Isolation and Characterization of the Novel Bacteriophage AXL3 against Stenotrophomonas maltophilia



Abstract
:1. Introduction
2. Results and Discussion
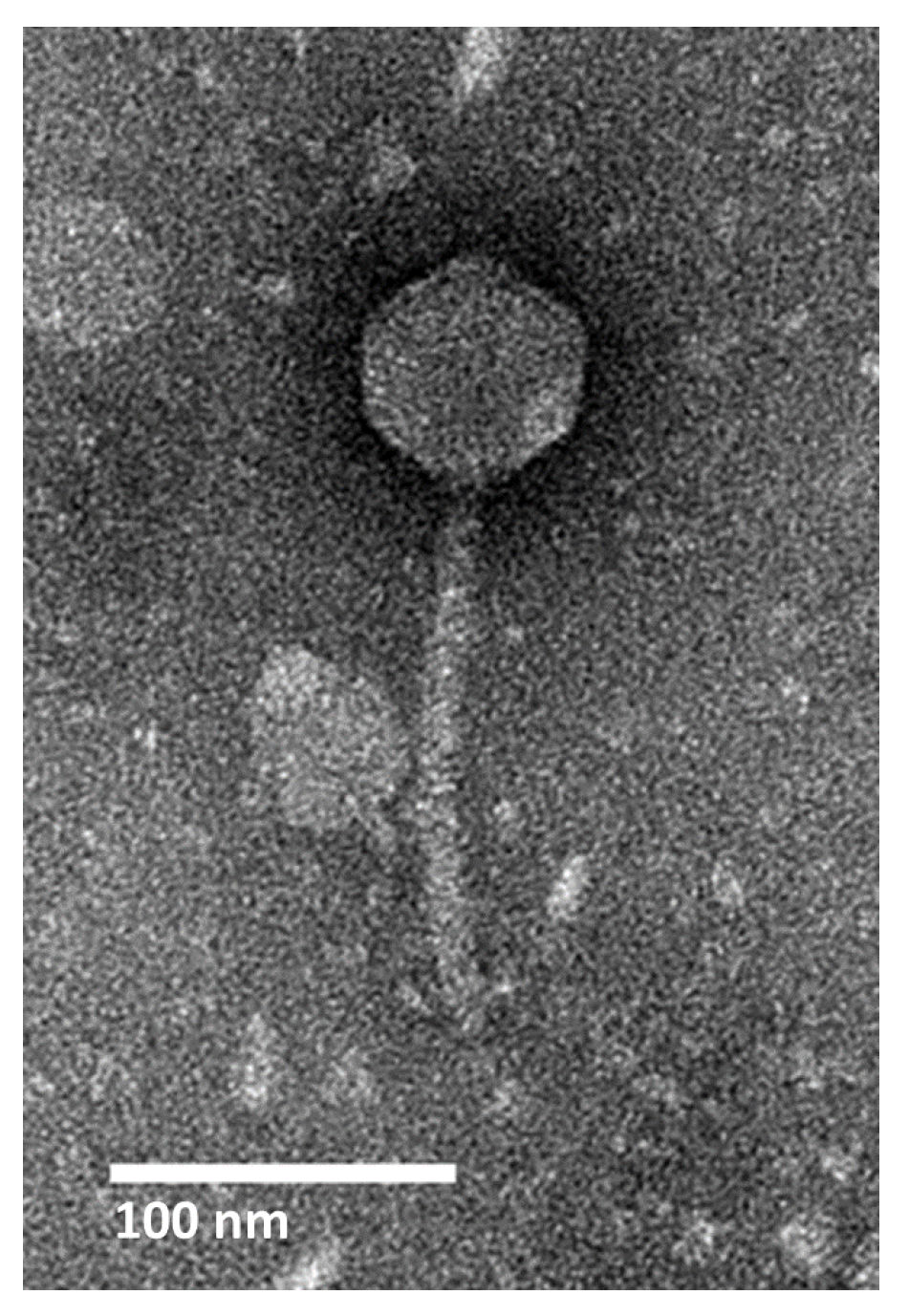
2.1. Isolation, Morphology, and Host Range
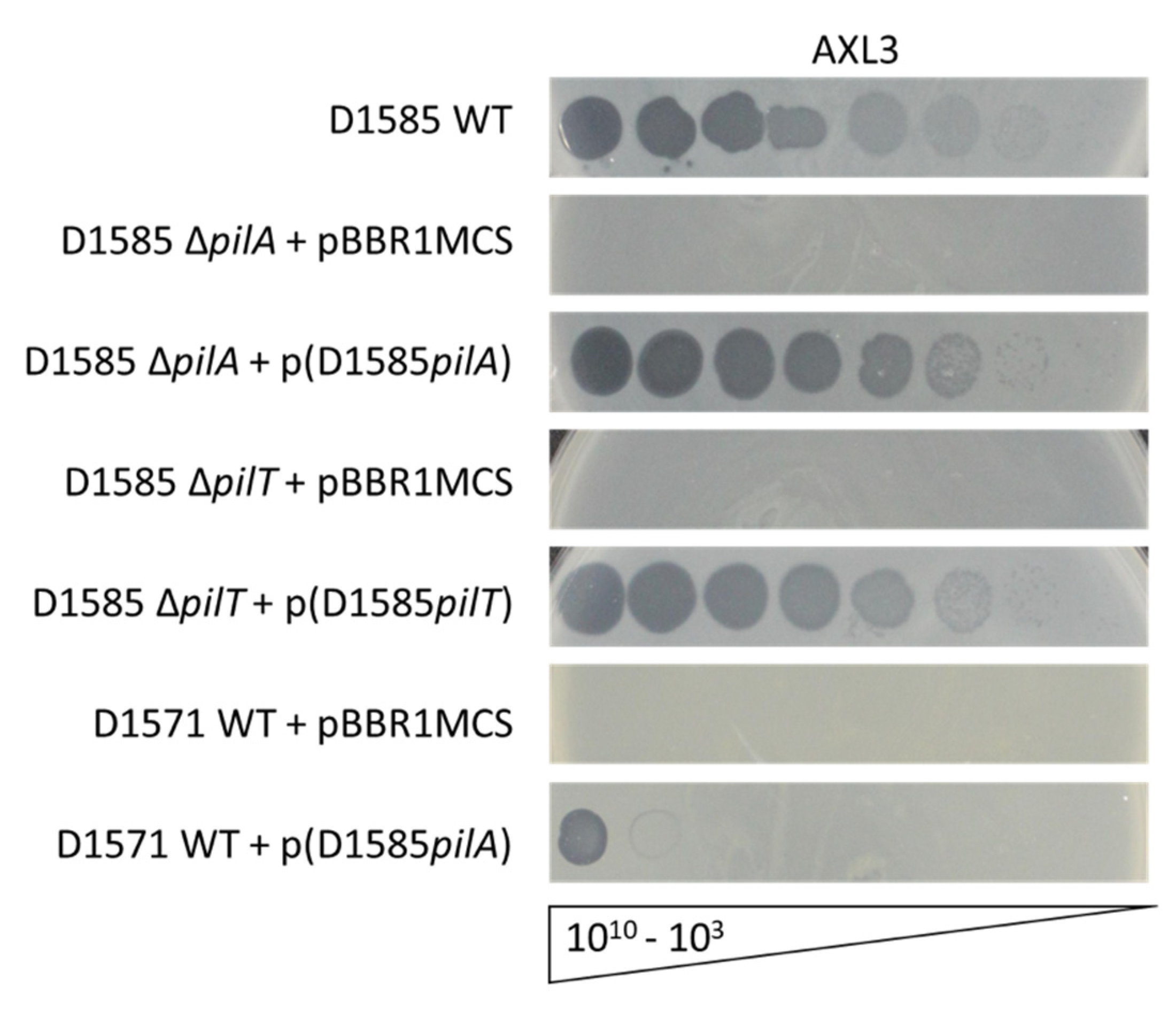
2.2. Receptor Identification
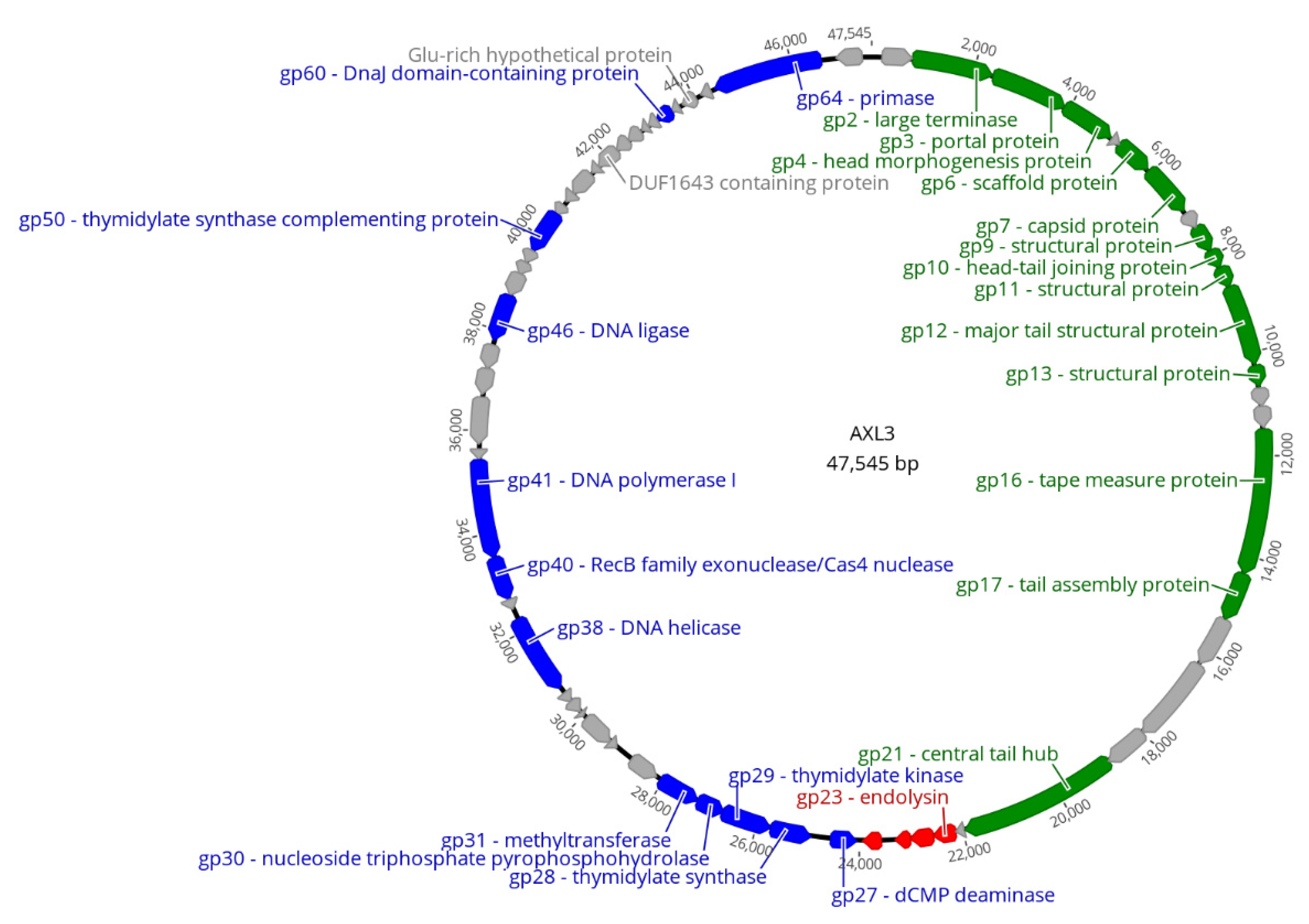
2.3. Genomic Characterization
2.3.1. DNA Replication and Repair Module
2.3.2. Virion Morphogenesis Module
2.3.3. Lysis Module
3. Conclusions
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Phage Isolation, Propagation, and Host Range
4.3. Electron Microscopy
4.4. Determining Phage Lifecycle
4.5. One-Step Growth Curve
4.6. Phage Plaquing Assays
4.7. Phage DNA Isolation, RFLP Analysis, and Sequencing
4.8. Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berg, G.; Eberl, L.; Hartmann, A. The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 2005, 7, 1673–1685. [Google Scholar] [CrossRef]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Brooke, J.S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Martinez, J.L. Friends or foes: Can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front. Microbiol. 2015, 6, 241. [Google Scholar] [CrossRef] [PubMed]
- Trifonova, A.; Strateva, T. Stenotrophomonas maltophilia—A low-grade pathogen with numerous virulence factors. Infect. Dis. 2019, 51, 168–178. [Google Scholar] [CrossRef]
- Waters, V.; Atenafu, E.G.; Lu, A.; Yau, Y.; Tullis, E.; Ratjen, F. Chronic Stenotrophomonas maltophilia infection and mortality or lung transplantation in cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [Green Version]
- Roach, D.R.; Debarbieux, L. Phage therapy: Awakening a sleeping giant. Emerg. Top. Life Sci. 2017, 1, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.L.; Lynch, K.H.; Stothard, P.; Dennis, J.J. The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.L.; Stothard, P.; Dennis, J.J. The isolation and characterization of Stenotrophomonas maltophilia T4-like bacteriophage DLP6. PLoS ONE 2017, 12, e0173341. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.L.; Dennis, J.J. Complete genome sequence of temperate Stenotrophomonas maltophilia bacteriophage DLP5. Genome Announc. 2018, 6, e00073-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, D.L.; McCutcheon, J.G.; Stothard, P.; Dennis, J.J. Novel Stenotrophomonas maltophilia temperate phage DLP4 is capable of lysogenic conversion. BMC Genom. 2019, 20, 300. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.L.; McCutcheon, J.G.; Dennis, J.J. Characterization of novel broad-host-range bacteriophage DLP3 specific to Stenotrophomonas maltophilia as a potential therapeutic agent. Front. Microbiol. 2020, 11, 1358. [Google Scholar] [CrossRef]
