mRNA Post-Transcriptional Regulation by AU-Rich Element-Binding Proteins in Liver Inflammation and Cancer

 ,
,  and
and
Abstract
:1. Introduction
2. AU-Rich Element-Binding Proteins
2.1. Functions of AUBPs
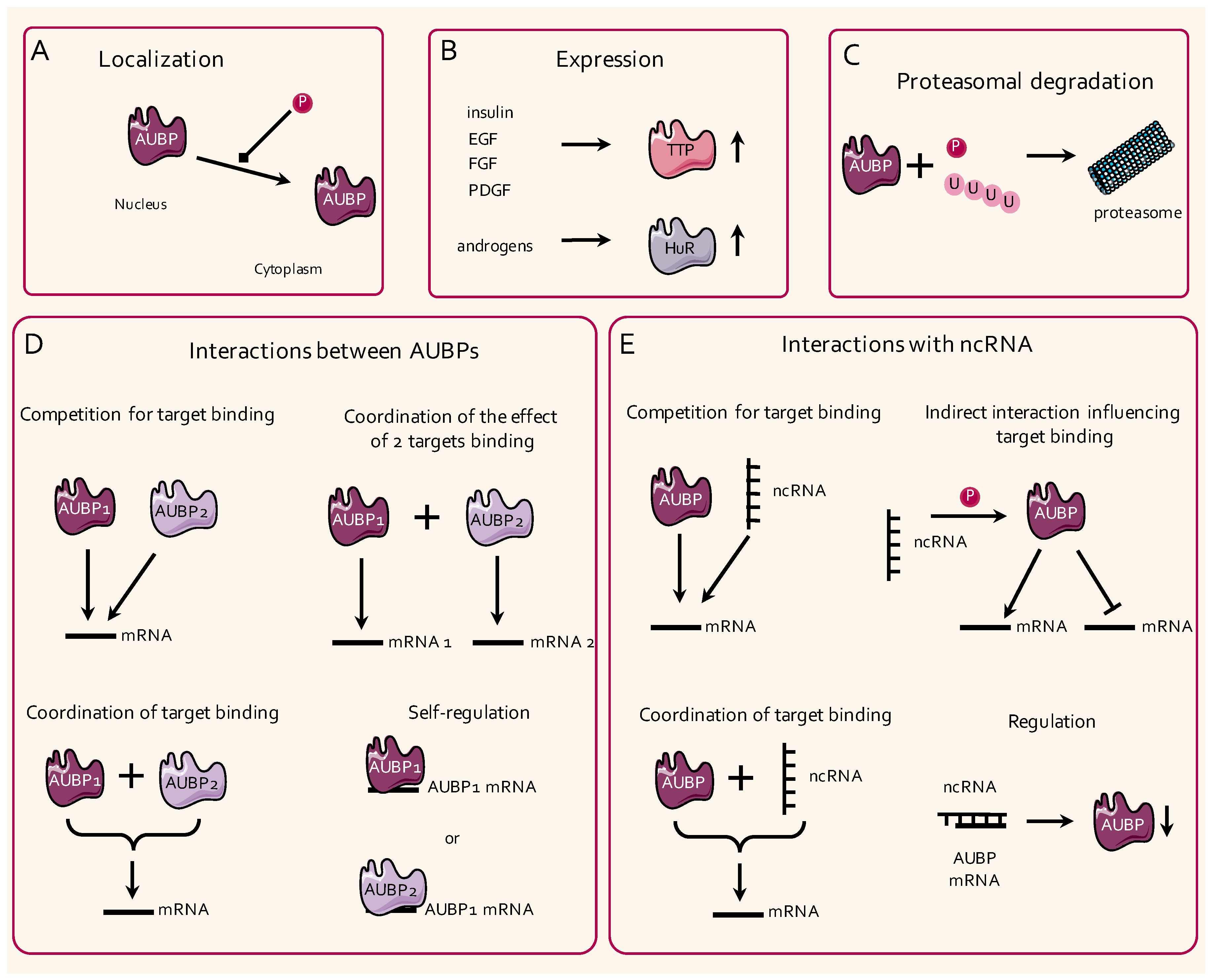
2.2. Regulation of the Activity of AUBPs
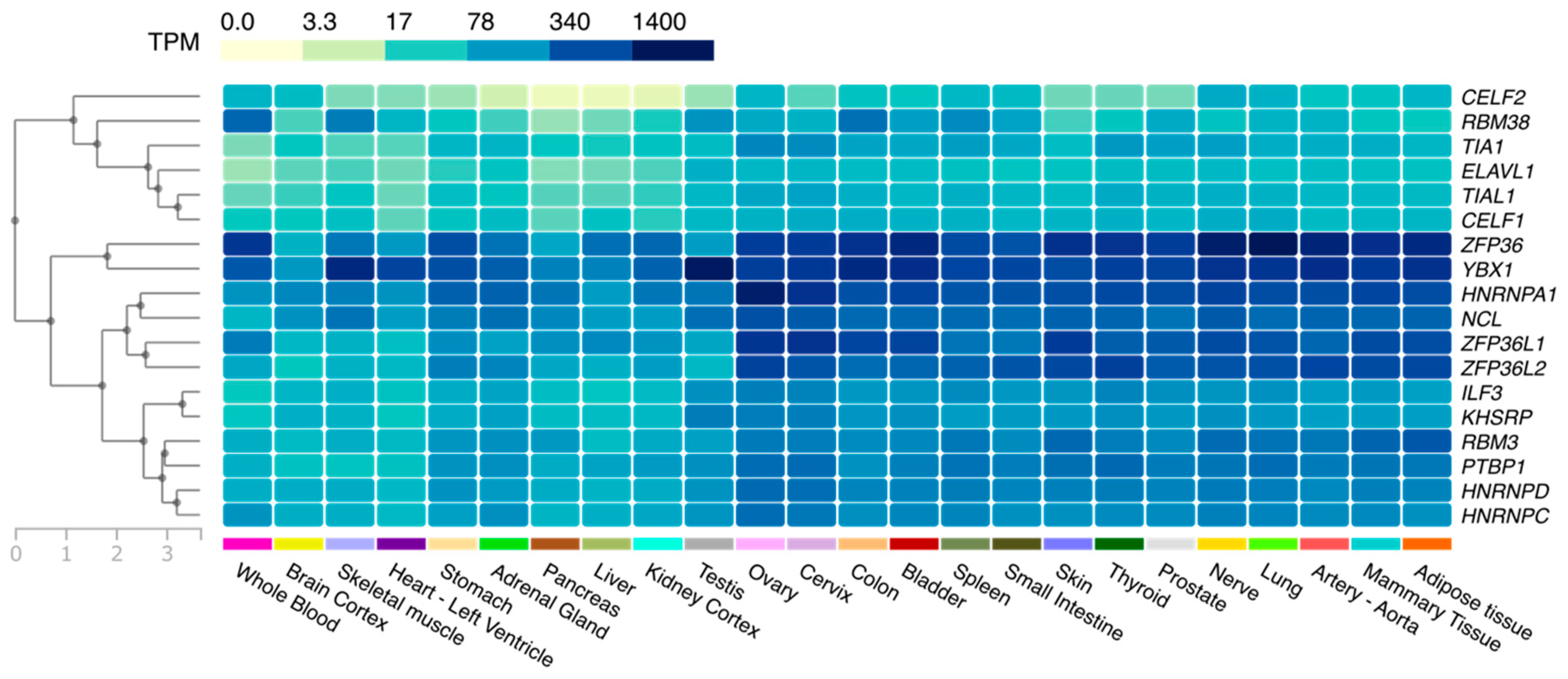
2.2.1. Temporal and Spatial Regulation of the Activity of AUBPs
2.2.2. Post-Translational Modifications
2.2.3. Competition and Self-Regulation of the Activity of AUBPs
2.2.4. Interactions with Noncoding RNAs
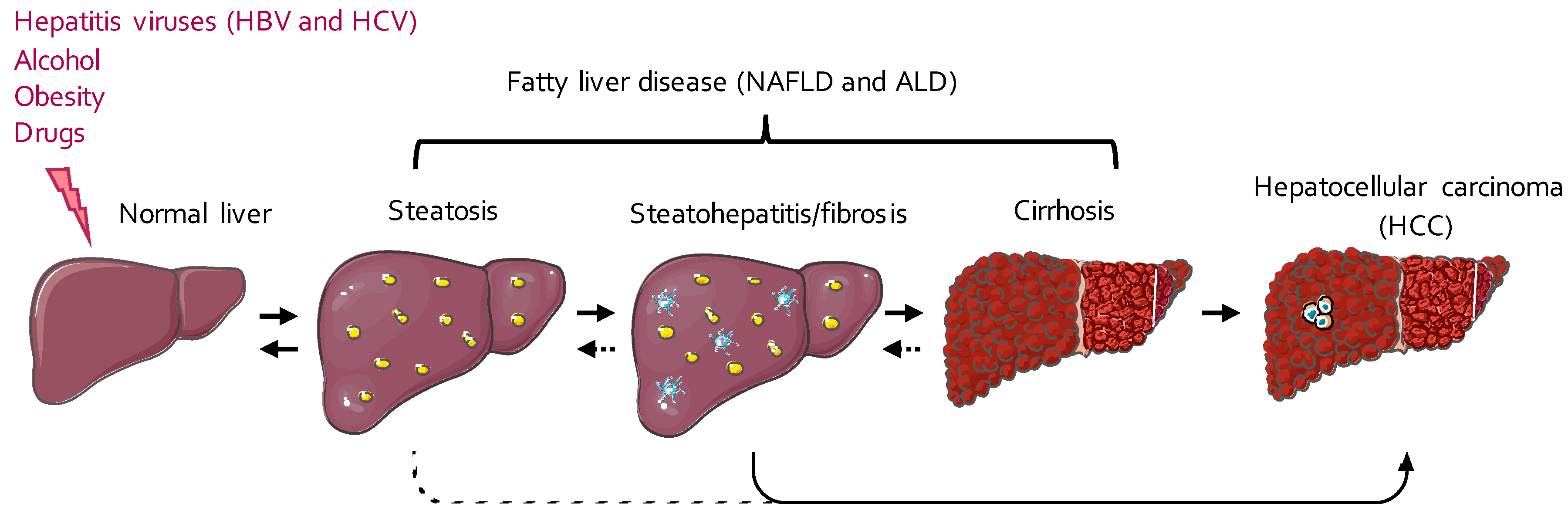
3. AUBPs in Liver Inflammation and Fibrosis
4. Modulation of Immune Responses by AUBPs in Inflammation and Cancer
5. AUBPs in the Hallmarks of Liver Cancer
5.1. Cell Cycle Regulation and Cell Proliferation
5.2. Apoptosis
5.3. Migration/Invasion/Metastasis
5.4. Angiogenesis
5.5. Metabolic Reprogramming
6. AUBPs as Biomarkers and Potential Therapeutic Targets
6.1. AUBPs as Biomarkers for Chronic Liver Diseases and HCC
6.2. Therapeutic Targeting of AUBPs and SG Formation in Liver Diseases and HCC
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AFP | alpha-fetoprotein |
| ARE | AU-rich element |
| ATG16L1 | Autophagy-related 16-like 1 |
| ATO | arsenic trioxide |
| AUBP | AU-rich element-binding protein |
| AUF1 | AU-rich element RNA-binding protein 1 |
| BMDM | bone marrow-derived macrophage |
| BMSC | bone marrow-derived stem cell |
| BRF1 | butyrate response factor 1 |
| CBF-A | CArG-binding factor A |
| circRNA | circular RNA |
| CUGBP1 | CUG triplet repeat RNA-binding protein 1 |
| CYP2A6 | cytochrome P450 2A6 |
| DEN | diethylnitrosamine |
| DHTS | dihydrotanshinone |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| eIF | eukaryotic initiation factor |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FLD | fatty liver disease |
| FGF21 | fibroblast growth factor 21 |
| H-AS1 | hedgehog-interacting protein antisense RNA-1 |
| HSC | hepatic stellate cell |
| HCC | hepatocellular carcinoma |
| HAUSP | herpesvirus-associated ubiquitin-specific protease |
| hnRNP | heterogeneous nuclear ribonucleoprotein |
| HK2 | hexokinase 2 |
| HFD | high-fat diet |
| HIF1α | hypoxia-induced factor 1α |
| HuD | Hu-antigen D |
| HULC | highly upregulated in liver cancer |
| HuR | Hu-antigen R |
| ICC | intrahepatic cholangiocarcinoma |
| IFNγ | interferon γ |
| IGF | insulin-like growth factor |
| IR | insulin resistance |
| KSRP | KH-type splicing regulatory protein |
| lncRNA | long noncoding RNA |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| miRNA | microRNA |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NCL | nucleolin |
| P-body | processing body |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed death-ligand 1 |
| PDGF | platelet-derived growth factor |
| PK-M2 | pyruvate kinase M2 |
| RBM3 | RNA-binding protein 3 |
| RBP | RNA-binding protein |
| SAM | S-adenosylmethionine |
| α-SMA | α-smooth muscle actin |
| SCD | stearoyl-CoA desaturase |
| SG | stress granule |
| TGF-β | transforming growth factor β |
| TGIF | TG-interacting factor |
| TIA1 | T-cell-restricted intracellular antigen-1 |
| TIAR | TIA-1-related protein |
| TNFα | tumor necrosis factor α |
| TTP | tristetraprolin |
| T2D | type 2 diabetes |
| VEGF | vascular endothelial growth factor |
| YB-1 | Y-box-binding protein 1 |
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Metze, D.; Cury, V.F.; Gomez, R.S.; Marco, L.; Robinson, D.; Melamed, E.; Leung, A.K.C.; Nam, J.-H.; Matsubara, Y.; Tada, K.; et al. Hepatocellular Carcinoma. Encycl. Mol. Mech. Dis. 2009, 380, 821–824. [Google Scholar]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16019. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sandhu, S.; Lai, J.-P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Mayr, C. Regulation by 3′-Untranslated Regions. Ann. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasuri, F.; Visani, M.; Acquaviva, G.; Brand, T.; Fiorentino, M.; Pession, A.; Tallini, G.; D’Errico, A.; De Biase, D. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 2647–2660. [Google Scholar] [CrossRef]
- Bakheet, T. ARED 3.0: The large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006, 34, D111–D114. [Google Scholar] [CrossRef]
- Chen, C.-Y.A.; Shyu, A.-B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005, 33, 7138–7150. [Google Scholar] [CrossRef]
- Sawicki, K.T.; Chang, H.-C.; Shapiro, J.S.; Bayeva, M.; De Jesus, A.; Finck, B.N.; Wertheim, J.A.; Blackshear, P.J.; Ardehali, H. Hepatic tristetraprolin promotes insulin resistance through RNA destabilization of FGF21. JCI Insight 2018, 3, e95948. [Google Scholar] [CrossRef] [Green Version]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Wagner, E. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005, 19, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratacós, F.M.; Brewer, G. The role of AUF1 in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 2010, 1, 457–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherzi, R.; Lee, K.-Y.; Briata, P.; Wegmüller, D.; Moroni, C.; Karin, M.; Chen, C.-Y. A KH Domain RNA Binding Protein, KSRP, Promotes ARE-Directed mRNA Turnover by Recruiting the Degradation Machinery. Mol. Cell 2004, 14, 571–583. [Google Scholar] [CrossRef]
- Piecyk, M.; Wax, S.; Beck, A.R.; Kedersha, N.; Gupta, M.; Maritim, B.; Chen, S.; Gueydan, C.; Kruys, V.; Streuli, M.; et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-α. EMBO J. 2000, 19, 4154–4163. [Google Scholar] [CrossRef] [Green Version]
- Sheth, U.; Parker, R. Decapping and Decay of Messenger RNA Occur in Cytoplasmic Processing Bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Sen, G.L.; Blau, H.M. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat. Cell Biol. 2005, 7, 633–636. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N.; Ivanov, P. Stress granules, P-bodies and cancer. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2014, 1849, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Franks, T.M.; Lykke-Andersen, J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 2007, 21, 719–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedje, C.; Ronkina, N.; Tehrani, M.; Dhamija, S.; Laass, K.; Holtmann, H.; Kotlyarov, A.; Gaestel, M. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 2012, 8, e1002977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative Regulatory Mapping Indicates that the RNA-Binding Protein HuR Couples Pre-mRNA Processing and mRNA Stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.; Anderson, P. Mammalian Stress Granules and Processing Bodies. Enzyme Eng. Evol. Gen. Methods 2007, 431, 61–81. [Google Scholar] [CrossRef]
