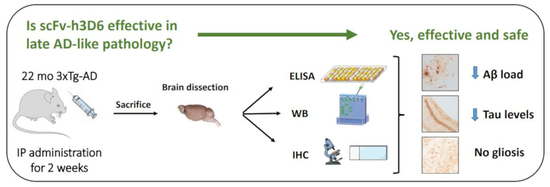
Both Amyloid-β Peptide and Tau Protein Are Affected by an Anti-Amyloid-β Antibody Fragment in Elderly 3xTg-AD Mice


Abstract
:
1. Introduction
2. Results
2.1. ScFv-h3D6 Accumulates in the Hippocampus
2.2. ScFv-h3D6 Reduces the Area of 6E10 Staining
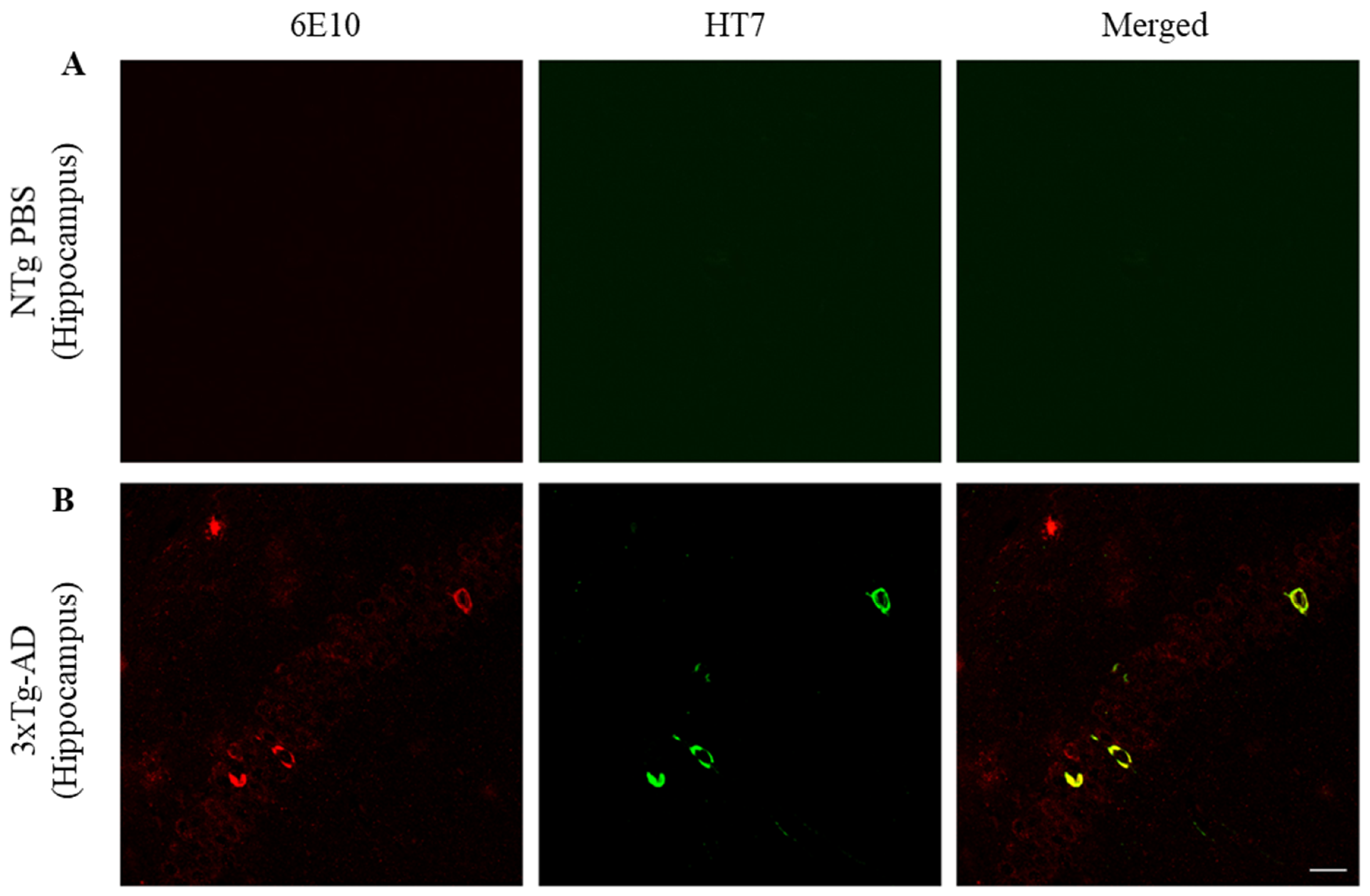
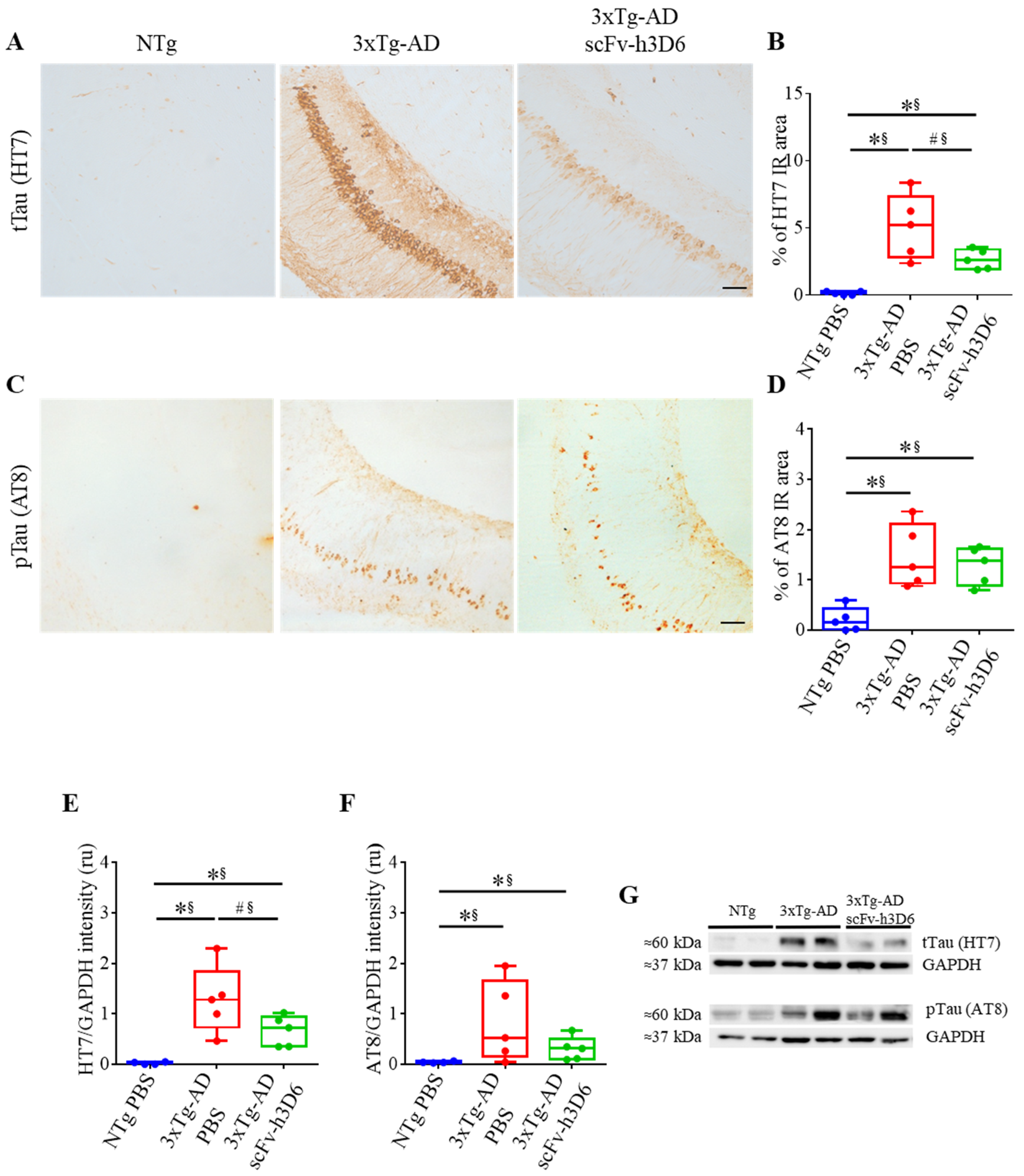
2.3. ScFv-h3D6 Decreases Total Tau Levels
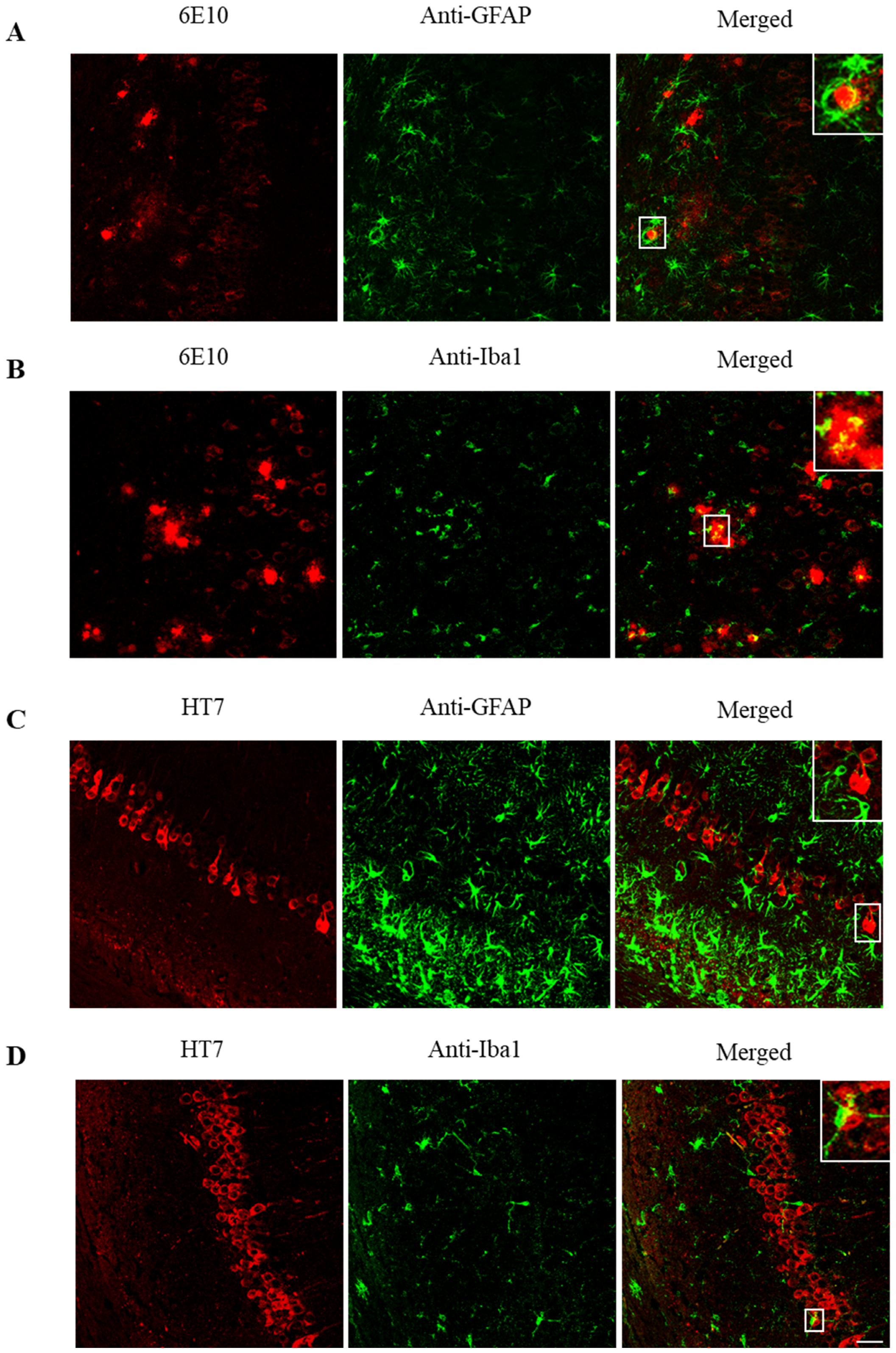
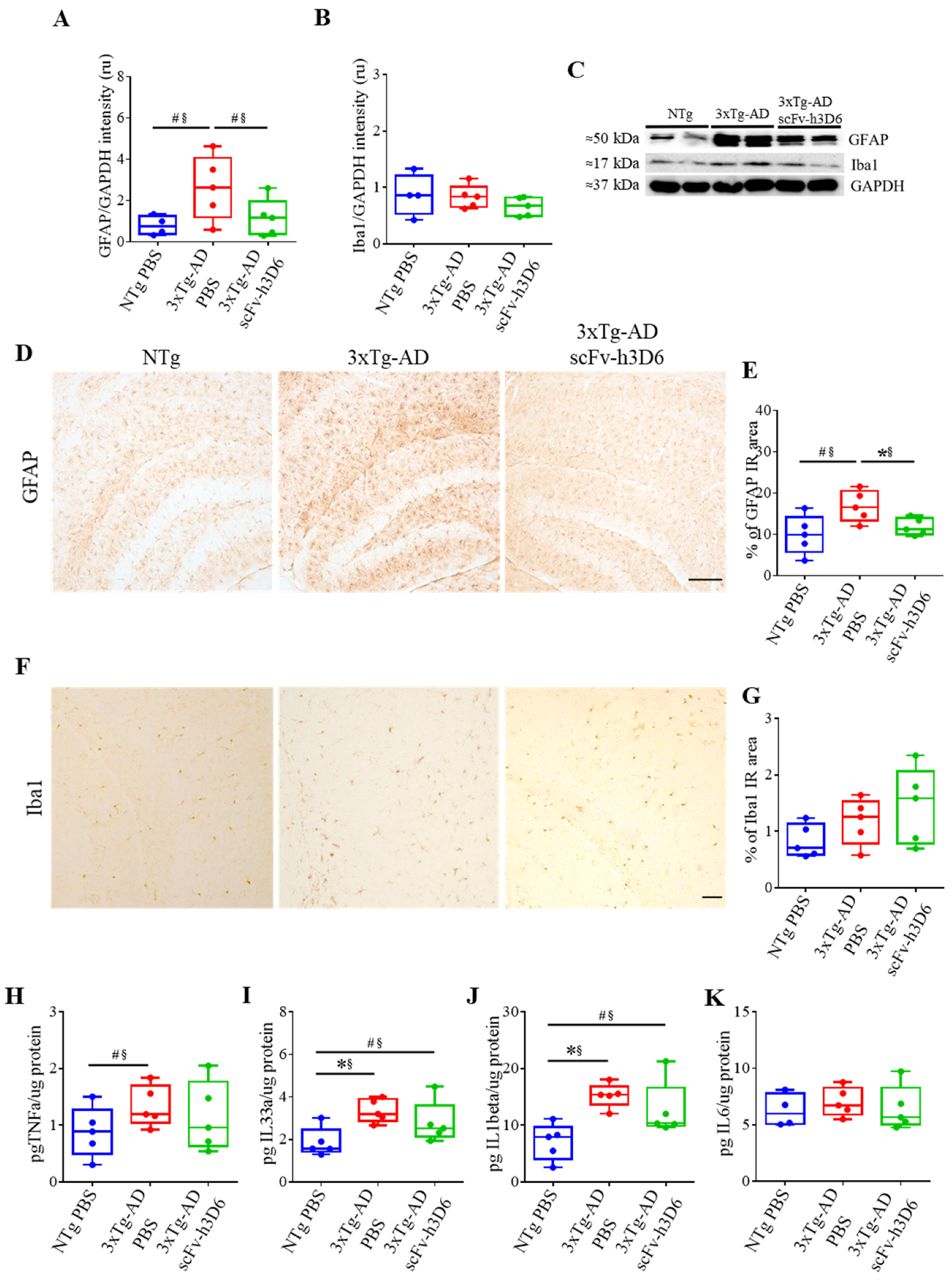
2.4. ScFv-h3D6 Slightly Ameliorates the Pro-Inflammatory Status
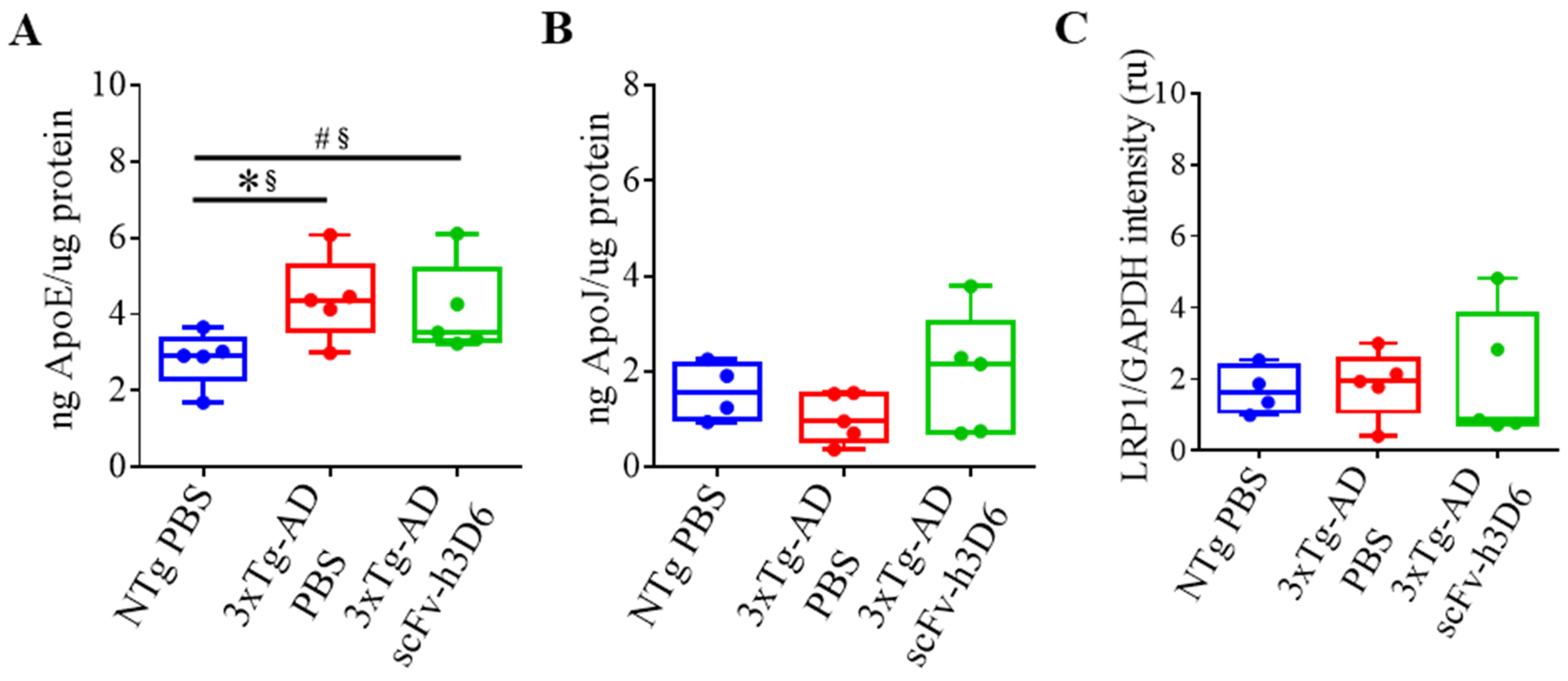
2.5. Effect of ScFv-h3D6 on Apolipoproteins’ Levels
3. Discussion
4. Materials and Methods
4.1. ScFv-h3D6 Production
4.2. Animals
4.3. Experimental Design and Statistical Analysis
4.4. Sample Collection and Processing
4.5. Immunohistochemistry
4.6. Immunofluorescence
4.7. Image Capture and Processing
4.8. Western Blotting
4.9. Enzyme-Linked Immunosorbent Assays (ELISAs)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Significance Statement
Abbreviations
| AD | Alzheimer’s disease |
| apoE | apolipoprotein E |
| apoJ | apolipoprotein J |
| ARIAs | amyloid-related imaging abnormalities |
| Aβ | amyloid-β |
| BPSD | behavioral and psychological symptoms of dementia |
| CA | cornus amonis |
| GFAP | glial fibrillary acidic protein |
| Iba1 | ionized calcium-binding adaptor molecule 1 |
| LRP1 | lipoprotein receptor-related protein 1 |
| mAbs | monoclonal antibodies |
| NFT | neurofibrillary tangle |
| NTg | non-transgenic mice |
| scFv | single-chain variable fragment |
| 3xTg-AD | triple-transgenic mouse model of AD |
References
- Dementia Statistics. Alzheimer’s Disease International. Available online: https://www.alz.co.uk/research/statistics (accessed on 9 August 2020).
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Álvarez-Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia 2017, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Comas-Herrera, A.; Knapp, M.; Guerchet, M.; Karagiannidou, M. World Alzheimer Report 2016 Improving Healthcare for People Living with Dementia. Coverage, Quality and Costs Now and in the Future; Alzheimer’s Disease International: London, UK, 2016. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panza, F.; Lozupone, M.; Seripa, D.; Imbimbo, B.P. Amyloid-β immunotherapy for alzheimer disease: Is it now a long shot? Ann. Neurol. 2019, 85, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Welikovitch, L.A.; Do Carmo, S.; Maglóczky, Z.; Szocsics, P.; Lőke, J.; Freund, T.; Cuello, A.C. Evidence of intraneuronal Aβ accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol. 2018, 136, 901–917. [Google Scholar] [CrossRef]