- Chang, H.C.; Chen, C.R.; Lin, J.W.; Shen, G.H.; Chang, K.M.; Tseng, Y.H.; Weng, S.F. Isolation and characterization of novel giant Stenotrophomonas maltophilia phage ΦSMA5. Appl. Environ. Microbiol. 2005, 71, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.R.; Lin, C.H.; Lin, J.W.; Chang, C.I.; Tseng, Y.H.; Weng, S.F. Characterization of a novel T4-type Stenotrophomonas maltophilia virulent phage Smp14. Arch. Microbiol. 2007, 188, 191–197. [Google Scholar] [CrossRef]
- García, P.; Monjardín, C.; Martín, R.; Madera, C.; Soberón, N.; Garcia, E.; Meana, Á.; Suárez, J.E. Isolation of new Stenotrophomonas bacteriophages and genomic characterization of temperate phage S1. Appl. Environ. Microbiol. 2008, 74, 7552–7560. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Huang, Y.; Mi, Z.; Yin, X.; Wang, L.; Fan, H.; Zhang, Z.; An, X.; Chen, J.; Tong, Y. Complete genome sequence of IME13, a Stenotrophomonas maltophilia bacteriophage with large burst size and unique plaque polymorphism. J. Virol. 2012, 86, 11392–11393. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Fan, H.; Pei, G.; Fan, H.; Zhang, Z.; An, X.; Mi, Z.; Shi, T.; Tong, Y. Complete genome sequence of IME15, the first T7-Like bacteriophage lytic to pan-antibiotic-resistant Stenotrophomonas maltophilia. J. Virol. 2012, 86, 13839–13840. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, X. Biological characteristics of phage SM1 for Stenotrophomonas maltophilia and its effect in animal infection model. Zhejiang Da Xue Xue Bao Yi Xue Ban 2013, 42, 331–336. [Google Scholar]
- Hayden, A.; Martinez, N.; Moreland, R.; Liu, M.; Gonzalez, C.F.; Gill, J.J.; Ramsey, J. Complete genome sequence of Stenotrophomonas phage Pokken. Microbiol. Resour. Announc. 2019, 8, e01095-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garza, K.D.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J.; Leavitt, J. Complete genome sequence of Stenotrophomonas phage Mendera. Microbiol. Resour. Announc. 2020, 9, e01411-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez, A.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J. Complete genome sequence of Stenotrophomonas maltophilia podophage Ponderosa. Microbiol. Resour. Announc. 2019, 8, e01032-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicary, A.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J.; Leavitt, J. Complete genome sequence of Stenotrophomonas maltophilia myophage Moby. Microbiol. Resour. Announc. 2020, 9, e01422-19. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, H.W.; Eisenstark, A. The present state of phage taxonomy. Intervirology 1974, 3, 201–219. [Google Scholar] [CrossRef]
- McCutcheon, J.G.; Peters, D.L.; Dennis, J.J. Identification and characterization of type IV pili as the cellular receptor of broad host range Stenotrophomonas maltophilia bacteriophages DLP1 and DLP2. Viruses 2018, 10, 338. [Google Scholar] [CrossRef] [Green Version]
- Burrows, L.L. Weapons of mass retraction. Mol. Microbiol. 2005, 57, 878–888. [Google Scholar] [CrossRef]