- Kedersha, N.; Chen, S.; Gilks, N.; Li, W.; Miller, I.J.; Stahl, J.; Anderson, P. Evidence That Ternary Complex (eIF2-GTP-tRNAi Met)–Deficient Preinitiation Complexes Are Core Constituents of Mammalian Stress Granules. Mol. Biol. Cell 2002, 13, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.S.; Stumpo, D.J.; Blackshear, P.J. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J. Biol. Chem. 1990, 265, 16556–16563. [Google Scholar]
- Dubois, R.N.; McLane, M.W.; Ryder, K.; Lau, L.F.; Nathans, D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J. Biol. Chem. 1990, 265, 19185–19191. [Google Scholar]
- Sheflin, L.G.; Zhang, W.; Spaulding, S.W. Androgen regulates the level and subcellular distribution of the AU-rich ribonucleic acid-binding protein HuR both in vitro and in vivo. Endocrinology 2001, 142, 2361–2368. [Google Scholar] [CrossRef]
- Kim, H.H.; Abdelmohsen, K.; Lal, A.; Pullmann, J.R.; Yang, X.; Galbán, S.; Srikantan, S.; Martindale, J.L.; Blethrow, J.; Shokat, K.M.; et al. Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 2008, 22, 1804–1815. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.F.; Sanduja, S.; Deane, N.G.; Blackshear, P.J.; Dixon, D.A. Transforming growth factor beta regulates P-body formation through induction of the mRNA decay factor tristetraprolin. Mol. Cell Biol. 2014, 34, 180–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Chen, F.; Kim, Y.-J.; Chen, Y. Transcriptional Regulation of Tristetraprolin by Transforming Growth Factor-β in Human T Cells. J. Biol. Chem. 2003, 278, 30373–30381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Morita, N.; Hikita, K.; Kiuchi, K.; Kiuchi, K.; Kaneda, N. Recruitment of mRNA-destabilizing protein TIS11 to stress granules is mediated by its zinc finger domain. Exp. Cell Res. 2005, 303, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Yoon, J.-H.; Parker, R. Stress-specific composition, assembly and kinetics of stress granules in Saccharomyces cerevisiae. J. Cell Sci. 2010, 124, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Decker, C.J.; Parker, R. P-Bodies and Stress Granules: Possible Roles in the Control of Translation and mRNA Degradation. Cold Spring Harb. Perspect. Biol. 2012, 4, a012286. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Schmidlin, M.; Min, L.; Gross, B.; Moroni, C. BRF1 Protein Turnover and mRNA Decay Activity Are Regulated by Protein Kinase B at the Same Phosphorylation Sites. Mol. Cell. Biol. 2006, 26, 9497–9507. [Google Scholar] [CrossRef] [Green Version]
- Blum, J.L.; Samarel, A.M.; Mestril, R. Phosphorylation and binding of AUF1 to the 3′-untranslated region of cardiomyocyte SERCA2a mRNA. Am. J. Physiol. Circ. Physiol. 2005, 289, H2543–H2550. [Google Scholar] [CrossRef]
- Hitti, E.; Iakovleva, T.; Brook, M.; Deppenmeier, S.; Gruber, A.D.; Radzioch, D.; Clark, A.R.; Blackshear, P.J.; Kotlyarov, A.; Gaestel, M. Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 Regulates Tumor Necrosis Factor mRNA Stability and Translation Mainly by Altering Tristetraprolin Expression, Stability, and Binding to Adenine/Uridine-Rich Element. Mol. Cell. Biol. 2006, 26, 2399–2407. [Google Scholar] [CrossRef] [Green Version]
- Filippova, N.; Yang, X.; King, P.; Nabors, B. Phosphoregulation of the RNA-binding Protein Hu Antigen R (HuR) by Cdk5 Affects Centrosome Function*. J. Biol. Chem. 2012, 287, 32277–32287. [Google Scholar] [CrossRef] [Green Version]
- Doller, A.; Winkler, C.; Azrilian, I.; Schulz, S.; Hartmann, S.; Pfeilschifter, J.; Eberhardt, W. High-constitutive HuR phosphorylation at Ser 318 by PKC{delta} propagates tumor relevant functions in colon carcinoma cells. Carcinogenesis 2011, 32, 676–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.L.; Defren, J.; Brewer, G. Hsp27 and F-box protein beta-TrCP promote degradation of mRNA decay factor AUF1. Mol. Cell Biol. 2013, 33, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Yu, Z.; Zhang, Z.; Ma, W.; Song, S.; Huang, G. Interaction with Pyruvate Kinase M2 Destabilizes Tristetraprolin by Proteasome Degradation and Regulates Cell Proliferation in Breast Cancer. Sci. Rep. 2016, 6, 22449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, A.; Ahuja, D.; Das, S.; Parasar, B.; Deyasi, K.; Roy, D.; Sharma, V.; Willard, B.; Ghosh, A.; Ray, P.S. Integrated Regulation of HuR by Translation Repression and Protein Degradation Determines Pulsatile Expression of p53 Under DNA Damage. iScience 2019, 15, 342–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cok, S.J.; Acton, S.J.; Sexton, A.E.; Morrison, A.R. Identification of RNA-binding Proteins in RAW 264.7 Cells That Recognize a Lipopolysaccharide-responsive Element in the 3-Untranslated Region of the Murine Cyclooxygenase-2 mRNA. J. Biol. Chem. 2003, 279, 8196–8205. [Google Scholar] [CrossRef] [Green Version]
- García-Mauriño, S.M.; Rivero-Rodríguez, F.; Velázquez-Cruz, A.; Hernández-Vellisca, M.; Díaz-Quintana, A.; De La Rosa, M.A.; Díaz-Moreno, I. RNA Binding Protein Regulation and Cross-Talk in the Control of AU-rich mRNA Fate. Front. Mol. Biosci. 2017, 4, 71. [Google Scholar] [CrossRef]
- Dassi, E. Handshakes and Fights: The Regulatory Interplay of RNA-Binding Proteins. Front. Mol. Biosci. 2017, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Shwetha, S.; Kumar, A.; Mullick, R.; Vasudevan, D.; Mukherjee, N.; Das, S. HuR Displaces Polypyrimidine Tract Binding Protein To Facilitate La Binding to the 3′ Untranslated Region and Enhances Hepatitis C Virus Replication. J. Virol. 2015, 89, 11356–11371. [Google Scholar] [CrossRef] [Green Version]
- Komnenov, D.; Scipione, C.; Bazzi, Z.; Garabon, J.J.W.; Koschinsky, M.; Boffa, M.B. Pro-inflammatory cytokines reduce human TAFI expression via tristetraprolin-mediated mRNA destabilisation and decreased binding of HuR. Thromb. Haemost. 2015, 114, 337–349. [Google Scholar] [CrossRef]
- Pan, Y.-X.; Chen, H.; Kilberg, M.S. Interaction of RNA-binding Proteins HuR and AUF1 with the Human ATF3 mRNA 3′-Untranslated Region Regulates Its Amino Acid Limitation-induced Stabilization. J. Biol. Chem. 2005, 280, 34609–34616. [Google Scholar] [CrossRef] [Green Version]
