Hydride Abstraction as the Rate-Limiting Step of the Irreversible Inhibition of Monoamine Oxidase B by Rasagiline and Selegiline: A Computational Empirical Valence Bond Study



Abstract
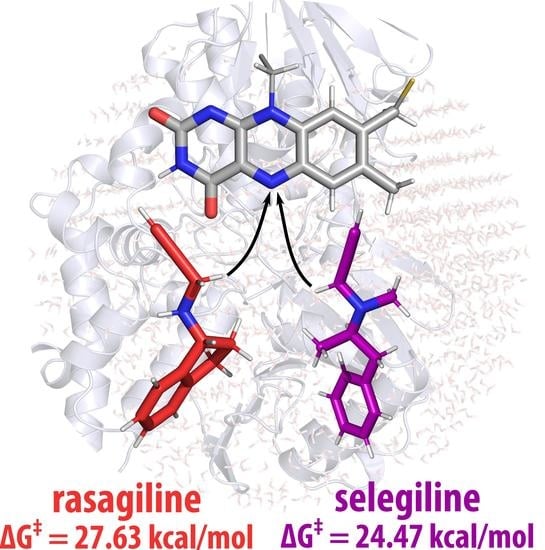
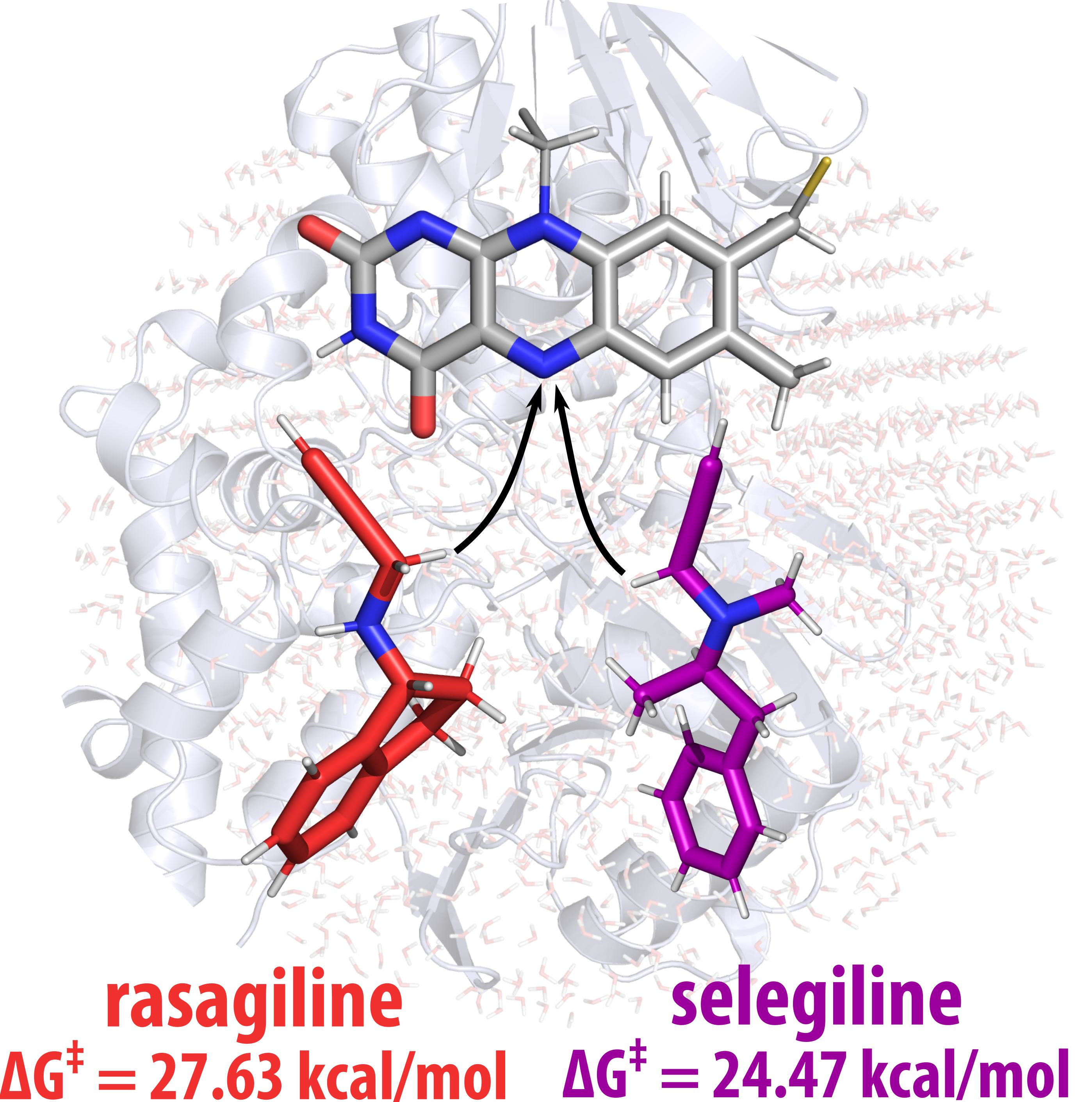
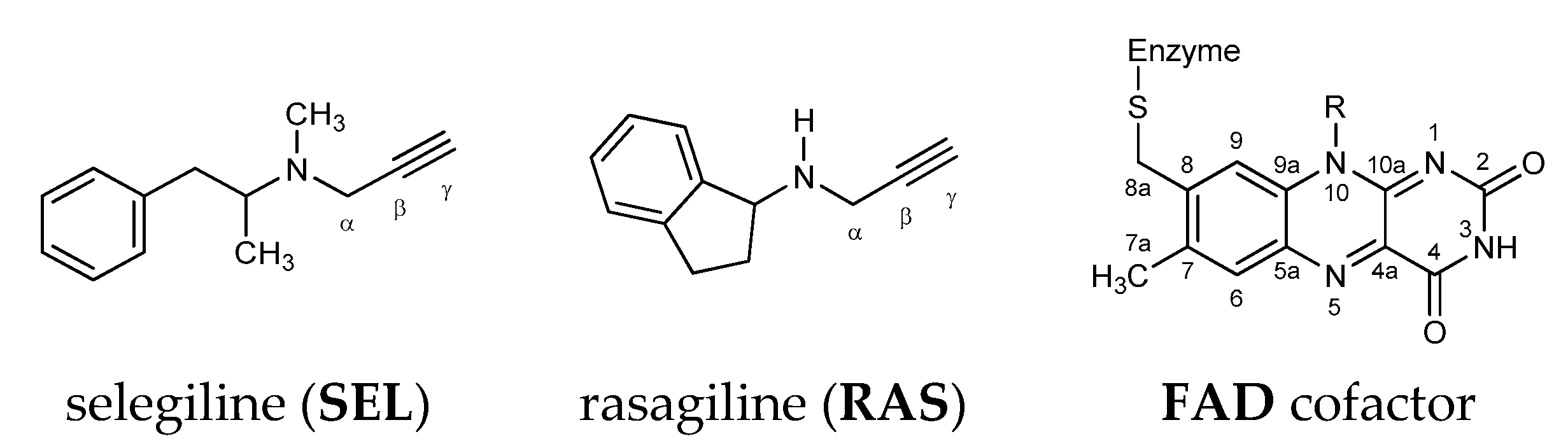
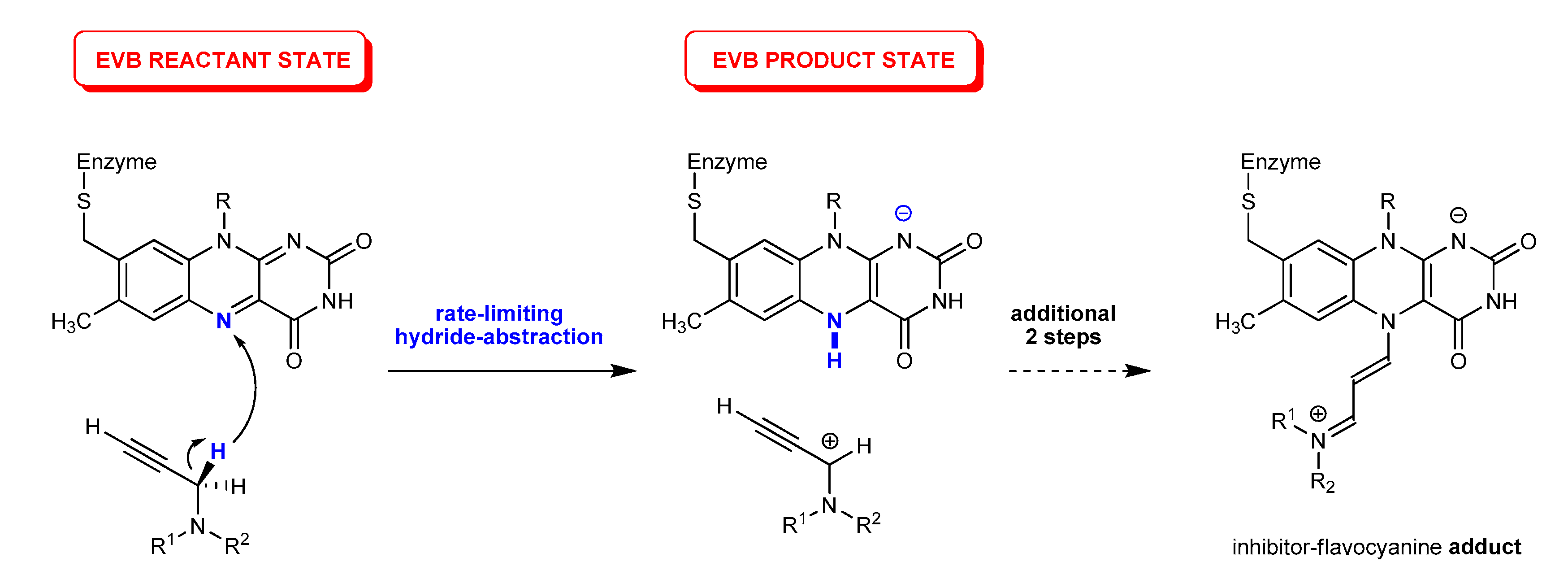
:
1. Introduction

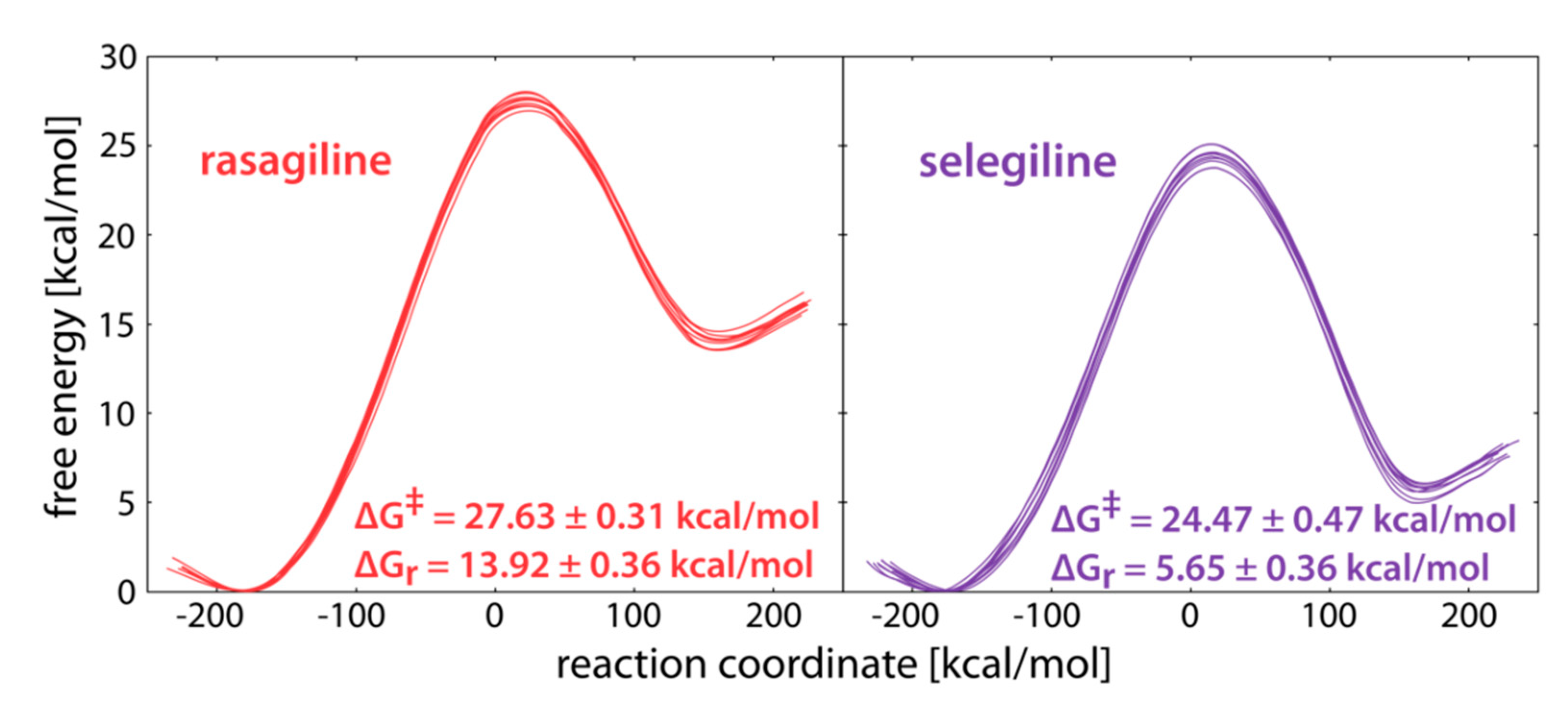
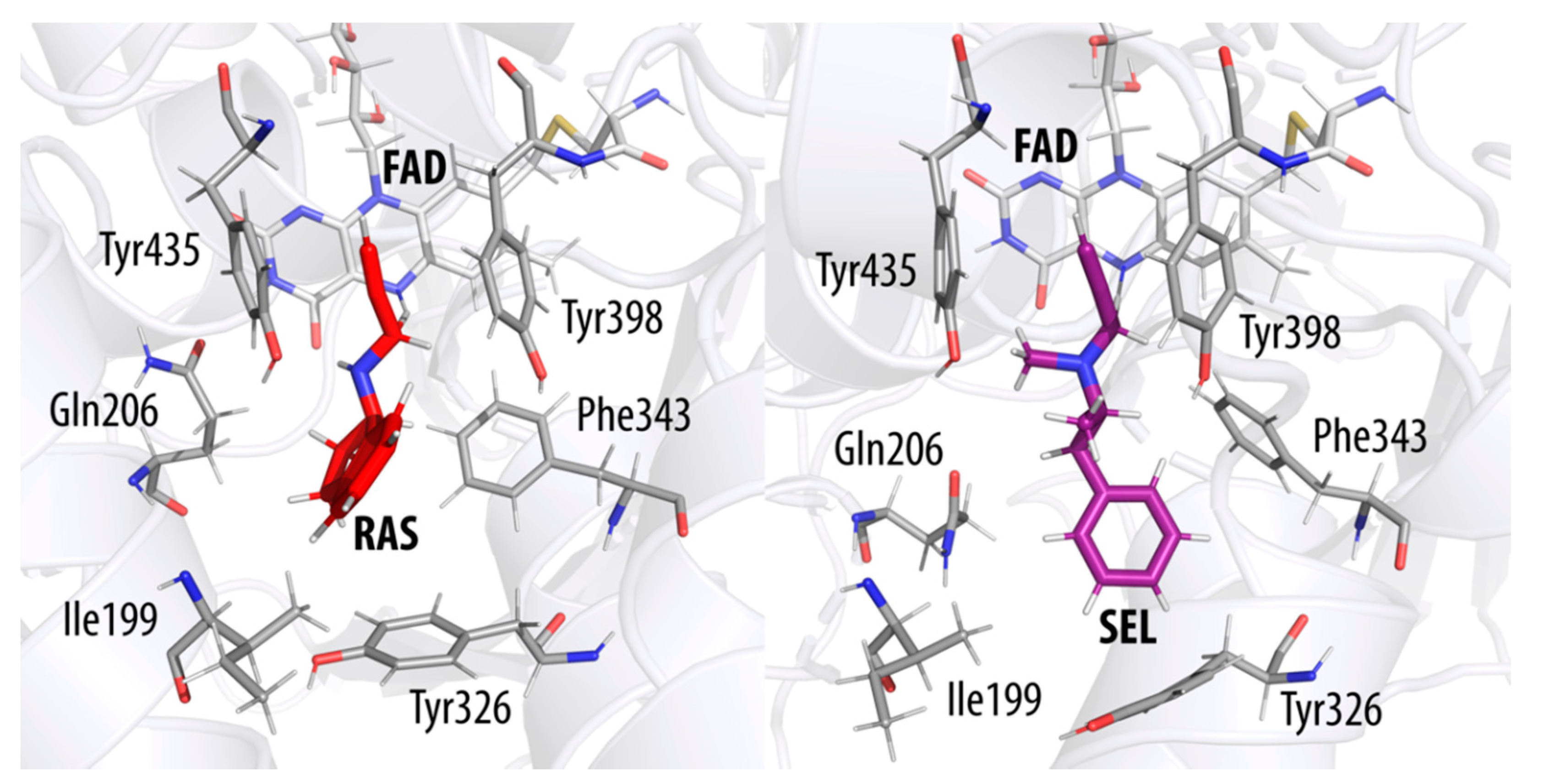
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| DFT | density functional theory |
| EVB | empirical valence bond |
| FAD | flavin adenin dinucleotide |
| FEP | free energy perturbation |
| LRF | local reaction field |
| MAO | monoamine oxidase |
| MM-GBSA | molecular mechanics-generalized Born and surface area |
| PD | Parkinson’s disease |
| QM | quantum mechanics |
| QM/MM | quantum mechanics/molecular mechanics |
| RAS | rasagiline |
| SEL | selegiline |
| US | umbrella sampling |
References
- Grimsby, J.; Lan, N.C.; Neve, R.; Chen, K.; Shih, J.C. Tissue distribution of human monoamine oxidase A and B mRNA. J. Neurochem. 1990, 55, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to hehavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westlund, K.N.; Denney, R.M.; Kochersperger, L.M.; Rose, R.M.; Abell, C.W. Distinct monoamine oxidase A and B populations in primate brain. Science 1985, 230, 181–183. [Google Scholar] [CrossRef]
- Weyler, W.; Hsu, Y.P.P.; Breakafield, X.O. Biochemistry and genetics of monoamine-oxidase. Pharmacol. Ther. 1990, 47, 391–417. [Google Scholar] [CrossRef]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-dimensional structure of human monoamine oxidase A: Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. USA 2005, 102, 12684–12689. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.J.; Tipton, K.F. On the substrate specificities of 2 forms of monoamine oxidase. J. Pharm. Pharmacol. 1984, 36, 111–115. [Google Scholar] [CrossRef]
- Gaweska, H.; Fitzpatrick, P.F. Structures and mechanism of the monoamine oxidase family. Biomol. Concepts 2011, 2, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. 2017, 33, 64–69. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef]