- Dunger, G.; Llontop, E.; Guzzo, C.R.; Farah, C.S. The Xanthomonas type IV pilus. Curr. Opin. Microbiol. 2016, 30, 88–97. [Google Scholar] [CrossRef]
- Craig, L.; Forest, K.T.; Maier, B. Type IV pili: Dynamics, biophysics and functional consequences. Nat. Rev. Microbiol. 2019, 17, 429–440. [Google Scholar] [CrossRef]
- Giltner, C.L.; Rana, N.; Lunardo, M.N.; Hussain, A.Q.; Burrows, L.L. Evolutionary and functional diversity of the Pseudomonas type IVa pilin island. Environ. Microbiol. 2011, 13, 250–264. [Google Scholar] [CrossRef]
- Burdman, S.; Bahar, O.; Parker, J.K.; de la Fuente, L. Involvement of type IV pili in pathogenicity of plant pathogenic bacteria. Genes 2011, 2, 706–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedramfar, A.; Maal, K.B.; Mirdamadian, S.H. Phage therapy of corrosion-producing bacterium Stenotrophomonas maltophilia using isolated lytic bacteriophages. Anti-Corros. Methods Mater. 2017, 64, 607–612. [Google Scholar] [CrossRef]
- Leon, M.; Bastias, R. Virulence reduction in bacteriophage resistant bacteria. Front. Microbiol. 2015, 6, 343. [Google Scholar] [CrossRef] [Green Version]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Faux, N.G.; Bottomley, S.P.; Lesk, A.M.; Irving, J.A.; Morrison, J.R.; De La Banda, M.G.; Whisstock, J.C. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 2005, 15, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.C.; Wang, A.H.J. Structural D/E-rich repeats play multiple roles especially in gene regulation through DNA/RNA mimicry. Mol. Biosyst. 2015, 11, 2144–2151. [Google Scholar] [CrossRef]
- Maley, F.; Maley, G.F. A tale of two enzymes, deoxycytidylate deaminase and thymidylate synthase. Prog. Nucleic Acid Res. Mol. Biol. 1990, 39, 49–80. [Google Scholar] [CrossRef]
- Hardy, L.W.; Finer-Moore, J.S.; Montfort, W.R.; Jones, M.O.; Santi, D.V.; Stroud, R.M. Atomic structure of thymidylate synthase: Target for rational drug design. Science 1987, 235, 448–455. [Google Scholar] [CrossRef]
- Mathews, I.I.; Deacon, A.M.; Canaves, J.M.; McMullan, D.; Lesley, S.A.; Agarwalla, S.; Kuhn, P. Functional analysis of substrate and cofactor complex structures of a thymidylate synthase-complementing protein. Structure 2003, 11, 677–690. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Lücker, S.; Albertsen, M.; Kitzinger, K.; Herbold, C.; Spieck, E.; Nielsen, H.; Wagner, M.; Daims, H. Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira. Proc. Natl. Acad. Sci. USA 2015, 112, 11371–11376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kasciukovic, T.; White, M.F. The CRISPR associated protein Cas4 is a 5′ to 3′ DNA exonuclease with an iron-sulfur cluster. PLoS ONE 2012, 7, e47232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooton, S.P.T.; Connerton, I.F. Campylobacter jejuni acquire new host-derived CRISPR spacers when in association with bacteriophages harboring a CRISPR-like Cas4 protein. Front. Microbiol. 2015, 5, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudaiberdiev, S.; Shmakov, S.; Wolf, Y.I.; Terns, M.P.; Makarova, K.S.; Koonin, E.V. Phylogenomics of Cas4 family nucleases. BMC Evol. Biol. 2017, 17, 232. [Google Scholar] [CrossRef] [Green Version]