- Esquerda-Canals, G.; Martí-Clúa, J.; Roda, A.R.; Villegas, S. An Intracellular Amyloid-β/AβPP Epitope Correlates with Neurodegeneration in those Neuronal Populations Early Involved in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 59, 1079–1096. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Villegas, S. Aβ-Immunotherapeutic strategies: A wide range of approaches for Alzheimer’s disease treatment. Expert Rev. Mol. Med. 2016, 18, e13. [Google Scholar] [CrossRef]
- Panza, F.; Frisardi, V.; Imbimbo, B.P.; Seripa, D.; Solfrizzi, V.; Pilotto, A. Monoclonal antibodies against β-amyloid (Aβ) for the treatment of Alzheimer’s disease: The Aβ target at a crossroads. Expert Opin. Biol. Ther. 2011, 11, 679–686. [Google Scholar] [CrossRef]
- Schroeter, S.; Khan, K.; Barbour, R.; Doan, M.; Chen, M.; Guido, T.; Gill, D.; Basi, G.; Schenk, D.; Seubert, P.; et al. Immunotherapy Reduces Vascular Amyloid-β in PDAPP Mice. J. Neurosci. 2008, 28, 6787–6793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, J.P.; Stavenhagen, J.B.; Teeling, J.L. New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer’s Disease. Front. Neurosci. 2014, 8, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Argany, M.; Rivera-Hernández, G.; Martí, J.; Villegas, S. An anti-Aβ (amyloid β) single-chain variable fragment prevents amyloid fibril formation and cytotoxicity by withdrawing Aβ oligomers from the amyloid pathway. Biochem. J. 2011, 437, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Hernández, G.; Marin-Argany, M.; Blasco-Moreno, B.; Bonet, J.; Oliva, B.; Villegas, S. Elongation of the C-terminal domain of an anti-amyloid β single-chain variable fragment increases its thermodynamic stability and decreases its aggregation tendency. MAbs 2013, 5, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Montoliu-Gaya, L.; Esquerda-Canals, G.; Bronsoms, S.; Villegas, S. Production of an anti-Aβ antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic effect. PLoS ONE 2017, 12, 8. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Murciano-Calles, J.; Martinez, J.C.; Villegas, S. Towards the improvement in stability of an anti-Aβ single-chain variable fragment, scFv-h3D6, as a way to enhance its therapeutic potential. Amyloid 2017, 24, 167–175. [Google Scholar] [CrossRef]