- Vázquez–Chantada, M.; Fernández–Ramos, D.; Embade, N.; Martínez–Lopez, N.; Varela–Rey, M.; Woodhoo, A.; Luka, Z.; Wagner, C.; Anglim, P.P.; Finnell, R.H.; et al. HuR/Methyl-HuR and AUF1 Regulate the MAT Expressed During Liver Proliferation, Differentiation, and Carcinogenesis. Gastroenterology 2010, 138, 1943–1953.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieu, N.A.; Motiño, O.; Mayoral, R.; Izquierdo, C.L.; Fernández-Alvarez, A.J.; Boscá, L.; Casado, M.; Martín-Sanz, P. Cyclooxygenase-2 Is a Target of MicroRNA-16 in Human Hepatoma Cells. PLoS ONE 2012, 7, e50935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, D.; Ramalingam, S.; Linehan, D.C.; Dieckgraefe, B.K.; Postier, R.G.; Houchen, C.W.; Jensen, R.A.; Anant, S. RNA binding protein CUGBP2/CELF2 mediates curcumin-induced mitotic catastrophe of pancreatic cancer cells. PLoS ONE 2011, 6, e16958. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; Murmu, N.; Rodriguez, P.; May, R.; Maheshwari, R.; Dieckgraefe, B.K.; Houchen, C.W.; Anant, S. Functional Antagonism Between RNA Binding Proteins HuR and CUGBP2 Determines the Fate of COX-2 mRNA Translation. Gastroenterology 2007, 132, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Wigington, C.P.; Jung, J.; Rye, E.A.; Belauret, S.L.; Philpot, A.M.; Feng, Y.; Santangelo, P.J.; Corbett, A.H. Post-transcriptional Regulation of Programmed Cell Death 4 (PDCD4) mRNA by the RNA-binding Proteins Human Antigen R (HuR) and T-cell Intracellular Antigen 1 (TIA1)*. J. Biol. Chem. 2014, 290, 3468–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsanou, V.; Papadaki, O.; Milatos, S.; Blackshear, P.J.; Anderson, P.; Kollias, G.; Kontoyiannis, D.L. HuR as a Negative Posttranscriptional Modulator in Inflammation. Mol. Cell 2005, 19, 777–789. [Google Scholar] [CrossRef]
- Tchen, C.R.; Brook, M.; Saklatvala, J.; Clark, A.R. The Stability of Tristetraprolin mRNA Is Regulated by Mitogen-activated Protein Kinase p38 and by Tristetraprolin Itself. J. Biol. Chem. 2004, 279, 32393–32400. [Google Scholar] [CrossRef] [Green Version]
- Brooks, S.A.; Connolly, J.E.; Rigby, W.F. The role of mRNA turnover in the regulation of tristetraprolin expression: Evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J. Immunol. 2004, 172, 7263–7271. [Google Scholar] [CrossRef] [Green Version]
- Al-Ahmadi, W.; Al-Ghamdi, M.; Al-Haj, L.; Al-Saif, M.; Khabar, K.S.A. Alternative polyadenylation variants of the RNA binding protein, HuR: Abundance, role of AU-rich elements and auto-Regulation. Nucleic Acids Res. 2009, 37, 3612–3624. [Google Scholar] [CrossRef] [Green Version]
- Pullmann, R.; Kim, H.H.; Abdelmohsen, K.; Lal, A.; Martindale, J.L.; Yang, X.; Gorospe, M. Analysis of Turnover and Translation Regulatory RNA-Binding Protein Expression through Binding to Cognate mRNAs. Mol. Cell. Biol. 2007, 27, 6265–6278. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yang, Z.; Trottier, J.; Barbier, O.; Wang, L. Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology 2016, 65, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, Y.; Yu, S. Long noncoding RNA AWPPH promotes hepatocellular carcinoma progression through YBX1 and serves as a prognostic biomarker. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Luo, S.-Z.; Liu, T.; Lu, L.-G.; Xu, M.-Y. linc-SCRG1 accelerates liver fibrosis by decreasing RNA-binding protein tristetraprolin. FASEB J. 2018, 33, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Cheng, S.; Yang, Z.; Lv, Z.; Xiao, H.; Du, C.; Peng, C.; Xie, H.; Zhou, L.; Wu, J.; et al. Long Non-Coding RNA HOTAIR Promotes Cell Migration and Invasion via Down-Regulation of RNA Binding Motif Protein 38 in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2014, 15, 4060–4076. [Google Scholar] [CrossRef] [Green Version]
- Ke, R.-S.; Zhang, K.; Lv, L.-Z.; Dong, Y.-P.; Pan, F.; Yang, F.; Cai, Q.-C.; Jiang, Y. Prognostic value and oncogene function of heterogeneous nuclear ribonucleoprotein A1 overexpression in HBV-related hepatocellular carcinoma. Int. J. Biol. Macromol. 2019, 129, 140–151. [Google Scholar] [CrossRef]
- Xiao, Z.; Chow, S.C.; Li, C.H.; Tang, S.C.; Tsui, S.K.-W.; Lin, Z.; Chen, Y. Role of microRNA-95 in the anticancer activity of Brucein D in hepatocellular carcinoma. Eur. J. Pharmacol. 2014, 728, 141–150. [Google Scholar] [CrossRef]
- Cheng, L.; Zhu, Y.; Han, H.; Zhang, Q.; Cui, K.; Shen, H.; Zhang, J.; Yan, J.; Prochownik, E.; Li, Y. MicroRNA-148a deficiency promotes hepatic lipid metabolism and hepatocarcinogenesis in mice. Cell Death Dis. 2017, 8, e2916. [Google Scholar] [CrossRef]
- Li, D.; Liu, X.; Zhou, J.; Hu, J.; Zhang, D.; Liu, J.; Qiao, Y.; Zhan, Q. Long noncoding RNA HULC modulates the phosphorylation of YB-1 through serving as a scaffold of extracellular signal-regulated kinase and YB-1 to enhance hepatocarcinogenesis. Hepatology 2017, 65, 1612–1627. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-Y.; Zou, X.-J.; Cao, C.-H.; Zhang, T.; Lei, L.; Qi, X.-L.; Liu, L.; Wu, D. Identification and Functional Characterization of Long Non-coding RNA MIR22HG as a Tumor Suppressor for Hepatocellular Carcinoma. Theranostics 2018, 8, 3751–3765. [Google Scholar] [CrossRef]
- Huang, J.-F.; Jiang, H.; Cai, H.; Liu, Y.; Zhu, Y.-Q.; Lin, S.-S.; Hu, T.-T.; Wang, T.-T.; Yang, W.-J.; Xiao, B.; et al. Genome-wide screening identifies oncofetal lncRNA Ptn-dt promoting the proliferation of hepatocellular carcinoma cells by regulating the Ptn receptor. Oncogene 2019, 38, 3428–3445. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, G.; Wang, X.; Liu, J.; Lu, Z.; Wang, Q.; Xu, B.; Liu, Z.; Li, J. CircBACH1 (hsa_circ_0061395) promotes hepatocellular carcinoma growth by regulating p27 repression via HuR. J. Cell. Physiol. 2020, 235, 6929–6941. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; De Sousa, M.C.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, V.; Jakymiw, A.; Van Tubergen, E.A.; D’Silva, N.J.; Kirkwood, K.L. Control of cytokine mRNA expression by RNA-binding proteins and microRNAs. J. Dent. Res. 2012, 91, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourou, N.; Gavriilidis, M.; Kontoyiannis, D.L. Lessons from studying the AU-rich elements in chronic inflammation and autoimmunity. J. Autoimmun. 2019, 104, 102334. [Google Scholar] [CrossRef]
- Chang, N.; Ge, J.; Xiu, L.; Zhao, Z.; Duan, X.; Tian, L.; Xie, J.; Yang, L.; Li, L. HuR mediates motility of human bone marrow-derived mesenchymal stem cells triggered by sphingosine 1-phosphate in liver fibrosis. J. Mol. Med. 2016, 95, 69–82. [Google Scholar] [CrossRef]
- Chang, N.; Duan, X.; Zhao, Z.; Tian, L.; Ji, X.; Yang, L.; Li, L. Both HuR and miR-29s regulate expression of CB1 involved in infiltration of bone marrow monocyte/macrophage in chronic liver injury. J. Cell. Physiol. 2019, 235, 2532–2544. [Google Scholar] [CrossRef]
- Woodhoo, A.; Iruarrizaga-Lejarreta, M.; Beraza, N.; García-Rodríguez, J.L.; Embade, N.; Fernández–Ramos, D.; Martínez-López, N.; Juan, V.G.-D.; Arteta, B.; Caballería, J.; et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology 2012, 56, 1870–1882. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [Green Version]