- Patil, P.P.; Midha, S.; Kumar, S.; Patil, P.B. Genome sequence of type strains of genus Stenotrophomonas. Front. Microbiol 2016, 7, 309. [Google Scholar] [CrossRef] [Green Version]
- Crippen, C.S.; Lee, Y.-J.; Hutinet, G.; Shajahan, A.; Sacher, J.C.; Azadi, P.; de Crécy-Lagard, V.; Weigele, P.R.; Szymanski, C.M. Deoxyinosine and 7-deaza-2-deoxyguanosine as carriers of genetic information in the DNA of Campylobacter viruses. J. Virol. 2019, 93, e01111-19. [Google Scholar] [CrossRef]
- Tsai, R.; Corrêa, I.R.; Xu, M.Y.; Xu, S.Y. Restriction and modification of deoxyarchaeosine (dG+)-containing phage 9 g DNA. Sci. Rep. 2017, 7, 8348. [Google Scholar] [CrossRef]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Ahern, S.J.; Das, M.; Bhowmick, T.S.; Young, R.; Gonzalez, C.F. Characterization of novel virulent broad-host-range phages of Xylella fastidiosa and Xanthomonas. J. Bacteriol. 2014, 196, 459–471. [Google Scholar] [CrossRef]
- Loessner, M.J.; Wendlinger, G.; Scherer, S. Heterogeneous endolysins in Listeria monocytogenes bacteriophages: A new class of enzymes and evidence for conserved holin genes within the siphoviral lysis cassettes. Mol. Microbiol. 1995, 16, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Mikoulinskaia, G.V.; Odinokova, I.V.; Zimin, A.A.; Lysanskaya, V.Y.; Feofanov, S.A.; Stepnaya, O.A. Identification and characterization of the metal ion-dependent l-alanoyl-d-glutamate peptidase encoded by bacteriophage T5. FEBS J. 2009, 276, 7329–7342. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Kovach, M.E.; Phillips, R.W.; Elzer, P.H.; Roop, R.M., 2nd; Peterson, K.M. pBBR1MCS: A broad-host-range cloning vector. Biotechniques 1994, 16, 800–802. [Google Scholar] [PubMed]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Charcterization, and Interactions; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: New Delhi, India, 2009; pp. 69–76. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Carlson, K. Working with bacteriophages: Common techniques and methodological approaches. In Bacteriophages: Biology and Applications; Kutter, E.M., Sulakvelidze, A., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 437–494. [Google Scholar]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 December 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juncker, A.S.; Willenbrock, H.; Von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction: Methods and Protocols, Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| S. maltophilia Strain | Efficiency of Plating |