- Pavlin, M.; Repič, M.; Vianello, R.; Mavri, J. The chemistry of neurodegeneration: Kinetic data and their implications. Mol Neurobiol. 2016, 53, 3400–3415. [Google Scholar] [CrossRef]
- Youdim, M.B.; Edmondson, D.E.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Dubrovina, N.I.; Popova, N.K.; Gilinskii, M.A.; Tomilenko, R.A.; Seif, I. Acquisition and extinction of a conditioned passive avoidance reflex in mice with genetic knockout of monoamine oxidase A. Neurosci. Behav. Physiol. 2006, 36, 335–339. [Google Scholar] [CrossRef]
- Lee, M.; Chen, K.; Shih, J.C.; Hiroi, N. MAO-B knockout mice exhibit deficient habituation of locomotor activity but normal nicotine intake. Genes Brain Behav. 2004, 3, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Shih, J.C. Behavioral outcomes of monoamine oxidase deficiency: Preclinical and clinical evidence. Int. Rev. Neurobiol. 2011, 100, 13–42. [Google Scholar] [PubMed] [Green Version]
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem 2018, 145, 445–497. [Google Scholar] [CrossRef]
- Monte, C.; D’Ascenzio, M.; Guglielmi, P.; Mancini, V.; Carradori, S. Opening new scenarios for human MAO inhibitors. Cent. Nerv. Syst. Agents Med. Chem. 2016, 16, 98–104. [Google Scholar] [CrossRef]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front. Pharmacol 2016, 7, 340. [Google Scholar] [CrossRef] [Green Version]
- Di Giovanni, G.; Švob Štrac, D.; Sole, M.; Unzeta, M.; Tipton, K.F.; Mück-Šeler, D.; Bolea, I.; Della Corte, L.; Nikolac Perković, M.; Pivac, N.; et al. Monoaminergic and histaminergic strategies and treatments in brain diseases. Front. Neurosci. 2016, 10, 541. [Google Scholar] [CrossRef] [Green Version]
- Riederer, P.; Müller, T. Monoamine oxidase-B inhibitors in the treatment of Parkinson’s disease: Clinical-Pharmacological aspects. J. Neural Transm. 2018, 125, 1751–1757. [Google Scholar] [CrossRef]
- Tipton, K.F.; Davey, G.P.; McDonald, A.G. Kinetic behavior and reversible inhibition of monoamine oxidases-enzymes that many want dead. Int. Rev. Neurobiol. 2011, 100, 43–64. [Google Scholar]
- Fox, H.H.; Gibas, J.T. Synthetic tuberculostats. V. Alkylidene derivatives of isonicotinylhydrazine. J. Org. Chem. 1953, 18, 983–989. [Google Scholar] [CrossRef]
- Saura, J.; Nadal, E.; van den Berg, B.; Vila, M.; Bombi, J.A.; Mahy, N. Localization of monoamine oxidases in human peripheral tissues. Life Sci. 1996, 59, 1341–1349. [Google Scholar] [CrossRef]