- Esquerda-Canals, G.; Marti, J.; Rivera-Hernández, G.; Giménez-Llort, L.; Villegas, S. Loss of deep cerebellar nuclei neurons in the 3xTg-AD mice and protection by an anti-amyloid β antibody fragment. MAbs 2013, 5, 660–664. [Google Scholar] [CrossRef]
- Giménez-Llort, L.; Rivera-Hernández, G.; Marín-Argany, M.; Sánchez-Quesada, J.L.; Villegas, S. Early intervention in the 3xTg-AD mice with an amyloid β-antibody fragment ameliorates first hallmarks of Alzheimer disease. MAbs 2013, 5, 665–864. [Google Scholar] [CrossRef] [Green Version]
- Esquerda-Canals, G.; Roda, A.R.; Martí-Clúa, J.; Montoliu-Gaya, L.; Rivera-Hernández, G.; Villegas, S. Treatment with scFv-h3D6 Prevented Neuronal Loss and Improved Spatial Memory in Young 3xTg-AD Mice by Reducing the Intracellular Amyloid-β Burden. J. Alzheimer’s Dis. 2019, 70, 1069–1091. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Güell-Bosch, J.; Esquerda-Canals, G.; Roda, A.R.; Serra-Mir, G.; Lope-Piedrafita, S.; Sánchez-Quesada, J.L.; Villegas, S. Differential effects of apoE and apoJ mimetic peptides on the action of an anti-Aβ scFv in 3xTg-AD mice. Biochem. Pharmacol. 2018, 155, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Güell-Bosch, J.; Lope-Piedrafita, S.; Esquerda-Canals, G.; Montoliu-Gaya, L.; Villegas, S. Progression of Alzheimer’s disease and effect of scFv-h3D6 immunotherapy in the 3xTg-AD mouse model: An in vivo longitudinal study using Magnetic Resonance Imaging and Spectroscopy. NMR Biomed. 2020, 33, e4263. [Google Scholar] [CrossRef] [PubMed]
- Esquerda-Canals, G.; Martí-Clúa, J.; Villegas, S. Pharmacokinetic parameters and mechanism of action of an efficient anti-Aβ single chain antibody fragment. PLoS ONE 2019, 14, e0217793. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.L.; Tung, Y.C.; Liu, F.; Gong, C.X.; Iqbal, K. Tau passive immunization inhibits not only tau but also Aβ pathology. Alzheimer’s Res. Ther. 2017, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhamron, S.; Rozenstein-Tsalkovich, L.; Nitzan, K.; Abramsky, O.; Rosenmann, H. Phos-tau peptide immunization of amyloid-tg-mice reduced non-mutant phos-tau pathology, improved cognition and reduced amyloid plaques. Exp. Neurol. 