|---|---|
| 101 | − |
| 102 | − |
| 103 | − |
| 152 | − |
| 155 | − |
| 174 | − |
| 176 | − |
| 213 | ++++ |
| 214 | − |
| 217 | − |
| 218 | − |
| 219 | − |
| 230 | − |
| 236 | − |
| 242 | − |
| 249 | − |
| 278 | − |
| 280 | + |
| 282 | − |
| 287 | − |
| 446 | − |
| 667 | − |
| D1585 a | ++++ |
| D1571 a | − |
| D1614 a | − |
| D1576 a | + |
| D1568 a | +++ |
| SMDP92 | − |
| ATCC13637 | − |
| CDS | Coding Region | Strand | Length (AA) | Putative Function | Hit | Species | Coverage (%) | E-Value | Identity (%) | Accession |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 204–806 | + | 200 | hypothetical protein | hypothetical protein | Burkholderia sp. SRS-W-2-2016 | 40 | 3.00 × 10−4 | 35.56 | WP_143752284.1 |
| 2 | 806–2368 | + | 520 | large terminase | terminase large subunit | Microcystic phage Me-ZS1 | 95 | 0 | 59.96 | AZF88145.1 |
| 3 | 2379–3887 | + | 502 | portal protein | putative portal protein | Prokaryotic dsDNA virus sp. | 97 | 2.00 × 10−141 | 47.7 | QDP56576.1 |
| 4 | 3891–4952 | + | 353 | head morphogenesis protein | hypothetical protein | uncultured Caudovirales phage | 98 | 1.00 × 10−91 | 44.17 | ASN68636.1 |
| 5 | 4998–5156 | + | 52 | hypothetical protein | - | |||||
| 6 | 5177–5911 | + | 244 | scaffold protein | scaffold protein | Pseudomonas phage vB_PaeS_SCH_Ab26 | 99 | 6.00 × 10−41 | 41.3 | YP_009044344.1 |
| 7 | 5946–6962 | + | 338 | capsid protein | capsid protein | Salmonella phage PMBT28 | 98 | 8.00 × 10−124 | 51.46 | AUZ95497.1 |
| 8 | 7041–7340 | + | 99 | hypothetical protein | - | |||||
| 9 | 7347–7871 | + | 174 | structural protein | structural protein | Achromobacter phage vB_Ade_ART | 99 | 1.00 × 10−32 | 45.56 | AYD82587.1 |
| 10 | 7876–8259 | + | 127 | head-tail joining protein | hypothetical protein | Pseudomonas phage PaSz-4 | 100 | 4.00 × 10−14 | 32.56 | QAX99460.1 |
| 11 | 8256–8687 | + | 143 | structural protein | putative structural protein | Pseudomonas phage PAE1 | 94 | 3.00 × 10−33 | 41.55 | YP_009215709.1 |
| 12 | 8692–10,245 | + | 517 | major tail structural protein | major tail structural protein | Pseudomonas phage NP1 | 98 | 0 | 65.36 | YP_009285827.1 |
| 13 | 10,272–10,691 | + | 139 | structural protein | putative structural protein | Pseudomonas phage NP1 | 100 | 4.00 × 10−56 | 57.55 | YP_009285828.1 |
| 14 | 10,712–11,071 | + | 119 | hypothetical protein | hypothetical protein | Pseudomonas phage LKO4 | 73 | 9.00 × 10−27 | 54.02 | YP_009601866.1 |
| 15 | 11,068–11,496 | + | 142 | hypothetical protein | hypothetical protein | Bordetella phage FP1 | 98 | 2.00 × 10−37 | 50 | YP_009794086.1 |
| 16 | 11,501–14,341 | + | 946 | tape measure protein | hypothetical protein | Pseudomonas virus M6 | 98 | 0 | 47.08 | YP_001294539.1 |
| 17 | 14,352–15,296 | + | 314 | tail assembly protein | tail assembly protein | Xylella phage Salvo | 99 | 6.00 × 10−133 | 58.72 | YP_009639180.1 |