- Dery, K.J.; Nakamura, K.; Kadono, K.; Hirao, H.; Kageyama, S.; Ito, T.; Kojima, H.; Kaldas, F.M.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Human Antigen R (HuR): A New Regulator of Heme Oxygenase-1 Cytoprotection in Mouse and Human Liver Transplant Injury [published online ahead of print (27.12.2019)]. Hepatology 2019. [Google Scholar] [CrossRef]
- Liu, R.; Wu, K.; Li, Y.; Sun, R.; Li, X. Human antigen R: A potential therapeutic target for liver diseases. Pharmacol. Res. 2020, 155, 104684. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Chen, Q.; Tang, D.; Ou, W.; Wang, J.; Mo, Z.; Peng, L. Activation of liver X receptors promotes inflammatory cytokine mRNA degradation by upregulation of tristetraprolin. Acta Biochim. Biophys. Sin. 2017, 49, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Yoon, N.A.; Vo, M.-T.; Kim, C.W.; Woo, J.M.; Cha, H.J.; Cho, Y.W.; Lee, B.J.; Cho, W.J.; Park, J.W. Tristetraprolin down-regulates IL-17 through mRNA destabilization. FEBS Lett. 2011, 586, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-X.; Su, X.; Xu, J.; Zhang, W.-Y.; Shi, Y. HuR post-transcriptionally regulates TNF-α-induced IL-6 expression in human pulmonary microvascular endothelial cells mainly via tristetraprolin. Respir. Physiol. Neurobiol. 2012, 181, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.; Kedersha, N.; Shen, L.; Blackshear, P.J.; Anderson, P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor α, cyclooxygenase 2, and inflammatory arthritis. Proc. Natl. Acad. Sci. USA 2004, 101, 2011–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kröhler, T.; Kessler, S.M.; Hosseini, K.; List, M.; Barghash, A.; Patial, S.; Laggai, S.; Gemperlein, K.; Haybaeck, J.; Müller, R.; et al. The mRNA-binding Protein TTP/ZFP36 in Hepatocarcinogenesis and Hepatocellular Carcinoma. Cancers 2019, 11, 1754. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cao, M.; Wu, D.; Li, N.; Peng, J.; Song, L.; Qi, P.; Zhang, M.; Zhao, J. KH-type splicing regulatory protein mediate inflammatory response in gastric epithelial cells induced by lipopolysaccharide. Cell Biol. Int. 2017, 41, 871–878. [Google Scholar] [CrossRef]
- Xia, Z.; Lu, Y.; Li, X.; Mao, T.; Chen, X.-M.; Zhou, R. Upregulation of KSRP by miR-27b provides IFN-γ-induced post-transcriptional regulation of CX3CL1 in liver epithelial cells. Sci. Rep. 2015, 5, 17590. [Google Scholar] [CrossRef] [Green Version]
- Zuo, M.; Wang, A.; Tian, Y.; Mao, L.; Song, G.; Yang, Z. Oxymatrine ameliorates insulin resistance in rats with type 2 diabetes by regulating the expression of KSRP, PETN, and AKT in the liver. J. Cell. Biochem. 2019, 120, 16185–16194. [Google Scholar] [CrossRef]
- Chou, C.-F.; Zhu, X.; Lin, Y.-Y.; Gamble, K.L.; Garvey, W.T.; Chen, C.-Y. KSRP is critical in governing hepatic lipid metabolism through controlling Per2 expression. J. Lipid Res. 2014, 56, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Xiong, P.; Zhang, J.; Xu, D.; Zhu, J.; Li, W.; Liu, J.; Liu, F. Positive feedback loop of YB-1 interacting with Smad2 promotes liver fibrosis. Biochem. Biophys. Res. Commun. 2017, 484, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Wieland, S.; Chisari, F.V. Hepatitis C Virus (HCV) Induces Formation of Stress Granules Whose Proteins Regulate HCV RNA Replication and Virus Assembly and Egress. J. Virol. 2012, 86, 11043–11056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-T.; Tsai, T.-Y.; Chao, C.-H.; Lai, B.-Y.; Lee, Y.-H.W. Y-Box Binding Protein 1 Stabilizes Hepatitis C Virus NS5A via Phosphorylation-Mediated Interaction with NS5A To Regulate Viral Propagation. J. Virol. 2015, 89, 11584–11602. [Google Scholar] [CrossRef] [Green Version]
- Makokha, G.N.; Abe-Chayama, H.; Chowdhury, S.; Hayes, C.N.; Tsuge, M.; Yoshima, T.; Ishida, Y.; Zhang, Y.; Uchida, T.; Tateno, C.; et al. Regulation of the Hepatitis B virus replication and gene expression by the multi-functional protein TARDBP. Sci. Rep. 2019, 9, 8462. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Luedde, T. The transition from inflammation to cancer in the liver. Clin. Liver Dis. 2016, 8, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Ann. Rev. Pathol. Mech. Dis. 2016, 12, 153–186. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Zhang, H.; Sun, B.; Karin, M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J. Hepatol. 2020, 72, 167–182. [Google Scholar] [CrossRef]
- Roderburg, C.; Wree, A.; Demir, M.; Schmelzle, M.; Tacke, F. The role of the innate immune system in the development and treatment of hepatocellular carcinoma. Hepatic Oncol. 2020, 7, HEP17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Bao, X.; Wang, D.; You, L.; Li, X.; Yang, H.; Bian, J.-S.; Wang, Y.; Yang, Y. APOBEC3B and IL-6 form a positive feedback loop in hepatocellular carcinoma cells. Sci. China Life Sci. 2017, 60, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, T.O.; Butler, N.S.; Monick, M.M.; Hunninghake, G.W. Early exposure to IL-4 stabilizes IL-4 mRNA in CD4+ T cells via RNA-binding protein HuR. J. Immunol. 2006, 177, 4426–4435. [Google Scholar] [CrossRef] [Green Version]
- Casolaro, V.; Fang, X.; Tancowny, B.; Fan, J.; Wu, F.; Srikantan, S.; Asaki, S.Y.; De Fanis, U.; Huang, S.-K.; Gorospe, M.; et al. Posttranscriptional regulation of IL-13 in T cells: Role of the RNA-binding protein HuR. J. Allergy Clin. Immunol. 2008, 121, 853–859.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadaki, O.; Milatos, S.; Grammenoudi, S.; Mukherjee, N.; Keene, J.D.; Kontoyiannis, D.L. Control of Thymic T Cell Maturation, Deletion and Egress by the RNA-Binding Protein HuR. J. Immunol. 2009, 182, 6779–6788. [Google Scholar] [CrossRef] [Green Version]
- Techasintana, P.; Ellis, J.S.; Glascock, J.; Gubin, M.M.; Ridenhour, S.E.; Magee, J.D.; Hart, M.L.; Yao, P.; Zhou, H.; Whitney, M.S.; et al. The RNA-Binding Protein HuR Posttranscriptionally Regulates IL-2 Homeostasis and CD4+ Th2 Differentiation. ImmunoHorizons 2017, 1, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cascio, J.; Magee, J.D.; Techasintana, P.; Gubin, M.M.; Dahm, G.M.; Calaluce, R.; Yu, S.; Atasoy, U. Posttranscriptional Gene Regulation of IL-17 by the RNA-Binding Protein HuR Is Required for Initiation of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2013, 191, 5441–5450. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Techasintana, P.; Magee, J.D.; Dahm, G.M.; Calaluce, R.; Martindale, J.L.; Whitney, M.S.; Franklin, C.L.; Besch-Williford, C.; Hollingsworth, J.W.; et al. Conditional knockout of the RNA-binding protein HuR in CD4+ T cells reveals a gene dosage effect on cytokine production. Mol. Med. 2014, 20, 93–108. [Google Scholar] [CrossRef]