2018, 303, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Syvänen, S.; Hultqvist, G.; Gustavsson, T.; Gumucio, A.; Laudon, H.; Söderberg, L.; Ingelsson, M.; Lannfelt, L.; Sehlin, D. Efficient clearance of Aβ protofibrils in AβPP-transgenic mice treated with a brain-penetrating bifunctional antibody. Alzheimer’s Res. Ther. 2018, 10, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Zetterberg, H.; Rinne, J.O.; Salloway, S.; Wei, J.; Black, R.; Grundman, M.; Liu, E.; AAB-001 201/202 Investigators. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1002–1010. [Google Scholar] [CrossRef]
- Czirr, E.; Wyss-Coray, T. The immunology of neurodegeneration. J. Clin. Investig. 2012, 122, 1156–1163. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.; Salloway, S.; Brooks, D.J.; Tampieri, D.; Barakos, J.; Fox, N.C.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: A retrospective analysis. Lancet Neurol. 2012, 11, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Kerchner, G.A.; Boxer, A.L. Bapineuzumab. Expert Opin. Biol. Ther. 2010, 10, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Montoliu-Gaya, L.; Mulder, S.D.; Veerhuis, R.; Villegas, S. Effects of an Aβ-antibody fragment on Aβ aggregation and astrocytic uptake are modulated by apolipoprotein E and J mimetic peptides. PLoS ONE 2017, 12, e0188191. [Google Scholar] [CrossRef] [Green Version]
- Montoliu-Gaya, L.; Mulder, S.D.; Herrebout, M.A.C.; Baayen, J.C.; Villegas, S.; Veerhuis, R. Aβ-oligomer uptake and the resulting inflammatory response in adult human astrocytes are precluded by an anti-Aβ single chain variable fragment in combination with an apoE mimetic peptide. Mol. Cell. Neurosci. 2018, 89, 49–59. [Google Scholar] [CrossRef]
- Youmans, K.L.; Tai, L.M.; Kanekiyo, T.; Stine, W.B.; Michon, S.C.; Nwabuisi-Heath, E.; Manelli, A.M.; Fu, Y.; Riordan, S.; Eimer, W.A.; et al. Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol. Neurodegener. 2012, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Rasool, S.; Martinez-Coria, H.; Wu, J.W.; Laferla, F.; Glabe, C.G. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Aβ deposition and tau pathology in 3xTg-AD mice. J. Neurochem. 