| 18 | 15,296–16,279 | + | 327 | hypothetical protein | hypothetical protein | Stenotrophomonas phage vB_SmaS-DLP_1 | 98 | 1.00 × 10−72 | 42.04 | AKI28800.1 |
| 19 | 16,282–18,018 | + | 578 | hypothetical protein | hypothetical protein | Pseudomonas phage vB_PaeS_C1 | 91 | 2.00 × 10−115 | 36.84 | AVJ48097.1 |
| 20 | 18,015–18,854 | + | 279 | hypothetical protein | hypothetical protein | Burkholderia phage vB_BceS_KL1 | 99 | 3.00 × 10−80 | 45.68 | YP_006560776.1 |
| 21 | 18,858–21,968 | + | 1036 | central tail hub | tail protein | Burkholderia phage vB_BceS_KL1 | 77 | 0 | 46.73 | YP_006560777.1 |
| 22 | 21,965–22,138 | + | 57 | hypothetical protein | - | |||||
| 23 | 22,135–22,584 | + | 149 | endolysin | endolysin | Xanthomonas phage Xp15 | 98 | 2.00 × 10−53 | 53.9 | YP_239293.1 |
| 24 | 22,590–23,021 | + | 143 | hypothetical protein | - | |||||
| 25 | 23,018–23,323 | + | 101 | hypothetical protein | hypothetical protein | Sinobacteraceae bacterium | 92 | 1.00 × 10−9 | 40 | TXH02718.1 |
| 26 | 23,579–23,941 | + | 120 | hypothetical protein | - | |||||
| 27 | 24,050–24,571 | − | 173 | dCMP deaminase | dCMP deaminase | uncultured Mediterranean phage uvMED | 91 | 6.00 × 10−43 | 47.17 | YP_009778145.1 |
| 28 | 24,942–25,751 | − | 269 | thymidylate synthase | thymidylate synthase | Pelagibacter phage HTVC200P | 87 | 6.00 × 10−65 | 44.49 | AXH71582.1 |
| 29 | 25,732–26,715 | − | 327 | thymidylate kinase | thymidylate kinase | Caudovirales sp. ctOwN3 | 93 | 3.00 × 10−33 | 33.98 | QGH72154.1 |
| 30 | 26,696–27,250 | − | 184 | nucleoside triphosphate pyrophosphohydrolase | NTP-Ppase | Caudovirales sp. ctOwN3 | 69 | 5.00 × 10−32 | 46.21 | QGH72159.1 |
| 31 | 27,260–28,084 | − | 274 | methyltransferase | methyltransferase domain-containing protein | Nitrospira cf. moscoviensis SBR1015 | 94 | 5.00 × 10−133 | 73.08 | WP_087475488.1 |
| 32 | 28,162–28,761 | − | 199 | hypothetical protein | hypothetical protein | Nitrospira cf. moscoviensis SBR1015 | 74 | 3.00 × 10−26 | 44.13 | WP_087475489.1 |
| 33 | 29,100–29,273 | − | 57 | hypothetical protein | - | |||||
| 34 | 29,273–29,920 | − | 215 | hypothetical protein | - | |||||
| 35 | 29,997–30,089 | − | 30 | hypothetical protein | - | |||||
| 36 | 30,100–30,375 | − | 91 | hypothetical protein | hypothetical protein | Xanthomonas phage Xoo-sp2 | 93 | 4.00 × 10−20 | 48.24 | ANT45253.1 |
| 37 | 30,379–30,588 | − | 69 | hypothetical protein | - | |||||
| 38 | 30,700–32,256 | − | 518 | DNA helicase | hypothetical protein | Prokaryotic dsDNA virus sp. | 91 | 4.00 × 10−123 | 44.49 | QDP55885.1 |
| 39 | 32,463–32,690 | − | 75 | hypothetical protein | - | |||||
| 40 | 32,678–33,532 | − | 284 | RecB family exonuclease/Cas4 | putative RecB family exonuclease | Campylobacter phage vB_CjeM_Los1 | 84 | 1.00 × 10−10 | 27.31 | YP_009597155.1 |
