulding, D.; Barrell, B.; Rance, R.; Harris, D.; Wetter, M.; et al. A conserved acetyl esterase domain targets diverse bacteriophages to the Vi capsular receptor of Salmonella enterica serovar Typhi. J. Bacteriol. 2010, 192, 5746–5754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szu, S.C.; Li, X.R.; Stone, A.L.; Robbins, J.B. Relation between structure and immunologic properties of the Vi capsular polysaccharide. Infect. Immun. 1991, 59, 4555–4561. [Google Scholar] [CrossRef] [Green Version]
- Iwashita, S.; Kanegasaki, S. Deacetylation reaction catalyzed by Salmonella phage c341 and its baseplate parts. J. Biol. Chem. 1976, 251, 5361–5365. [Google Scholar]
- Verma, N.K.; Brandt, J.M.; Verma, D.J.; Lindberg, A.A. Molecular characterization of the O-acetyl transferase gene of converting bacteriophage SF6 that adds group antigen 6 to Shigella flexneri. Mol. Microbiol. 1991, 5, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.A.; Beltrame, J.; Manning, P.A. The oac gene encoding a lipopolysaccharide O-antigen acetylase maps adjacent to the integrase-encoding gene on the genome of Shigella flexneri bacteriophage Sf6. Gene 1991, 107, 43–52. [Google Scholar] [CrossRef]
- Lerouge, I.; Vanderleyden, J. O-antigen structural variation: Mechanisms and possible roles in animal/plant-microbe interactions. FEMS. Microbiol. Rev. 2002, 26, 17–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decewicz, P.; Dziewit, L.; Golec, P.; Kozlowska, P.; Bartosik, D.; Radlinska, M. Characterization of the virome of Paracoccus spp. (Alphaproteobacteria) by combined in silico and in vivo approaches. Sci. Rep. 2019, 9, 7899. [Google Scholar] [CrossRef] [PubMed]
- Chikova, A.K.; Schaaper, R.M. The bacteriophage P1 hot gene product can substitute for the Escherichia coli DNA polymerase III {theta} subunit. J. Bacteriol. 2005, 187, 5528–5536. [Google Scholar] [CrossRef] [Green Version]
- Chikova, A.K.; Schaaper, R.M. The bacteriophage P1 hot gene, encoding a homolog of the E. coli DNA polymerase III theta subunit, is expressed during both lysogenic and lytic growth stages. Mutat. Res. 2007, 624, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC. Genomics. 2014, 15, 1027. [Google Scholar] [CrossRef] [Green Version]
- Biswas, T.; Aihara, H.; Radman-Livaja, M.; Filman, D.; Landy, A.; Ellenberger, T. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005, 435, 1059–1066. [Google Scholar] [CrossRef]
- Yang, W. Nucleases: Diversity of structure, function and mechanism. Q Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Bleriot, I.; Trastoy, R.; Blasco, L.; Fernández-Cuenca, F.; Ambroa, A.; Fernández-García, L.; Pacios, O.; Perez-Nadales, E.; Torre-Cisneros, J.; Martínez-Martínez, L. Genomic analysis of 40 prophages located in the genomes of 16 carbapenemase-producing clinical strains of Klebsiella pneumoniae. Microb. Genom. 2020, 6, e000369. [Google Scholar] [CrossRef]
- Sharples, G.J.; Corbett, L.M.; Graham, I.R. lambda Rap protein is a structure-specific endonuclease involved in phage recombination. Proc. Natl. Acad. Sci. USA 1998, 95, 13507–13512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharples, G.J.; Curtis, F.A.; McGlynn, P.; Bolt, E.L. Holliday junction binding and resolution by the Rap structure-specific endonuclease of phage lambda. J. Mol. Biol. 2004, 340, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Markovski, M.; Wickner, S. Preventing bacterial suicide: A novel toxin-antitoxin strategy. Mol. Cell. 2013, 52, 611–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the β sliding clamp. Mol. Cell. 2013, 52, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramisetty, B.C.; Natarajan, B.; Santhosh, R.S. mazEF-mediated programmed cell death in bacteria: “What is this?”. Crit. Rev. Microbiol. 2015, 41, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II Toxin-Antitoxin Systems: Evolution and Revolutions. J. Bacteriol. 2020, 202, e0076319. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yuan, S.; Chen, L.; Liu, Q.; Zhang, H.; Ma, Y.; Wei, T.; Huang, S. Complete genome analysis of bacteriophage AsXd-1, a new member of the genus Hk97virus, family Siphoviridae. Arch. Virol. 2018, 163, 3195–3197. [Google Scholar] [CrossRef]