2013, 126, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, E.A.; Bouter, Y.; Richard, B.C.; Lannfelt, L.; Ingelsson, M.; Paetau, A.; Verkkoniemi-Ahola, A.; Wirths, O.; Bayer, T.A. Abundance of Aβ5-x like immunoreactivity in transgenic 5XFAD, APP/PS1KI and 3xTG mice, sporadic and familial Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Zhao, X.; Lu, W.; Zhang, Q.; Hua, Z. Intracellular A? and its Pathological Role in Alzheimer’s Disease: Lessons from Cellular to Animal Models. Curr. Alzheimer’s Res. 2016, 13, 621–630. [Google Scholar] [CrossRef]
- Pardridge, W.M. CSF, blood-brain barrier, and brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Limone, A.; Napolitano, F.; Cerchia, C.; Parisi, S.; Minopoli, G.; Montuori, N.; Lavecchia, A.; Sarnataro, D. APP maturation and intracellular localization are controlled by a specific inhibitor of 37/67 kda laminin-1 receptor in neuronal cells. Int. J. Mol. Sci. 2020, 21, 1738. [Google Scholar] [CrossRef] [Green Version]
- Brion, J.-P. Neurofibrillary Tangles and Alzheimer’s Disease. Eur. Neurol. 1998, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Muñoz, M.J.; Gerson, J.; Castillo-Carranza, D.L. Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 464. [Google Scholar] [CrossRef]
- Sarnataro, D. Attempt to untangle the prion-like misfolding mechanism for neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosal, K.; Fan, Q.; Dawson, H.N.; Pimplikar, S.W. Tau Protein Mediates APP Intracellular Domain (AICD)-Induced Alzheimer’s-Like Pathological Features in Mice. PLoS ONE 2016, 11, e0159435. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, R.N.; Fu, M.; Lambracht-Washington, D. Active full-length DNA Aβ42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimer’s Res. Ther. 2018, 10, 115. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Panza, F.; Frisardi, V.; Imbimbo, B.P.; D’Onofrio, G.; Pietrarossa, G.; Seripa, D.; Pilotto, A.; Solfrizzi, V. Bapineuzumab: Anti-β-amyloid monoclonal antibodies for the treatment of Alzheimer’s disease. Immunotherapy 2010, 2, 767–782. [Google Scholar] [CrossRef]
- Robert, R.; Wark, K.L. Engineered antibody approaches for Alzheimer’s disease immunotherapy. Arch. Biochem. Biophys. 2012, 526, 132–138. [Google Scholar] [CrossRef]