- Coelho, M.A.; Trécesson, S.D.C.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e6. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Qu, H.; Shan, T.; Chen, Y.; Chen, Y.; Xia, J. Tristetraprolin Overexpression in Gastric Cancer Cells Suppresses PD-L1 Expression and Inhibits Tumor Progression by Enhancing Antitumor Immunity. Mol. Cells 2018, 41, 653–664. [Google Scholar] [PubMed]
- Young, L.E.; Sanduja, S.; Bemis–Standoli, K.; Peña, E.A.; Price, R.L.; Dixon, D.A. The mRNA binding proteins HuR and tristetraprolin regulate cyclooxygenase 2 expression during colon carcinogenesis. Gastroenterology 2009, 136, 1669–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E 2 and T cells: Friends or foes? Immunol. Cell Biol. 2011, 90, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.-X.; Ling, Y.; Wang, H.-Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tao, Z.; Ruan, H.; Sun, L.; Kuang, D.; Song, Y.; Wang, Q.; Wang, T.; Hao, Y.; Chen, K. Targeting the YB-1/PD-L1 Axis to Enhance Chemotherapy and Antitumor Immunity. Cancer Immunol. Res. 2019, 7, 1135–1147. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Gherzi, R.; Andersen, J.S.; Gaietta, G.; Jürchott, K.; Royer, H.-D.; Mann, M.; Karin, M. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genome Res. 2000, 14, 1236–1248. [Google Scholar]
- Kang, S.; Lee, T.A.; Ra, E.A.; Lee, E.; Choi, H.J.; Lee, S.; Park, B. Differential Control of Interleukin-6 mRNA Levels by Cellular Distribution of YB-1. PLoS ONE 2014, 9, e112754. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Chi, M.; Morello, D. Aberrant regulation of mRNA 3’ untranslated region in cancers and inflammation. Med. Sci. 2008, 24, 290–296. [Google Scholar]
- Naz, S.; Battu, S.; Khan, R.A.; Afroz, S.; Giddaluru, J.; Vishwakarma, S.K.; Satti, V.; Habeeb, M.A.; Khan, A.A.; Khan, N. Activation of integrated stress response pathway regulates IL-1beta production through posttranscriptional and translational reprogramming in macrophages. Eur. J. Immunol. 2019, 49, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Tian, F.; Tang, L.; Wang, S.; Chen, G.; Duan, G.-J.; Yan, X. Local distribution analysis of cytotoxic molecules in liver allograft is helpful for the diagnosis of acute cellular rejection after orthotopic liver transplantation. Diagn. Pathol. 2012, 7, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlobec, I.; Karamitopoulou, E.; Terracciano, L.; Piscuoglio, S.; Iezzi, G.; Muraro, M.G.; Spagnoli, G.; Baker, K.; Tzankov, A.; Lugli, A. TIA-1 Cytotoxic Granule-Associated RNA Binding Protein Improves the Prognostic Performance of CD8 in Mismatch Repair-Proficient Colorectal Cancer. PLoS ONE 2010, 5, e14282. [Google Scholar] [CrossRef] [PubMed]
- Käfer, R.; Schmidtke, L.; Schrick, K.; Montermann, E.; Bros, M.; Kleinert, H.; Pautz, A. The RNA-Binding Protein KSRP Modulates Cytokine Expression of CD4+ T Cells. J. Immunol. Res. 2019, 2019, 4726532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legrand, N.; A Dixon, D.; Sobolewski, C. AU-rich element-binding proteins in colorectal cancer. World J. Gastrointest. Oncol. 2019, 11, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liang, R.; Bai, T.; Lin, Y.; Mai, R.; Wei, M.; Ye, X.; Li, L.-Q.; Wu, F. RBM38 plays a tumor-suppressor role via stabilizing the p53-mdm2 loop function in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 212. [Google Scholar] [CrossRef]
- Kang, H.; Heo, S.; Shin, J.J.; Ji, E.; Tak, H.; Ahn, S.; Lee, K.J.; Lee, E.K.; Kim, W. A miR-194/PTBP1/CCND3 axis regulates tumor growth in human hepatocellular carcinoma. J. Pathol. 2019, 249, 395–408. [Google Scholar] [CrossRef]
- Dong, W.; Dai, Z.H.; Liu, F.C.; Guo, X.G.; Ge, C.M.; Ding, J.; Liu, H.; Yang, F. The RNA-binding protein RBM3 promotes cell proliferation in hepatocellular carcinoma by regulating circular RNA SCD-circRNA 2 production. EBioMedicine 2019, 45, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Huang, H.; Ding, L.; Zhu, P.; Saiyin, H.; Ji, G.; Zuo, J.; Han, D.; Pan, Y.; Ding, D.; et al. Regulation of cell cycle of hepatocellular carcinoma by NF90 through modulation of cyclin E1 mRNA stability. Oncogene 2014, 34, 4460–4470. [Google Scholar] [CrossRef] [Green Version]
- Tak, H.; Kang, H.; Ji, E.; Hong, Y.; Kim, W.; Lee, E.K. Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27 as a novel marker for hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 1117–1122. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, A.; Zhang, A.; Zhou, C. HuR promotes breast cancer cell proliferation and survival via binding to CDK3 mRNA. Biomed. Pharmacother. 2017, 91, 788–795. [Google Scholar] [CrossRef]
- Wang, W.; Caldwell, M.; Lin, S.; Furneaux, H.; Gorospe, M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000, 19, 2340–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantar, M.L.M.; Vázquez–Chantada, M.; Garnacho, M.; Latasa, M.U.; Varela–Rey, M.; Dotor, J.; Santamaria, M.; Martínez-Cruz, L.A.; Parada, L.A.; Lu, S.C.; et al. S–Adenosylmethionine Regulates Cytoplasmic HuR Via AMP–Activated Kinase. Gastroenterology 2006, 131, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Sun, J.; Zhang, D.; Guo, X.; Xie, L.; Li, X.; Wu, D.; Liu, L. The long intergenic noncoding RNA UFC1, a target of MicroRNA 34a, interacts with the mRNA stabilizing protein HuR to increase levels of beta-catenin in HCC cells. Gastroenterology 2015, 148, 415–426.e18. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.M.; Huang, H.X.; Chang, P.H.; Tseng, K.C.; Miyajima, A.; Chern, E. Y-box binding protein-1 promotes hepatocellular carcinoma-initiating cell progression and tumorigenesis via Wnt/beta-catenin pathway. Oncotarget 2017, 8, 2604–2616. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Huang, H.; Yuan, B.; Luo, T.; Li, J.; Qin, X. Suppression of CUGBP1 inhibits growth of hepatocellular carcinoma cells. Clin. Investig. Med. 2014, 37, E10-18. [Google Scholar] [CrossRef] [Green Version]
- Sohn, B.H.; Park, I.Y.; Lee, J.J.; Yang, S.; Jang, Y.J.; Park, K.C.; Kim, D.J.; Lee, D.C.; Sohn, H.A.; Kim, T.W.; et al. Functional Switching of TGF-β1 Signaling in Liver Cancer via Epigenetic Modulation of a Single CpG Site in TTP Promoter. Gastroenterology 2010, 138, 1898–1908.e12. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Ariana, A.; Alturki, N.A.; Hajjar, S.; Stumpo, D.J.; Tiedje, C.; Alnemri, E.S.; Gaestel, M.; Blackshear, P.J.; Sad, S. Tristetraprolin regulates necroptosis during tonic Toll-like receptor 4 (TLR4) signaling in murine macrophages. J. Biol. Chem. 2020, 295, 4661–4672. [Google Scholar] [CrossRef]