- Asatoor, A.M.; Levi, A.J.; Milne, M.D. Tranylcypromine and cheese. Lancet 1963, 282, 733–734. [Google Scholar] [CrossRef]
- Szökö, É.; Riederer, P.; Vécsei, L.; Magyar, K. Pharmacological aspects of the neuroprotective effects of irreversible MAO-B inhibitors, selegiline and rasagiline, in Parkinson’s disease. J. Neural Transm. 2018, 125, 1735–1749. [Google Scholar] [CrossRef]
- Schapira, A.H. Progress in neuroprotection in Parkinson’s disease. Eur. J. Neurol. 2008, 15, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B. Rasagiline: A novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol. 2010, 92, 330–344. [Google Scholar] [CrossRef]
- Saura Marti, J.; Kettler, R.; Da Prada, M.; Richards, J.G. Molecular neuroanatomy of MAO-A and MAO-B. J. Neural Transm. Suppl. 1990, 32, 49–53. [Google Scholar]
- Binda, C.; Milczek, E.M.; Bonivento, D.; Wang, J.; Mattevi, A.; Edmondson, D.E. Lights and shadows on monoamine oxidase inhibition in neuroprotective pharmacological therapies. Curr. Top. Med. Chem. 2011, 11, 2788–2796. [Google Scholar] [CrossRef]
- Ramsay, R.R. Inhibitor design for monoamine oxidases. Curr. Pharm. Des. 2013, 19, 2529–2539. [Google Scholar] [CrossRef]
- Ramsay, R.R. Molecular aspects of monoamine oxidase B. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 69, 81–89. [Google Scholar] [CrossRef] [Green Version]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef] [PubMed]
- Knoll, J. (−) Deprenyl (Selegiline): Past, present and future. Neurobiology 2000, 8, 179–199. [Google Scholar]
- Chuang, H.Y.K.; Patek, D.R.; Hellerma, L. Mitochondrial monoamine oxidase-inactivation by pargyline–adduct formation. J. Biol. Chem. 1974, 249, 2381–2384. [Google Scholar]
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Binda, C.; Wang, J.; Upadhyay, A.K.; Mattevi, A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 2009, 48, 4220–4230. [Google Scholar] [CrossRef] [Green Version]
- Kamada, T.; Chow, T.; Hiroi, T.; Imaoka, S.; Morimoto, K.; Ohde, H.; Funae, Y. Metabolism of selegiline hydrochloride, a selective monoamine b-type inhibitor, in human liver microsomes. Drug Metab. Pharmacokinet. 2002, 17, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J.; Swope, D.M. Clinical pharmacology of rasagiline: A novel, second generation propargylamine for the treatment of Parkinson disease. J. Clin. Pharmacol. 2005, 45, 878–894. [Google Scholar] [CrossRef]