- Gadani, S.P.; Walsh, J.T.; Lukens, J.R.; Kipnis, J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron 2015, 87, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Waldkirch, S.; Meyer-Luehmann, M. The Role of Glial Cells and Synapse Loss in Mouse Models of Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 473. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J. Neuroinflamm. 2005, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Möller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.Y.; Benveniste, E.N. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J. Immunol. 1990, 144, 2999–3007. [Google Scholar] [PubMed]
- Fu, A.K.Y.; Hung, K.-W.; Yuen, M.Y.F.; Zhou, X.; Mak, D.S.Y.; Chan, I.C.W.; Cheung, T.H.; Zhang, B.; Fu, W.-Y.; Liew, F.Y.; et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc. Natl. Acad. Sci. USA 2016, 113, E2705–E2713. [Google Scholar] [CrossRef] [Green Version]
- Carlock, C.; Wu, J.; Shim, J.; Moreno-Gonzalez, I.; Pitcher, M.R.; Hicks, J.; Suzuki, A.; Iwata, J.; Quevado, J.; Lou, Y. Interleukin33 deficiency causes tau abnormality and neurodegeneration with Alzheimer-like symptoms in aged mice. Transl. Psychiatry 2017, 7, e1164. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Thangavel, R.; Kempuraj, D.; Yang, E.; Zaheer, S.; Zaheer, A. Alzheimer’s Disease: Evidence for the Expression of Interleukin-33 and Its Receptor ST2 in the Brain. J. Alzheimer’s Dis. 2014, 40, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudry, E.; Klickstein, J.; Cannavo, C.; Jackson, R.; Muzikansky, A.; Gandhi, S.; Urick, D.; Sargent, T.; Wrobleski, L.; Roe, A.D.; et al. Opposing Roles of apolipoprotein E in aging and neurodegeneration. Life Sci. Alliance 2019, 2, e201900325. [Google Scholar] [CrossRef] [PubMed]
- D’argenio, V.; Sarnataro, D. New insights into the molecular bases of familial alzheimer’s disease. J. Pers. Med. 2020, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y. Apolipoprotein E and Alzheimer disease. In Proceedings of the Neurology; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2006; Volume 66. [Google Scholar]
- Herline, K.; Prelli, F.; Mehta, P.; MacMurray, C.; Goñi, F.; Wisniewski, T. Immunotherapy to improve cognition and reduce pathological species in an Alzheimer’s disease mouse model. Alzheimer’s Res. Ther. 2018, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Jackson Laboratory. B6;129-Tg(APPSwe,tauP301L)1Lfa Psen1/Mmjax. Available online: https://www.jax.org/strain/004807 (accessed on 26 October 2018).