- Park, S.B.; Lee, J.H.; Jeong, W.W.; Kim, Y.H.; Cha, H.J.; Joe, Y.; Chung, H.T.; Cho, W.J.; Do, J.W.; Lee, B.J.; et al. TTP mediates cisplatin-induced apoptosis of head and neck cancer cells by down-regulating the expression ofBcl-2. J. Chemother. 2015, 27, 174–180. [Google Scholar] [CrossRef]
- Kim, T.W.; Yim, S.; Choi, B.J.; Jang, Y.; Lee, J.J.; Sohn, B.H.; Yoo, H.-S.; Yeom, Y.I.; Park, K.C. Tristetraprolin regulates the stability of HIF-1α mRNA during prolonged hypoxia. Biochem. Biophys. Res. Commun. 2010, 391, 963–968. [Google Scholar] [CrossRef]
- Tran, D.D.H.; Koch, A.; Allister, A.; Saran, S.; Ewald, F.; Koch, M.; Nashan, B.; Tamura, T. Treatment with MAPKAP2 (MK2) inhibitor and DNA methylation inhibitor, 5-aza dC, synergistically triggers apoptosis in hepatocellular carcinoma (HCC) via tristetraprolin (TTP). Cell. Signal. 2016, 28, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2011, 10, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Chen, J.; Lu, B.; Dong, L.; Wang, H.; Bi, C.; Wu, G.; Guo, H.; Wu, M.; Guo, Y. TIP30 Induces Apoptosis under Oxidative Stress through Stabilization of p53 Messenger RNA in Human Hepatocellular Carcinoma. Cancer Res. 2008, 68, 4133–4141. [Google Scholar] [CrossRef] [Green Version]
- Martínez-López, N.; Varela-Rey, M.; Fernández–Ramos, D.; Woodhoo, A.; Vázquez-Chantada, M.; Embade, N.; Espinosa-Hevia, L.; Bustamante, F.J.; Parada, L.A.; Rodriguez, M.S.; et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology 2010, 52, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef]
- Bo, C.; Li, X.; He, L.; Zhang, S.; Li, N.; An, Y. A novel long noncoding RNA HHIP-AS1 suppresses hepatocellular carcinoma progression through stabilizing HHIP mRNA. Biochem. Biophys. Res. Commun. 2019, 520, 333–340. [Google Scholar] [CrossRef]
- Liu, Z.; Tseng, J.T.; Hong, D.; Huang, H.-S. Suppression of TG-interacting factor sensitizes arsenic trioxide-induced apoptosis in human hepatocellular carcinoma cells. Biochem. J. 2011, 438, 349–358. [Google Scholar] [CrossRef]
- Hung, C.-M.; Huang, W.-C.; Pan, H.-L.; Chien, P.-H.; Lin, C.-W.; Chen, L.-C.; Chien, Y.-F.; Lin, C.-C.; Leow, K.-H.; Chen, W.-S.; et al. Hepatitis B Virus X Upregulates HuR Protein Level to Stabilize HER2 Expression in Hepatocellular Carcinoma Cells. BioMed Res. Int. 2014, 2014, 827415. [Google Scholar] [CrossRef]
- Subramaniam, K.; Ooi, L.L.P.; Hui, K.M. Transcriptional down-regulation of IGFBP-3 in human hepatocellular carcinoma cells is mediated by the binding of TIA-1 to its AT-rich element in the 3′-untranslated region. Cancer Lett. 2010, 297, 259–268. [Google Scholar] [CrossRef]
- Grimberg, A. p53 and IGFBP-3: Apoptosis and Cancer Protection. Mol. Genet. Metab. 2000, 70, 85–98. [Google Scholar] [CrossRef]
- Han, J.; Xue, D.-W.; Han, Q.-R.; Liang, X.-H.; Xie, L.; Li, S.; Wu, H.-Y.; Song, B. Induction of apoptosis by IGFBP3 overexpression in hepatocellular carcinoma cells. Asian Pac. J. Cancer Prev. 2014, 15, 10085–10089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Kwon, H.Y.; Ryu, D.H.; Nam, M.-H.; Shim, B.S.; Kim, J.H.; Lee, J.Y.; Kim, S. Inhibition of CUG-binding protein 1 and activation of caspases are critically involved in piperazine derivative BK10007S induced apoptosis in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0186490. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Huang, H.; Wang, J.; Zhao, Y.; Hu, X.; He, F.; Yu, L.; Wu, J. NF90 regulates PARP1 mRNA stability in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 488, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Y.; Huang, Y.; Chen, Y.; Wang, T.; Wu, S.; Tong, L.; Wang, Y.; Lin, L.; Hao, M.; et al. RNA-binding protein AUF1 suppresses miR-122 biogenesis by down-regulating Dicer1 in hepatocellular carcinoma. Oncotarget 2018, 9, 14815–14827. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Liu, H.; Li, X.; Liu, X. Over-expression of MiR-122 promotes apoptosis of hepatocellular carcinoma via targeting TLR4. Ann. Hepatol. 2019, 18, 869–878. [Google Scholar] [CrossRef]
- Cao, F.; Yin, L.X. miR-122 enhances sensitivity of hepatocellular carcinoma to oxaliplatin via inhibiting MDR1 by targeting Wnt/beta-catenin pathway. Exp. Mol. Pathol. 2019, 106, 34–43. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, J.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, L.; Luo, Y.; Yao, C.; Qian, C. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016, 371, 171–181. [Google Scholar] [CrossRef]
- Frau, M.; Tomasi, M.L.; Simile, M.M.; Demartis, M.I.; Salis, F.; Latte, G.; Calvisi, D.F.; Seddaiu, M.A.; Daino, L.M.E.; Feo, C.F.; et al. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology 2012, 56, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ramani, K.; Sun, Z.; Zee, C.; Grant, E.G.; Yang, H.; Xia, M.; Oh, P.; Ko, K.; Mato, J.M.; et al. Forced Expression of Methionine Adenosyltransferase 1A in Human Hepatoma Cells Suppresses in Vivo Tumorigenicity in Mice. Am. J. Pathol. 2010, 176, 2456–2466. [Google Scholar] [CrossRef]
- Zhu, H.; Berkova, Z.; Mathur, R.; Sehgal, L.; Khashab, T.; Tao, R.-H.; Ao, X.; Feng, L.; Sabichi, A.L.; Blechacz, B.; et al. HuR Suppresses Fas Expression and Correlates with Patient Outcome in Liver Cancer. Mol. Cancer Res. 2015, 13, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, M.; Li, Y.; Shen, M.; Kong, D.; Shao, J.; Ding, H.; Tan, S.; Chen, A.; Zhang, F.; et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 2020, 16, 1482–1505. [Google Scholar] [CrossRef] [PubMed]
- Ji, E.; Kim, C.; Kang, H.; Ahn, S.; Jung, M.; Hong, Y.; Tak, H.; Lee, S.; Kim, W.; Lee, E.K. RNA Binding Protein HuR Promotes Autophagosome Formation by Regulating Expression of Autophagy-Related Proteins 5, 12 and 16 in Human Hepatocellular Carcinoma Cells. Mol. Cell. Biol. 2019, 39, e00508-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Zheng, Y.; Zhang, Y.; Gan, Y.; Zhou, Y.; Liang, H.; Wu, D.; Ge, W.; Deng, J.; Xu, X. Long non-coding RNA AK058003, as a precursor of miR-15a, interacts with HuR to inhibit the expression of gamma-synuclein in hepatocellular carcinoma cells. Oncotarget 2017, 8, 9451–9465. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, S.; Zhang, M.; Ma, X.; Zhang, L.; Wang, Y.; Rong, R.; Ma, J.; Xia, S.; Du, M.; et al. Prostaglandin E2 promotes hepatocellular carcinoma cell invasion through upregulation of YB-1 protein expression. Int. J. Oncol. 2013, 44, 769–780. [Google Scholar] [CrossRef]