- Simmons, S.J.; Leyrer-Jackson, J.M.; Oliver, C.F.; Hicks, C.; Muschamp, J.W.; Rawls, S.M.; Olive, M.F. DARK classics in chemical neuroscience: cathinone-derived psychostimulants. ACS Chem. Neurosci. 2018, 9, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- dela Peña, I.; Gevorkiana, R.; Shi, W.-X. Psychostimulants affect dopamine transmission through both dopamine transporter-dependent and independent mechanisms. Eur. J. Pharmacol. 2015, 764, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umek, N.; Geršak, B.; Vintar, N.; Šoštarič, M.; Mavri, J. Dopamine autoxidation is controlled by acidic pH. Front. Mol. Neurosci. 2018, 11, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B. Mechanism of neuroprotective action of the anti-Parkinson drug rasagiline and its derivatives. Brain Res. Rev. 2005, 48, 379–387. [Google Scholar] [CrossRef]
- Azzaro, A.J.; Ziemniak, J.; Kemper, E.; Campbell, B.J.; VanDenBerg, C. Pharmacokinetics and absolute bioavailability of selegiline following treatment of healthy subjects with the selegiline transdermal system (6 mg/24 h): A comparison with oral selegiline capsules. J. Clin. Pharmacol. 2007, 47, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Zhu, X.; Jiang, R.; Ji, F.; Su, Z.; Xue, R.; Zhou, Y. Comparison for efficacy and tolerability among ten drugs for treatment of Parkinson’s disease: A network meta-analysis. Sci. Rep. 2017, 7, 45865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robakis, D.; Fahn, S. Defining the role of the monoamine oxidase-B inhibitors for Parkinson’s disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Farina, R.; Nicolotti, O.; Gadaleta, D.; Soto-Otero, R.; Catto, M.; Di Braccio, M.; Mendez-Alvarez, E.; Carotti, A. In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur. J. Med. Chem. 2015, 89, 98–105. [Google Scholar] [CrossRef]
- Pisani, L.; Farina, R.; Catto, M.; Iacobazzi, R.M.; Nicolotti, O.; Cellamare, S.; Mangiatordi, G.F.; Denora, N.; Soto-Otero, R.; Siragusa, L.; et al. Exploring basic tail modifications of coumarin based dual acetylcholinesterase-monoamineoxidase B inhibitors: Identification of water-soluble, brain permeant neuroprotective multitarget agents. J. Med. Chem. 2016, 59, 6791–6806. [Google Scholar] [CrossRef]