- Carroll, J.C.; Rosario, E.R.; Kreimer, S.; Villamagna, A.; Gentzschein, E.; Stanczyk, F.Z.; Pike, C.J. Sex differences in β-amyloid accumulation in 3xTg-AD mice: Role of neonatal sex steroid hormone exposure. Brain Res. 2010, 1366, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ Causes the Onset of Early Alzheimer’s Disease-Related Cognitive Deficits in Transgenic Mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Kane, A.E.; Shin, S.; Wong, A.A.; Fertan, E.; Faustova, N.S.; Howlett, S.E.; Brown, R.E. Sex differences in healthspan predict lifespan in the 3xTg-AD Mouse model of Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Amrhein, V.; Greenland, S.; McShane, B. Scientists rise up against statistical significance. Nature 2019, 567, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Wendt, H.W. Dealing with a common problem in Social science: A simplified rank-biserial coefficient of correlation based on the U statistic. Eur. J. Soc. Psychol. 1972, 2, 463–465. [Google Scholar] [CrossRef]
- Franklin, K.B.J.; Paxinos, G. Paxinos and Franklin’s The Mouse Brain in Stereotaxic Coordinates, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2012; ISBN 9780123910578. [Google Scholar]








{kind=link}
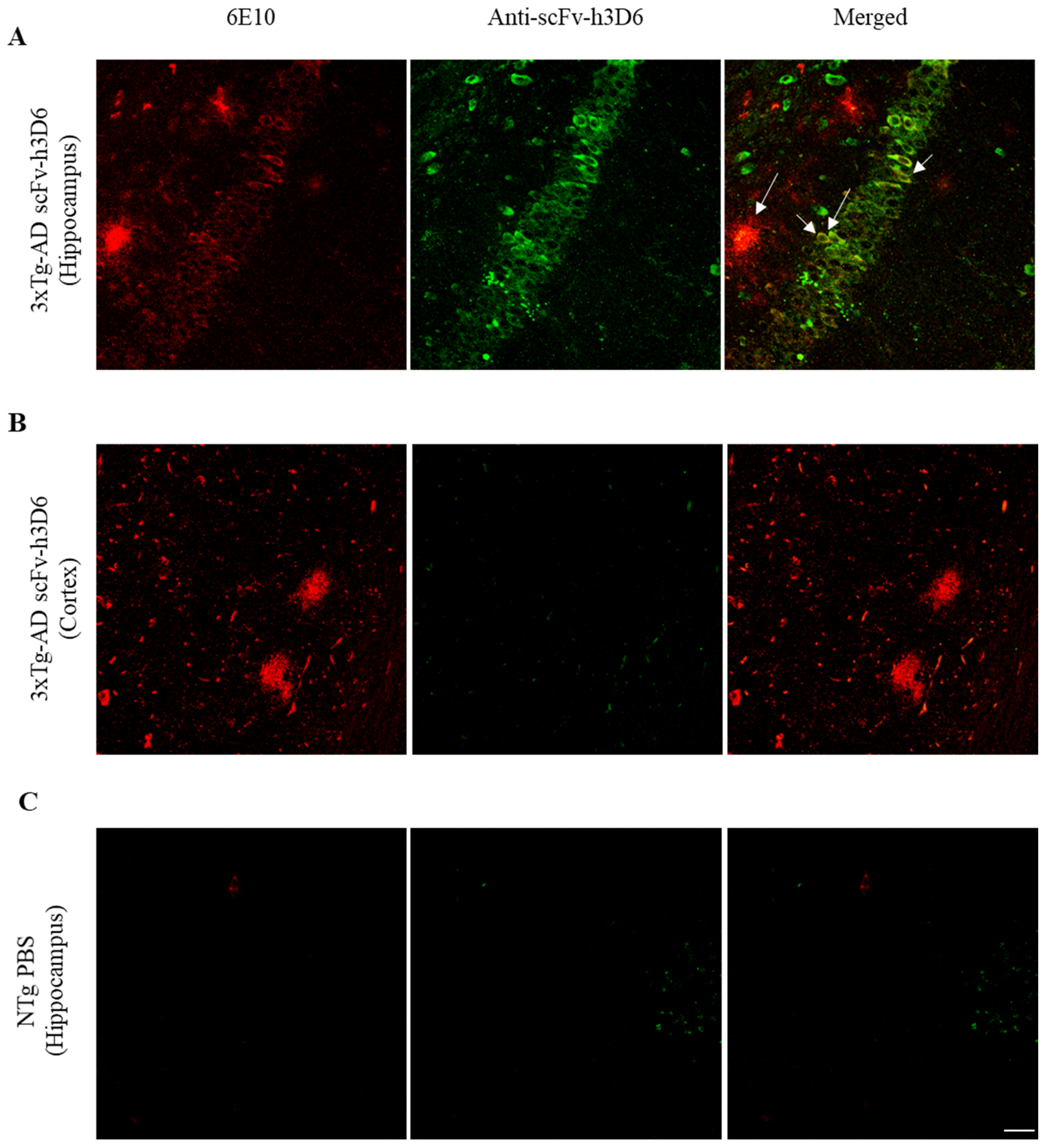{kind=link}
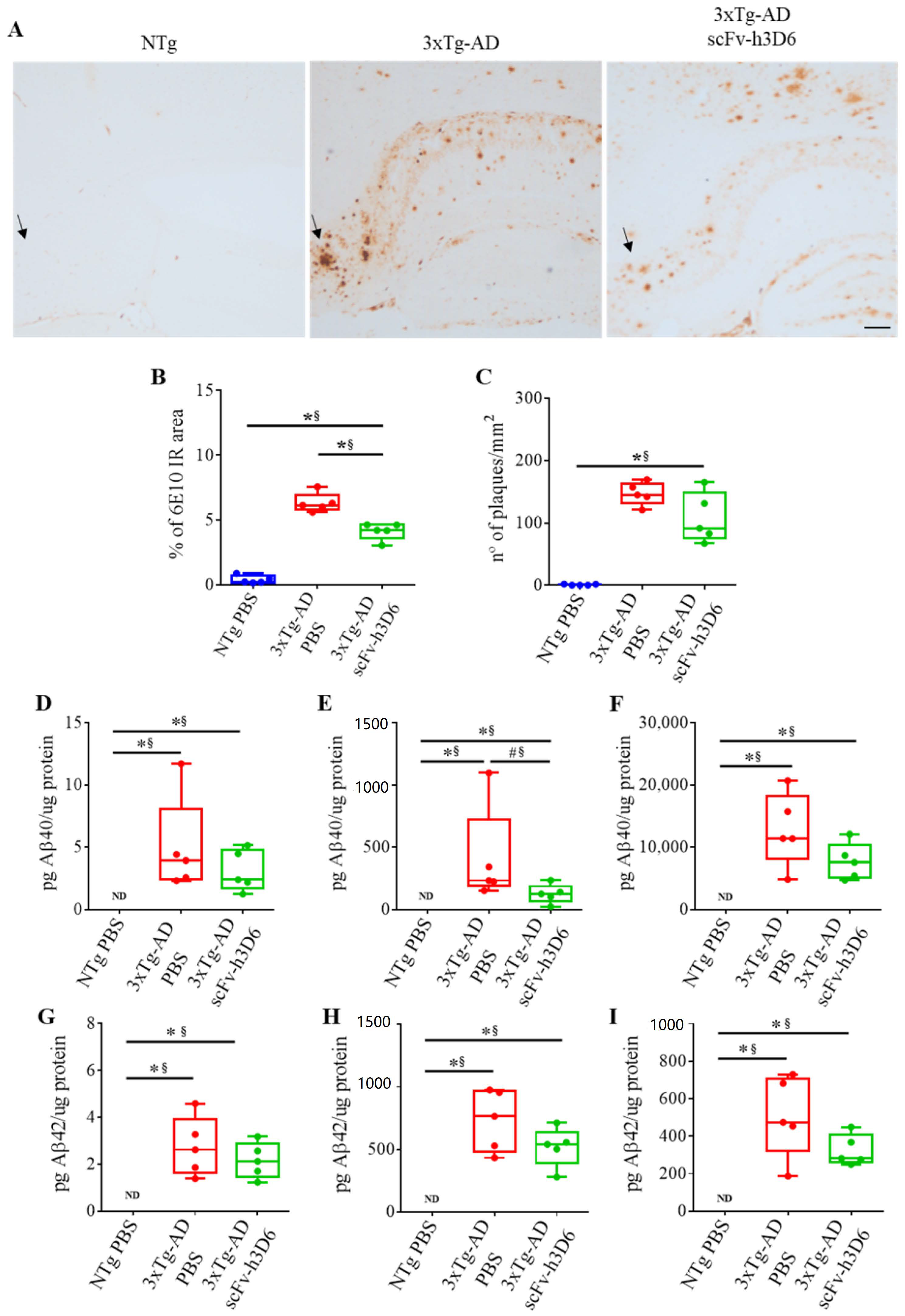{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NTg vs. 3xTg-AD | 3xTg-AD (−) vs. 3xTg-AD (+) | |||
|---|---|---|---|---|
| p-Value | r-Coeff. | p-Value | r-Coeff. | |
| % of 6e10 IR area (Figure 2B) | 0.0079 ** | 1 ** | 0.0079 ** | 1 ** |
| n° of plaques/mm2 (Figure 2C) | 0.0079 ** | 1 ** | 0.1508 | 0.60 |
| TBS-fraction pg Aβ40/µg protein (Figure 2D) | 0.0079 ** | 1 ** | 0.5317 | 0.28 |
| SDS-fraction pg Aβ40/µg protein (Figure 2E) | 0.0079 ** | 1 ** | 0.0556 # | 0.76 * |
| FA-fraction pg Aβ40/µg protein (Figure 2F) | 0.0079 ** | 1 ** | 0.2222 | 0.52 |
| TBS-fraction pg Aβ42/µg protein (Figure 2G) | 0.0079 ** | 1 ** | 0.4127 | 0.36 |
| SDS-fraction pg Aβ42/µg protein (Figure 2H) | 0.0079 ** | 1 ** | 0.3095 | 0.44 |