- Wen, Z.; Lian, L.; Ding, H.; Hu, Y.; Xiao, Z.; Xiong, K.; Yang, Q. LncRNA ANCR promotes hepatocellular carcinoma metastasis through upregulating HNRNPA1 expression. RNA Biol. 2020, 17, 381–394. [Google Scholar] [CrossRef]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbán, S.; Kuwano, Y.; Pullmann, R.; Martindale, J.L.; Kim, H.H.; Lal, A.; Abdelmohsen, K.; Yang, X.; Dang, Y.; Liu, J.O.; et al. RNA-Binding Proteins HuR and PTB Promote the Translation of Hypoxia-Inducible Factor 1α. Mol. Cell. Biol. 2007, 28, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lou, J.; Lu, M.; Gao, C.; Zhao, S.; Li, B.; Liang, S.; Li, Y.; Li, D.; Liu, M. Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J. 2015, 34, 2671–2685. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; He, L.; Zuo, D.; He, W.; Wang, Y.; Zhang, Y.; Liu, W.; Yuan, Y. Mutual Regulation of MiR-199a-5p and HIF-1α Modulates the Warburg Effect in Hepatocellular Carcinoma. J. Cancer 2017, 8, 940–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei-Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxidative Med. Cell. Longev. 2018, 2018, 7512159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agosti, P.; Sabbà, C.; Mazzocca, A. Emerging metabolic risk factors in hepatocellular carcinoma and their influence on the liver microenvironment. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, H.; Hong, H.; Zhang, Z. MiR-374b re-sensitizes hepatocellular carcinoma cells to sorafenib therapy by antagonizing PKM2-mediated glycolysis pathway. Am. J. Cancer Res. 2019, 9, 765–778. [Google Scholar]
- Christian, K.; Lang, M.; Maurel, P.; Raffalli-Mathieu, F. Interaction of Heterogeneous Nuclear Ribonucleoprotein A1 with Cytochrome P450 2A6 mRNA: Implications for Post-Transcriptional Regulation of the CYP2A6 Gene. Mol. Pharmacol. 2004, 65, 1405–1414. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Rosas, M.G.; Chávez, E.; Velasco-Loyden, G.; Domínguez-López, M.; Martínez-Pérez, L.; De Sánchez, V.C. Diminished S-adenosylmethionine biosynthesis and its metabolism in a model of hepatocellular carcinoma is recuperated by an adenosine derivative. Cancer Biol. Ther. 2019, 21, 81–94. [Google Scholar] [CrossRef]
- Jin, W.-J.; Chen, C.-F.; Liao, H.-Y.; Gong, L.-L.; Yuan, X.-H.; Zhao, B.-B.; Zhang, D.; Feng, X.; Liu, J.-J.; Wang, Y.; et al. Downregulation of the AU-Rich RNA-Binding Protein ZFP36 in Chronic HBV Patients: Implications for Anti-Inflammatory Therapy. PLoS ONE 2012, 7, e33356. [Google Scholar] [CrossRef] [Green Version]
- Roskams, T.; Kojiro, M. Pathology of Early Hepatocellular Carcinoma: Conventional and Molecular Diagnosis. Semin. Liver Dis. 2010, 30, 17–25. [Google Scholar] [CrossRef]
- Toyota, K.; Murakami, Y.; Kondo, N.; Uemura, K.; Nakagawa, N.; Takahashi, S.; Sueda, T. Cytoplasmic Hu-Antigen R (HuR) Expression is Associated with Poor Survival in Patients with Surgically Resected Cholangiocarcinoma Treated with Adjuvant Gemcitabine-Based Chemotherapy. Ann. Surg. Oncol. 2018, 25, 1202–1210. [Google Scholar] [CrossRef]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collén, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef] [Green Version]
- Ung, T.H.; Madsen, H.J.; Hellwinkel, J.E.; Lencioni, A.M.; Graner, M.W. Exosome proteomics reveals transcriptional regulator proteins with potential to mediate downstream pathways. Cancer Sci. 2014, 105, 1384–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, J.A.; Alam, J.M.; Mahmood, S.R.; Baig, M.; Shaheen, R.; Sultana, I.; Waheed, A. Hepatocellular carcinoma (HCC) and diagnostic significance of A-fetoprotein (AFP). J. Ayub Med. Coll. 2010, 21, 72–75. [Google Scholar]
- Schultz, C.W.; Preet, R.; Dhir, T.; Dixon, D.A.; Brody, J.R. Understanding and targeting the disease-related RNA binding protein human antigen R (HuR). Wiley Interdiscip. Rev. RNA 2020, 11, 1581. [Google Scholar] [CrossRef]
- Wu, X.; Lan, L.; Wilson, D.M.; Marquez, R.T.; Tsao, W.-C.; Gao, P.; Roy, A.; Turner, B.A.; McDonald, P.; A Tunge, J.; et al. Identification and Validation of Novel Small Molecule Disruptors of HuR-mRNA Interaction. ACS Chem. Biol. 2015, 10, 1476–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hjelmeland, A.B.; Nabors, L.B.; King, P. Anti-cancer effects of the HuR inhibitor, MS-444, in malignant glioma cells. Cancer Biol. Ther. 2019, 20, 979–988. [Google Scholar] [CrossRef]
- Blanco, F.F.; Preet, R.; Aguado, A.; Vishwakarma, V.; Stevens, L.E.; Vyas, A.; Padhye, S.; Xu, L.; Weir, S.J.; Anant, S.; et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget 2016, 7, 74043–74058. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, S.; Mazroui, R.; Dallaire, P.; Chittur, S.; Tenenbaum, S.A.; Radzioch, D.; Marette, A.; Gallouzi, I.E. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol. Cell Biol. 2005, 25, 6533–6545. [Google Scholar] [CrossRef] [Green Version]
- Yashiro, T.; Nanmoku, M.; Shimizu, M.; Inoue, J.; Sato, R. 5-Aminoimidazole-4-carboxamide ribonucleoside stabilizes low density lipoprotein receptor mRNA in hepatocytes via ERK-dependent HuR binding to an AU-rich element. Atherosclerosis 2013, 226, 95–101. [Google Scholar] [CrossRef]
- Doller, A.; Badawi, A.; Schmid, T.; Brauß, T.; Pleli, T.; Zu Heringdorf, D.M.; Piiper, A.; Pfeilschifter, J.; Eberhardt, W. The cytoskeletal inhibitors latrunculin A and blebbistatin exert antitumorigenic properties in human hepatocellular carcinoma cells by interfering with intracellular HuR trafficking. Exp. Cell Res. 2015, 330, 66–80. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chung, T.-W.; Choi, H.-J.; Lee, C.H.; Eun, J.S.; Han, Y.T.; Choi, J.Y.; Kim, S.-Y.; Han, C.; Jeong, H.-S.; et al. A novel cantharidin analog N-Benzylcantharidinamide reduces the expression of MMP-9 and invasive potentials of Hep3B via inhibiting cytosolic translocation of HuR. Biochem. Biophys. Res. Commun. 2014, 447, 371–377. [Google Scholar] [CrossRef]
- Muralidharan, R.; Mehta, M.; Ahmed, R.; Roy, S.; Xu, L.; Aubé, J.; Chen, A.; Zhao, Y.D.; Herman, T.; Ramesh, R.; et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci. Rep. 2017, 7, 9694. [Google Scholar] [CrossRef]