- Abrescia, N.G.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure unifies the viral universe. Annu. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Liljas, L.; Duda, R.L.; Tsuruta, H.; Hendrix, R.W.; Johnson, J.E. Topologically linked protein rings in the bacteriophage HK97 capsid. Science. 2000, 289, 2129–2133. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, H.; Matsuyama, S. Sorting of lipoproteins to the outer membrane in E. coli. Biochim. Biophys. Acta. 2004, 1693, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Wietzorrek, A.; Schwarz, H.; Herrmann, C.; Braun, V. The genome of the novel phage Rtp, with a rosette-like tail tip, is homologous to the genome of phage T1. J. Bacteriol. 2006, 188, 1419–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arguijo-Hernández, E.S.; Hernandez-Sanchez, J.; Briones-Peña, S.J.; Oviedo, N.; Mendoza-Hernández, G.; Guarneros, G.; Kameyama, L. Cor interacts with outer membrane proteins to exclude FhuA-dependent phages. Arch. Virol. 2018, 163, 2959–2969. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Eisaki, N.; Ohtsubo, E. Translational control in production of transposase and in transposition of insertion sequence IS3. J. Mol. Biol. 1994, 235, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Rak, B. Escherichia coli insertion sequence IS150: Transposition via circular and linear intermediates. J. Bacteriol. 2002, 184, 5833–5841. [Google Scholar] [CrossRef] [Green Version]
- Sekine, Y.; Nagasawa, H.; Ohtsubo, E. Identification of the site of translational frameshifting required for production of the transposase encoded by insertion sequence IS 1. Mol Gen. Genet. 1992, 235, 317–324. [Google Scholar] [CrossRef]
- Polard, P.; Chandler, M. Bacterial transposases and retroviral integrases. Mol. Microbiol. 1995, 15, 13–23. [Google Scholar] [CrossRef]
- Leclercq, S.; Gilbert, C.; Cordaux, R. Cargo capacity of phages and plasmids and other factors influencing horizontal transfers of prokaryote transposable elements. Mob. Genet. Elements. 2012, 2, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, S.; Cordaux, R. Do phages efficiently shuttle transposable elements among prokaryotes? Evolution 2011, 65, 3327–3331. [Google Scholar] [CrossRef]
- Casjens, S.R.; Gilcrease, E.B.; Huang, W.M.; Bunny, K.L.; Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Hatfull, G.F.; Hendrix, R.W. The pKO2 linear plasmid prophage of Klebsiella oxytoca. J. Bacteriol. 2004, 186, 1818–1832. [Google Scholar] [CrossRef] [Green Version]
- Lemire, S.; Figueroa-Bossi, N.; Bossi, L. Bacteriophage crosstalk: Coordination of prophage induction by trans-acting antirepressors. PLoS Genet. 2011, 7, e1002149. [Google Scholar] [CrossRef] [Green Version]
- Koehler, B.T.; Hopson, H.; Kongari, R.; Bonasera, R.; Hernandez-Morales, A.C.; Liu, M. Complete Genome Sequence of Serratia marcescens Siphophage Scapp. Microbiol. Resour. Announc. 2019, 8, e00417–e00419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.A.; Hopson, H.; Rodriguez, A.; Kongari, R.; Bonasera, R.; Hernandez-Morales, A.C.; Liu, M. Complete Genome Sequence of Serratia marcescens Siphophage Serbin. Microbiol. Resour. Announc. 2019, 8, e00422-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, A.M.; Askora, A.; Soliman, A.M.; Nariya, H.; Kawasaki, T.; Fujie, M.; Shimamoto, T.; Yamada, T. Full genome sequence of a polyvalent bacteriophage infecting strains of Shigella, Salmonella, and Escherichia. Arch. Virol. 2018, 163, 3207–3210. [Google Scholar]
- Hamdi, S.; Rousseau, G.M.; Labrie, S.J.; Tremblay, D.M.; Kourda, R.S.; Slama, K.B.; Moineau, S. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci. Rep. 2017, 7, 40349. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Askora, A.; Barakat, A.B.; Rabie, O.E.; Hassan, S.E. Isolation and characterization of polyvalent bacteriophages infecting multi drug resistant Salmonella serovars isolated from broilers in Egypt. Int. J. Food Microbiol. 2018, 266, 8–13. [Google Scholar] [CrossRef]
- Park, M.; Lee, J.-H.; Shin, H.; Kim, M.; Choi, J.; Kang, D.-H.; Heu, S.; Ryu, S. Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl. Environ. Microbiol. 2012, 78, 58–69. [Google Scholar] [CrossRef] [Green Version]
- El Haddad, L.; Ben Abdallah, N.; Plante, P.L.; Dumaresq, J.; Katsarava, R.; Labrie, S.; Corbeil, J.; St-Gelais, D.; Moineau, S. Improving the safety of Staphylococcus aureus polyvalent phages by their production on a Staphylococcus xylosus strain. PLoS ONE 2014, 9, e102600. [Google Scholar] [CrossRef] [Green Version]
- Matilla, M.A.; Salmond, G.P. Bacteriophage ϕMAM1, a viunalikevirus, is a broad-host-range, high-efficiency generalized transducer that infects environmental and clinical isolates of the enterobacterial genera Serratia and Kluyvera. Appl. Environ. Microbiol. 2014, 80, 6446–6457. [Google Scholar] [CrossRef] [Green Version]