- Gökhan-Kelekçi, N.; Koyunoğlu, S.; Yabanoğlu, S.; Yelekçi, K.; Özgen, Ö.; Uçar, G.; Erol, K.; Kendi, E.; Yeşilada, A. Newpyrazoline bearing 4(3H)-quinazolinone inhibitors of monoamine oxidase: Synthesis, biological evaluation, and structural determinants of MAO-A and MAO-B selectivity. Bioorg. Med. Chem. 2009, 17, 675–689. [Google Scholar] [CrossRef]
- Wang, Z.-M.; Li, X.-M.; Xue, G.-M.; Xu, W.; Wang, X.-B.; Kong, L.-Y. Synthesis and evaluation of 6-substituted 3-arylcoumarin derivatives as multifunctional acetylcholinesterase/ monoamine oxidase B dual inhibitors for the treatment of Alzheimer’s disease. RSC Adv. 2015, 5, 104122–104137. [Google Scholar] [CrossRef]
- Matos, M.J.; Viña, D.; Quezada, E.; Picciau, C.; Delogu, G.; Orallo, F.; Santana, L.; Uriarte, E. A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3268–3270. [Google Scholar] [CrossRef]
- Matos, M.J.; Viña, D.; Janeiro, P.; Borges, F.; Santana, L.; Uriarte, E. New halogenated 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 5157–5160. [Google Scholar] [CrossRef]
- Mangiatordi, G.F.; Alberga, D.; Pisani, L.; Gadaleta, D.; Trisciuzzi, D.; Farina, R.; Carotti, A.; Lattanzi, G.; Catto, M.; Nicolotti, O. A rational approach to elucidate human monoamine oxidase molecular selectivity. Eur. J. Pharm. Sci. 2017, 101, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Tandarić, T.; Vianello, R. Computational insight into the mechanism of the irreversible inhibition of monoamine oxidase enzymes by the antiparkinsonian propargylamine inhibitors rasagiline and selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Hofer, T.S.; de Visser, S.P. Quantum mechanical/molecular mechanical approaches for the investigation of chemical systems–recent developments and advanced applications. Front. Chem. 2018, 6, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vianello, R.; Repič, M.; Mavri, J. How are biogenic amines metabolized by monoamine oxidases? Eur. J. Org. Chem. 2012, 7057–7065. [Google Scholar] [CrossRef]