- Lee, Y.W.; Liu, K.W.; Yeung, J.H. Reactive oxygen species-mediated kinase activation by dihydrotanshinone in tanshinones-induced apoptosis in HepG2 cells. Cancer Lett. 2009, 285, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Ye, D.; Zhang, X.; Jiang, Z.; Xiao, B.; Guo, J. Inhibitory effect ofHuRgene small interfering RNA segment on laryngeal carcinoma Hep-2 cell growth. J. Laryngol. Otol. 2010, 124, 1183–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan, R.; Babu, A.; Amreddy, N.; Basalingappa, K.; Mehta, M.; Chen, A.; Zhao, Y.D.; Kompella, U.B.; Munshi, A.; Ramesh, R. Folate receptor-targeted nanoparticle delivery of HuR-RNAi suppresses lung cancer cell proliferation and migration. J. Nanobiotechnol. 2016, 14, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Micalizzi, D.; Truesdell, S.S.; Bukhari, S.I.A.; Boukhali, M.; Lombardi-Story, J.; Kato, Y.; Choo, M.-K.; Dey-Guha, I.; Ji, F.; et al. A post-transcriptional program of chemoresistance by AU-rich elements and TTP in quiescent leukemic cells. Genome Biol. 2020, 21, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-R.; Jin, H.; Kim, W.-T.; Kim, W.-J.; Kim, S.Z.; Leem, S.-H.; Kim, S.M. Tristetraprolin activation by resveratrol inhibits the proliferation and metastasis of colorectal cancer cells. Int. J. Oncol. 2018, 53, 1269–1278. [Google Scholar] [CrossRef]
- Cao, H.; A Kelly, M.; Kari, F.; Dawson, H.D.; Urban, J.F.; Coves, S.; Roussel, A.-M.; Anderson, R.A. Green tea increases anti-inflammatory tristetraprolin and decreases pro-inflammatory tumor necrosis factor mRNA levels in rats. J. Inflamm. 2007, 4, 1. [Google Scholar] [CrossRef]
- Li, S.; Wang, W.; Ding, H.; Xu, H.; Zhao, Q.; Li, J.; Li, H.; Xia, W.; Su, X.; Chen, Y.; et al. Aptamer BC15 Against Heterogeneous Nuclear Ribonucleoprotein A1 Has Potential Value in Diagnosis and Therapy of Hepatocarcinoma. Nucleic Acid Ther. 2012, 22, 391–398. [Google Scholar] [CrossRef]
- Cho, Y.; Bin Lee, Y.; Lee, J.-H.; Lee, D.H.; Cho, E.J.; Yu, S.J.; Kim, Y.J.; Kim, J.I.; Im, J.H.; Lee, J.H.; et al. Modified AS1411 Aptamer Suppresses Hepatocellular Carcinoma by Up-Regulating Galectin-14. PLoS ONE 2016, 11, e0160822. [Google Scholar] [CrossRef]
- Le Trinh, T.; Zhu, G.; Xiao, X.; Puszyk, W.; Sefah, K.; Wu, Q.; Tan, W.; Liu, C. A Synthetic Aptamer-Drug Adduct for Targeted Liver Cancer Therapy. PLoS ONE 2015, 10, e0136673. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AUBP | HCC | Target | Interaction | Model/Cell Type | Process | PMID |
|---|---|---|---|---|---|---|
| HuR | Up | ACTA1 | Direct | HSCs | Fibrosis, migration | 22576182 |
| Col1a1 | Unclear | HSCs | Fibrosis | 22576182 | ||
| MMP9 | Direct | HSCs | Fibrosis, migration | 22576182 | ||
| MCP1 | Direct | HSCs | Liver Fibrosis | 22576182 | ||
| CCND1 | Direct | HSCs | Fibrosis, proliferation | 22576182 | ||
| Tgfb | Direct | HSCs | Fibrosis | 22576182 | ||
| CCNB1 | Direct | HSCs | Fibrosis, proliferation | 22576182 | ||
| S1PR3 | Direct | BMSCs | Fibrosis, migration | 27543493 | ||
| CNR1 | Direct | BMDMs | BMDM recruitment | 31495934 | ||
| HMOX1 | Unclear | Hepatocytes | Oxidative stress | 31879990 | ||
| TP53 | Direct | HCC cells | Proliferation, apoptosis | 18519672 | ||
| FAS | Direct | HCC cells | Apoptosis | 25678597 | ||
| MAT2A | Direct | HCC cells | Proliferation, differentiation | 20102719 | ||
| Ptn-dt | Direct | HCC cells | Cell proliferation | 30643194 | ||
| BECN1 | Direct | HSCs | Autophagy/Ferroptosis | 30081711 | ||
| ATG5 | Direct | HCC cells | Autophagy | 30602494 | ||
| ATG12 | Direct | HCC cells | Autophagy | 30602494 | ||
| ATG16 | Direct | HCC cells | Autophagy | 30602494 | ||
| HIF1A | Direct | HCC cells | Hypoxia/Angiogenesis | 30083257 | ||
| IL6 | Direct | HCC cells | Inflammation | 28646470 | ||
| CCNA2 | Direct | Hepatocytes | Proliferation | 16831604 | ||
| CCND1 | Direct | Hepatocytes | Proliferation | 16831604 | ||
| ETS1 | Direct | HCC cells | Proliferation | 31438961 | ||
| UFC1 | Direct | HCC cells | Proliferation | 25449213 | ||
| HAUSP | Direct | HCC cells | P53 signaling | 20815019 | ||
| HHIP | Direct | HCC cells | Proliferation, apoptosis, migration | 31604528 | ||
| TGIF | Direct | HCC cells | Apoptosis | 21649584 | ||
| CTNNB1 | Direct | HCC cells | Apoptosis | 25449213 | ||
| TTP | Down | PTGS2 | Unclear | Hepatocytes | Inflammation | 26876787 |
| DUSP1 | Unclear | Hepatocytes | Proliferation | 26876787 | ||
| FOS | Unclear | Hepatocytes | Proliferation | 26876787 | ||
| MYC | Direct | Hepatocytes | Proliferation | 20038433 | ||
| MMP2 | Direct | HSCs | Migration | 30226813 | ||
| TNFa | Direct | HSCs | Inflammation | 30226813 | ||
| FGF21 | Direct | Hepatocytes | Insulin sensitivity | 29997282 | ||
| VEGFA | Unclear | HCC cells | Angiogenesis | 31717307 | ||
| ATG16L1 | Direct | HSCs | Autophagy, ferroptosis | 31679460 | ||
| IER3 | Unclear | HCC cells | Apoptosis | 27619201 | ||
| AKT1 | Unclear | HCC cells | Apoptosis | 27619201 | ||
| AUF1 | Up | MAT1A | Direct | HCC cells | Proliferation, differentiation | 20102719 |
| Dicer | Unclear | HCC cells | MicroRNA maturation | 29599909 | ||
| TIA1 | Up | IGFBP3 | Direct | HCC cells | Apoptosis | 20599318 |
| MFF | Direct | HCC cells | Mitochondrial metabolism | 27612012 | ||
| YB-1 | Up | SMAD2 | Direct | HSCs | TGFβ signaling | 28153731 |
| EGFR | Unclear | HCC cells | Proliferation | 24378923 | ||
| CCNA | Unclear | HCC cells | Proliferation | 27911878 | ||
| CCNB1 | Unclear | HCC cells | Proliferation | 27911878 | ||
| PCNA | Unclear | HCC cells | Proliferation | 27911878 | ||
| TP53 | Unclear | HCC cells | Proliferation, apoptosis | 27911878 | ||
| SNAI1 | Direct | HCC cells | Migration | 28428004 | ||
| KSRP | Unclear | Per2 | Unclear | Hepatocytes | Metabolism | 25514904 |
| PTBP1 | Unclear | CCND3 | Direct | HCC cells | Proliferation | 31301177 |
| RBM38 | Down | MDM2 | Direct | HCC cells | Proliferation, apoptosis | 30176896 |
| RBM3 | Down | SCD-circRNA 2 | Direct | HCC cells | Proliferation | 31235426 |
| CUGBP1 | Up | CCNB1 | Unclear | HCC cells | Proliferation | 24502807 |
| CCND1 | Unclear | HCC cells | Proliferation | 24502807 | ||
| HNRNPA1 | Up | PKM2 | Unclear | HCC cells | Metabolism | 31106002 |
| CYP2A6 | Direct | HCC cells | Metabolism | 15155834 | ||
| ILF3 | Up | PARP1 | Direct | HCC cells | Apoptosis | 28487110 |
| CCNE1 | Direct | HCC cells | Proliferation | 25399696 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolicka, D.; Sobolewski, C.; Correia de Sousa, M.; Gjorgjieva, M.; Foti, M. mRNA Post-Transcriptional Regulation by AU-Rich Element-Binding Proteins in Liver Inflammation and Cancer. Int. J. Mol. Sci. 2020, 21, 6648. https://doi.org/10.3390/ijms21186648
Dolicka D, Sobolewski C, Correia de Sousa M, Gjorgjieva M, Foti M. mRNA Post-Transcriptional Regulation by AU-Rich Element-Binding Proteins in Liver Inflammation and Cancer. International Journal of Molecular Sciences. 2020; 21(18):6648. https://doi.org/10.3390/ijms21186648
Chicago/Turabian StyleDolicka, Dobrochna, Cyril Sobolewski, Marta Correia de Sousa, Monika Gjorgjieva, and Michelangelo Foti. 2020. "mRNA Post-Transcriptional Regulation by AU-Rich Element-Binding Proteins in Liver Inflammation and Cancer" International Journal of Molecular Sciences 21, no. 18: 6648. https://doi.org/10.3390/ijms21186648