- Evans, T.J.; Crow, M.A.; Williamson, N.R.; Orme, W.; Thomson, N.R.; Komitopoulou, E.; Salmond, G.P.C. Characterization of a broad-host-range flagellum-dependent phage that mediates high-efficiency generalized transduction in, and between, Serratia and Pantoea. Microbiology (Reading) 2010, 156, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Zhao, J.; Zhang, Z.; Chen, X.; Wei, X.; Li, H.; Lin, W.; Ke, W.; Hu, L.; Jiang, A.; et al. Identification and molecular characterization of Serratia marcescens phages vB_SmaA_2050H1 and vB_SmaM_2050HW. Arch. Virol. 2019, 164, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Akusobi, C.; Fang, X.; Salmond, G.P.C. Environmental T4-Family Bacteriophages Evolve to Escape Abortive Infection via Multiple Routes in a Bacterial Host Employing “Altruistic Suicide” through Type III Toxin-Antitoxin Systems. Front. Microbiol. 2017, 8, 1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drewniak, L.; Styczek, A.; Majder-Lopatka, M.; Sklodowska, A. Bacteria, hypertolerant to arsenic in the rocks of an ancient gold mine, and their potential role in dissemination of arsenic pollution. Environ. Pollut. 2008, 156, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.F.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Siwek, W.; Czapinska, H.; Bochtler, M.; Bujnicki, J.M.; Skowronek, K. Crystal structure and mechanism of action of the N6-methyladenine-dependent type IIM restriction endonuclease R.DpnI. Nucleic. Acids. Res. 2012, 40, 7563–7572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, K.M.; Polz, M.F. Streamlining standard bacteriophage methods for higher throughput. MethodsX 2018, 5, 159–172. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [Green Version]
- Claudel-Renard, C.; Chevalet, C.; Faraut, T.; Kahn, D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids. Res. 2003, 31, 6633–6639. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ning, M.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; De Weese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, A.; Remmert, M.; Biegert, A.; Söding, J. Fast and accurate automatic structure prediction with HHpred. Proteins 2009, 77 (Suppl. 9), 128–132. [Google Scholar] [CrossRef] [Green Version]
- Hulo, N.; Sigrist, C.J.; Saux, V.L.; Petra, S. Langendijk-Genevaux, Lorenza Bordoli, Alexandre Gattiker, Edouard De Castro, Philipp Bucher, Amos Bairoch. Recent improvements to the PROSITE database. Nucleic Acids Res. 2004, 32, D134–D137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, P.; Kanehisa, M.; DeLisi, C. The detection and classification of membrane-spanning proteins. Biochim. Biophys. Acta 1985, 815, 468–476. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.E.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS ONE 2014, 9, e98679. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, J.; Kittas, A.; Bennett, L.; Tsoka, S. DyCoNet: A Gephi plugin for community detection in dynamic complex networks. PLoS ONE 2014, 9, e101357. [Google Scholar] [CrossRef] [PubMed]
- Drozdz, M.; Piekarowicz, A.; Bujnicki, J.M.; Radlinska, M. Novel non-specific DNA adenine methyltransferases. Nucleic Acids Res. 2012, 40, 2119–2130. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Characterization of DNA condensates induced by poly(ethylene oxide) and polylysine. Proc. Natl. Acad. Sci. USA 1975, 72, 4288–4292. [Google Scholar] [CrossRef] [Green Version]
- Golec, P.; Karczewska-Golec, J.; Łoś, M.; Węgrzyn, G. Bacteriophage T4 can produce progeny virions in extremely slowly growing Escherichia coli host: Comparison of a mathematical model with the experimental data. FEMS Microbiol. Lett. 2014, 351, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Golec, P.; Karczewska-Golec, J.; Voigt, B.; Albrecht, D.; Schweder, T.; Hecker, M.; Węgrzyn, G.; Łoś, M. Proteomic profiles and kinetics of development of bacteriophage T4 and its rI and rIII mutants in slowly growing Escherichia coli. J. Gen. Virol. 2013, 94, 896–905. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bujak, K.; Decewicz, P.; Kaminski, J.; Radlinska, M. Identification, Characterization, and Genomic Analysis of Novel Serratia Temperate Phages from a Gold Mine. Int. J. Mol. Sci. 2020, 21, 6709. https://doi.org/10.3390/ijms21186709
Bujak K, Decewicz P, Kaminski J, Radlinska M. Identification, Characterization, and Genomic Analysis of Novel Serratia Temperate Phages from a Gold Mine. International Journal of Molecular Sciences. 2020; 21(18):6709. https://doi.org/10.3390/ijms21186709
Chicago/Turabian StyleBujak, Katarzyna, Przemyslaw Decewicz, Jerzy Kaminski, and Monika Radlinska. 2020. "Identification, Characterization, and Genomic Analysis of Novel Serratia Temperate Phages from a Gold Mine" International Journal of Molecular Sciences 21, no. 18: 6709. https://doi.org/10.3390/ijms21186709