| FA-fraction pg Aβ42/µg protein (Figure 2I) | 0.0079 ** | 1 ** | 0.1508 | 0.60 |
| % of HT7 IR area (Figure 4B) | 0.0079 ** | 1 ** | 0.0952 # | 0.68* |
| % of AT8 IR area (Figure 4D) | 0.0079 ** | 1 ** | 0.8016 | 0.12 |
| HT7/GAPDH intensity (U.A.) (Figure 4E) | 0.0079 ** | 1 ** | 0.0952 # | 0.68 * |
| AT8/GAPDH intensity (U.A.) (Figure 4F) | 0.0159 * | 0.92 ** | 0.5317 | 0.28 |
| GFAP/GAPDH intensity (U.A.) (Figure 6A) | 0.0556 # | 0.76 * | 0.0952 # | 0.68 * |
| Iba1/GAPDH intensity (U.A.) (Figure 6B) | 0.7857 | 0.12 | 0.1508 | 0.60 |
| % of GFAP IR area (Figure 6E) | 0.0556 # | 0.76 * | 0.0317 * | 0.84 * |
| % of Iba1 IR area (Figure 6G) | 0.2222 | 0.52 | 0.5317 | 0.28 |
| pg TNFα/µg protein (Figure 6H) | 0.0952 # | 0.68 * | 0.5317 | 0.28 |
| pg IL33α/µg protein (Figure 6i) | 0.0159 * | 0.92 ** | 0.2222 | 0.52 |
| pg IL1β/µg protein (Figure 6J) | 0.0079 ** | 1 ** | 0.1508 | 0.60 |
| pg IL6/µg protein (Figure 6K) | 0.6667 | 0.20 | 0.5317 | 0.28 |
| ng ApoE/µg protein (Figure 7A) | 0.0317 * | 0.84 * | 0.6667 | 0.20 |
| ng ApoJ/µg protein (Figure 7B) | 0.3889 | 0.36 | 0.2222 | 0.52 |
| LRP1/GAPDH intensity (U.A.) (Figure 7C) | 0.5159 | 0.28 | 0.9444 | 0.04 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roda, A.R.; Montoliu-Gaya, L.; Serra-Mir, G.; Villegas, S. Both Amyloid-β Peptide and Tau Protein Are Affected by an Anti-Amyloid-β Antibody Fragment in Elderly 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 6630. https://doi.org/10.3390/ijms21186630
Roda AR, Montoliu-Gaya L, Serra-Mir G, Villegas S. Both Amyloid-β Peptide and Tau Protein Are Affected by an Anti-Amyloid-β Antibody Fragment in Elderly 3xTg-AD Mice. International Journal of Molecular Sciences. 2020; 21(18):6630. https://doi.org/10.3390/ijms21186630
Chicago/Turabian StyleRoda, Alejandro R., Laia Montoliu-Gaya, Gabriel Serra-Mir, and Sandra Villegas. 2020. "Both Amyloid-β Peptide and Tau Protein Are Affected by an Anti-Amyloid-β Antibody Fragment in Elderly 3xTg-AD Mice" International Journal of Molecular Sciences 21, no. 18: 6630. https://doi.org/10.3390/ijms21186630