| 41 | 33,532–35,457 | − | 641 | DNA polymerase I | DNA polymerase A | Vibrio phage VpKK5 | 71 | 1.00 × 10−29 | 27.62 | YP_009126593.1 |
| 42 | 35,454–35,672 | − | 72 | hypothetical protein | hypothetical protein | Sandarakinorhabdus limnophila | 72 | 4.00 × 10−5 | 46.15 | WP_022681046.1 |
| 43 | 35,750–36,652 | − | 300 | hypothetical protein | hypothetical protein | Pseudomonas phage KPP25 | 72 | 4.00 × 10−12 | 24.55 | YP_009030602.1 |
| 44 | 36,720–37,232 | − | 170 | hypothetical protein | hypothetical protein | Nitrospira cf. moscoviensis SBR1015 | 76 | 9.00 × 10−35 | 52.31 | WP_087475496.1 |
| 45 | 37,232–37,702 | − | 156 | hypothetical protein | - | |||||
| 46 | 37,789–38,694 | − | 301 | DNA ligase | DNA ligase | Xanthomonas phage Xoo-sp2 | 99 | 3.00 × 10−81 | 45 | ANT45243.1 |
| 47 | 38,691–39,158 | − | 155 | hypothetical protein | hypothetical protein | Microcystic phage Me-ZS1 | 92 | 1.00 × 10−25 | 40.28 | AZF88158.1 |
| 48 | 39,155–39,403 | − | 82 | hypothetical protein | hypothetical protein | Cupriavidus sp. UYMSc13B | 100 | 3.00 × 10−9 | 37.35 | RWA55322.1 |
| 49 | 39,405–39,674 | − | 89 | hypothetical protein | - | |||||
| 50 | 39,667–40,515 | − | 282 | thymidylate synthase complementing protein | thimidilate synthase | Caulobacter phage Seuss | 98 | 3.00 × 10−66 | 43.77 | YP_009785564.1 |
| 51 | 40,505–40,747 | − | 80 | hypothetical protein | - | |||||
| 52 | 40,816–41,031 | − | 71 | hypothetical protein | - | |||||
| 53 | 41,028–41,546 | − | 172 | hypothetical protein | hypothetical protein | Mycobacterium phage OkiRoe | 56 | 4.00 × 10−9 | 36.61 | YP_009043654.1 |
| 54 | 41,548–41,745 | − | 65 | hypothetical protein | - | |||||
| 55 | 41,742–42,221 | − | 159 | DUF1643 containing protein | hypothetical protein | Pseudomonas phage Epa33 | 95 | 2.00 × 10−47 | 53.85 | QIQ65784.1 |
| 56 | 42,218–42,520 | − | 100 | hypothetical protein | hypothetical protein | Xylella phage Sano | 99 | 3.00 × 10−16 | 42.16 | YP_009639092.1 |
| 57 | 42,517–42,801 | − | 94 | hypothetical protein | - | |||||
| 58 | 42,830–42,973 | − | 47 | hypothetical protein | - | |||||
| 59 | 42,970–43,182 | − | 70 | hypothetical protein | - | |||||
| 60 | 43,179–43,514 | − | 111 | DnaJ domain-containing protein | DnaJ domain-containing protein | Pseudoalteromonas sp. FUC4 | 53 | 2.00 × 10−5 | 38.33 | WP_149594670.1 |
| 61 | 43,511–43,693 | − | 60 | hypothetical protein | - | |||||
| 62 | 43,756–44,055 | − | 99 | Glu-rich hypothetical protein | - | |||||
| 63 | 44,148–44,369 | − | 73 | hypothetical protein | - | |||||
| 64 | 44,463–46,577 | − | 704 | Primase | putative primase | Stenotrophomonas phage S1 | 33 | 4.00 × 10−28 | 32.91 | YP_002321451.1 |
| 65 | 46,876–47,385 | − | 169 | hypothetical protein | - |
| Gp | Hit Type | PSSM-ID | Interval | E-Value | Accession | Short Name | Superfamily |
|---|---|---|---|---|---|---|---|