- Repič, M.; Vianello, R.; Purg, M.; Duarte, F.; Bauer, P.; Kamerlin, S.C.L.; Mavri, J. Empirical valence bond simulations of the hydride transfer step in the monoamine oxidase B catalyzed metabolism of dopamine. Proteins Struct. Funct. Bioinform. 2014, 82, 3347–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maršavelski, A.; Vianello, R. What a difference a methyl group makes: The selectivity of monoamine oxidase B towards histamine and N-methylhistamine. Chem. Eur. J. 2017, 23, 2915–2925. [Google Scholar] [CrossRef]
- Poberžnik, M.; Purg, M.; Repič, M.; Mavri, J.; Vianello, R. Empirical valence bond simulations of the hydride- transfer step in the monoamine oxidase A catalyzed metabolism of noradrenaline. J. Phys. Chem. B 2016, 120, 11419–11427. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Himo, F. Recent developments of the quantum chemical cluster approach for modeling enzyme reactions. J. Biol. Inorg. Chem. 2009, 14, 643–651. [Google Scholar] [CrossRef]
- Himo, F. Recent trends in quantum chemical modeling of enzymatic reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef] [Green Version]
- Siegbahn, P.E.M.; Himo, F. The quantum chemical cluster approach for modeling enzyme reactions. WIREs Comput. Mol. Sci. 2011, 1, 323–336. [Google Scholar] [CrossRef]
- Patton, G.C.; Stenmark, P.; Gollapalli, D.R.; Sevastik, R.; Kursula, P.; Flodin, S.; Schuler, H.; Swales, C.T.; Eklund, H.; Himo, F.; et al. Cofactor mobility determines reaction outcome in the IMPDH and GMPR (β-α)8 barrel enzymes. Nat. Chem. Biol. 2011, 7, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modeling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Ribeiro, A.J.M.; Neves, R.P.P.; Brás, N.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. WIREs Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Quesne, M.G.; Silveri, F.; de Leeuw, N.H.; Catlow, C.R.A. Advances in sustainable catalysis: A computational perspective. Front. Chem. 2019, 7, 182. [Google Scholar] [CrossRef] [Green Version]
- Kamerlin, S.C.L.; Warshel, A. The EVB as a quantitative tool for formulating simulations and analyzing biological and chemical reactions. Faraday Discuss. 2010, 145, 71–106. [Google Scholar] [CrossRef] [Green Version]
- Völgyi, G.; Ruiz, R.; Box, K.; Comer, J.; Bosch, E.; Takacs-Novak, K. Potentiometric and spectrophotometric pKa determination of water-insoluble compounds: Validation study in a new cosolvent system. Anal. Chim. Acta 2007, 583, 418–428. [Google Scholar] [CrossRef]
- Borštnar, R.; Repič, M.; Kamerlin, S.C.L.; Vianello, R.; Mavri, J. Computational study of the pKa values of potential catalytic residues in the active site of monoamine oxidase, B. J. Chem. Theory Comput. 2012, 8, 3864–3870. [Google Scholar] [CrossRef] [Green Version]
- Pregeljc, D.; Jug, U.; Mavri, J.; Stare, J. Why does the Y326I mutant of monoamine oxidase B decompose an endogenous amphetamine at a slower rate than the wild type enzyme? Reaction step elucidated by multiscale molecular simulations. Phys. Chem. Chem. Phys. 2018, 20, 4181–4188. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R.; Albreht, A. Kinetics, mechanism, and inhibition of monoamine oxidase. J. Neural Transm. 2018, 125, 1659–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubalek, F.; Binda, C.; Li, M.; Herzig, Y.; Sterling, J.; Youdim, M.B.; Mattevi, A.; Edmondson, D.E. Inactivation of purified human recombinant monoamine oxidases A and B by rasagiline and its analogues. J. Med. Chem. 2004, 47, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; Rosta, E.; Warshel, A. Using the Constrained DFT Approach in generating diabatic surfaces and off diagonal empirical valence bond terms for modeling reactions in condensed phases. Phys. Chem. B 2006, 110, 39–19570. [Google Scholar] [CrossRef] [PubMed]
- Oanca, G.; Stare, J.; Vianello, R.; Mavri, J. Multiscale simulation of monoamine oxidase catalyzed decomposition of phenylethylamine analogs. Eur. J. Pharmacol. 2017, 817, 46–50. [Google Scholar] [CrossRef]
- Prah, A.; Frančišković, E.; Mavri, J.; Stare, J. Electrostatics as the driving force behind the catalytic function of the monoamine oxidase a enzyme confirmed by quantum computations. ACS Catal. 2019, 9, 1231–1240. [Google Scholar] [CrossRef]
- Oanca, G.; Stare, J.; Mavri, J. How fast monoamine oxidases decompose adrenaline? Kinetics of isoenzymes A and B evaluated by empirical valence bond simulation. Proteins 2017, 85, 2170–2178. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Fox. Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera–a visualization system for exploratory research and analysis. J. Comp. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-2: Maestro; Schrödinger, LLC: New York, NY, USA, 2020.