| 2 | Superfamily | 392065 | 31–274 | 1.96 × 10−3 | cl29365 | Terminase_6 superfamily | - |
| 3 | Specific | 372539 | 261–406 | 6.28 × 10−34 | pfam13264 | DUF4055 | cl16196 |
| 4 | Superfamily | 385666 | 186–261 | 3.11 × 10−12 | cl10072 | Phage_Mu_F superfamily | - |
| 4 | Superfamily | 225244 | 30–263 | 2.65 × 10−5 | cl26983 | COG2369 superfamily | - |
| 6 | Superfamily | 374274 | 41–106 | 2.85 × 10−3 | cl25765 | G_path_suppress superfamily | - |
| 7 | Superfamily | 391678 | 156–250 | 9.22 × 10−3 | cl27082 | Phage_capsid superfamily | - |
| 11 | Superfamily | 372633 | 16–94 | 1.13 × 10−4 | cl16304 | DUF4128 superfamily | - |
| 16 | Specific | 131723 | 128–202 | 7.49 × 10−19 | TIGR02675 | tap_meas_nterm | cl31236 |
| 16 | Specific | 227606 | 114–682 | 4.31 × 10−13 | COG5281 | COG5281 | cl34971 |
| 20 | Superfamily | 378160 | 211–271 | 7.98 × 10−11 | cl10710 | Phage_BR0599 superfamily | - |
| 21 | Specific | 379255 | 278–448 | 2.40 × 10−13 | pfam13550 | Phage-tail_3 | cl38419 |
| 21 | Superfamily | 389952 | 874–1024 | 9.63 × 10−4 | cl22861 | LamG superfamily | - |
| 23 | Specific | 350620 | 9–147 | 4.62 × 10−29 | cd14845 | L-Ala-D-Glu_peptidase_like | cl38918 |
| 27 | Superfamily | 381914 | 6–159 | 1.31 × 10−36 | cl00269 | cytidine_deaminase-like superfamily | - |
| 28 | Superfamily | 388507 | 30–219 | 1.84 × 10−31 | cl19097 | TS_Pyrimidine_HMase superfamily | - |
| 30 | Specific | 212137 | 34–122 | 1.32 × 10−21 | cd11530 | NTP-Ppase_DR2231_like | cl16941 |
| 31 | Superfamily | 225139 | 52–192 | 1.16 × 10−10 | cl34437 | Cfa superfamily | - |
| 38 | Specific | 223627 | 7–509 | 7.90 × 10−32 | COG0553 | HepA | cl33945 |
| Bacterial Strain | Genotype or Description | Source |
|---|---|---|
| D1585 | Wildtype, AXL3-sensitive | CBCCRRR * |
| D1585 ΔpilA | Clean deletion of pilA in D1585 | [26] |
| D1585 ΔpilT | Clean deletion of pilT in D1585 | [19] |
| D1571 | Wildtype, AXL3-resistant | CBCCRRR * |
| Plasmids | ||
| pBBR1MCS | Broad-host range cloning vector, CmR | [54] |
| pD1585pilA | pBBR1MCS carrying D1585 pilA, CmR | [26] |
| pD1585pilT | pBBR1MCS carrying D1585 pilT, CmR | [19] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCutcheon, J.G.; Lin, A.; Dennis, J.J. Isolation and Characterization of the Novel Bacteriophage AXL3 against Stenotrophomonas maltophilia. Int. J. Mol. Sci. 2020, 21, 6338. https://doi.org/10.3390/ijms21176338
McCutcheon JG, Lin A, Dennis JJ. Isolation and Characterization of the Novel Bacteriophage AXL3 against Stenotrophomonas maltophilia. International Journal of Molecular Sciences. 2020; 21(17):6338. https://doi.org/10.3390/ijms21176338
Chicago/Turabian StyleMcCutcheon, Jaclyn G., Andrea Lin, and Jonathan J. Dennis. 2020. "Isolation and Characterization of the Novel Bacteriophage AXL3 against Stenotrophomonas maltophilia" International Journal of Molecular Sciences 21, no. 17: 6338. https://doi.org/10.3390/ijms21176338