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Oanca, G.; Purg, M.; Mavri, J.; Shih, J.C.; Stare, J. Insights into enzyme point mutation effect by molecular simulation: Phenylethylamine oxidation catalyzed by monoamine oxidase. Phys. Chem. Chem. Phys. 2016, 18, 13346–13356. [Google Scholar] [CrossRef] [Green Version]
- Stare, J. Complete sampling of an enzyme reaction pathway: A lesson from gas phase simulations. RSC Adv. 2017, 7, 8740–8754. [Google Scholar] [CrossRef] [Green Version]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.B.; Olsson, M.H.M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef] [PubMed]
- Villà, J.; Warshel, A. Energetics and dynamics of enzymatic reactions. J. Phys. Chem. B 2001, 105, 7887–7907. [Google Scholar]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Aqvist, J. A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213–225. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Reactant Complex | Transition State | Intermediate | Hij | α0 |
|---|---|---|---|---|---|
| Rasagiline | 0.0 | 30.60 | 19.18 | 44.34 | 106.7 |
| Selegiline | 0.0 | 31.11 | 17.29 | 43.59 | 80.40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tandarić, T.; Prah, A.; Stare, J.; Mavri, J.; Vianello, R. Hydride Abstraction as the Rate-Limiting Step of the Irreversible Inhibition of Monoamine Oxidase B by Rasagiline and Selegiline: A Computational Empirical Valence Bond Study. Int. J. Mol. Sci. 2020, 21, 6151. https://doi.org/10.3390/ijms21176151
Tandarić T, Prah A, Stare J, Mavri J, Vianello R. Hydride Abstraction as the Rate-Limiting Step of the Irreversible Inhibition of Monoamine Oxidase B by Rasagiline and Selegiline: A Computational Empirical Valence Bond Study. International Journal of Molecular Sciences. 2020; 21(17):6151. https://doi.org/10.3390/ijms21176151
Chicago/Turabian StyleTandarić, Tana, Alja Prah, Jernej Stare, Janez Mavri, and Robert Vianello. 2020. "Hydride Abstraction as the Rate-Limiting Step of the Irreversible Inhibition of Monoamine Oxidase B by Rasagiline and Selegiline: A Computational Empirical Valence Bond Study" International Journal of Molecular Sciences 21, no. 17: 6151. https://doi.org/10.3390/ijms21176151