Are Myths and Preconceptions Preventing Us from Applying Ionic Liquid Forms of Antiviral Medicines to the Current Health Crisis?


Abstract
:
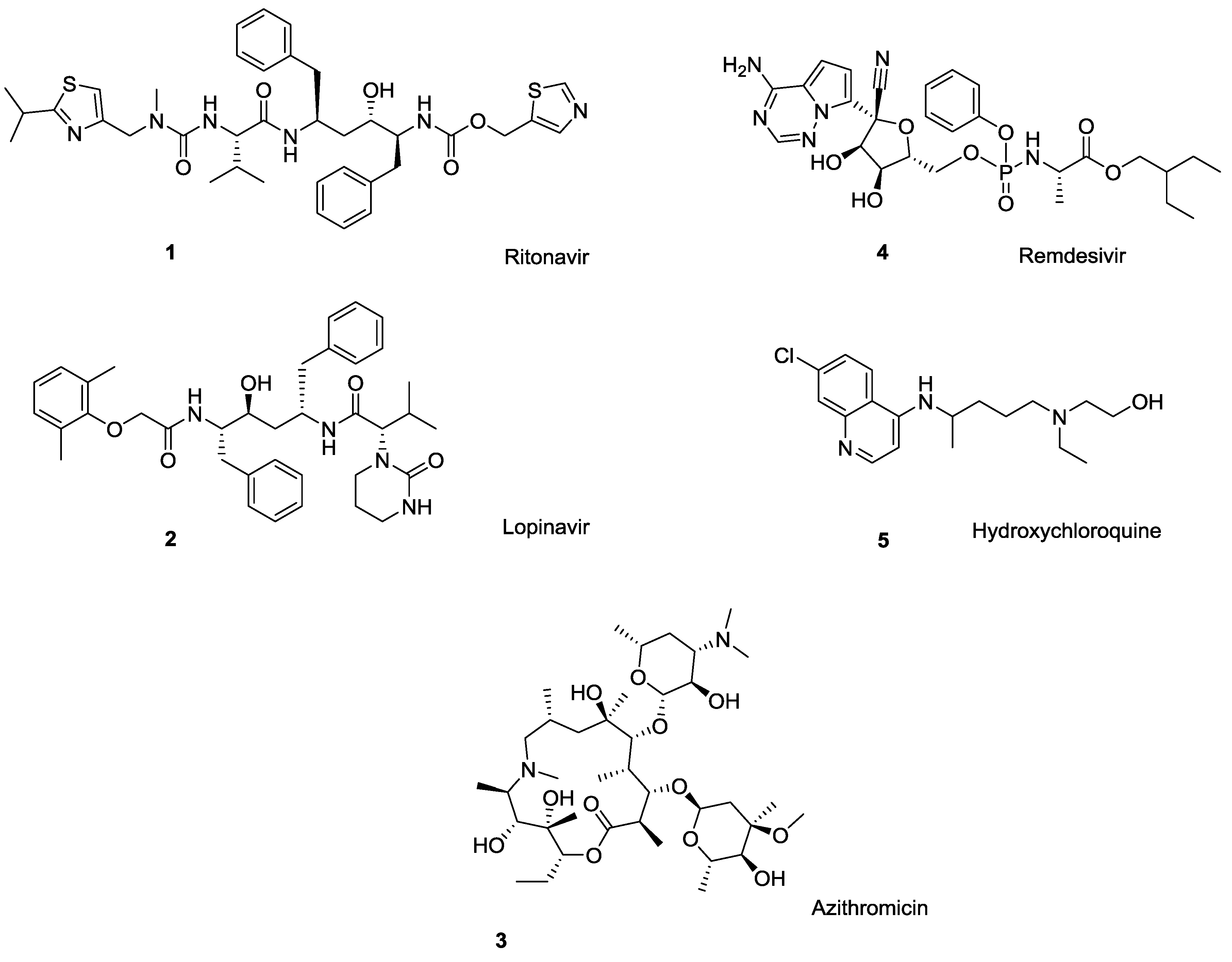
1. Difficulties with Current Pharmaceuticals
2. Current Tools to Reformulate APIs into More Bioavailable Forms
3. What about API Ionic Liquids (API-ILs) as Alternative to Common Strategies?
4. The Myths and Perceived Concerns Holding Back Implementation of API-ILs
4.1. Physical Properties: Viscosity, Hygroscopicity, and Stability
4.2. Toxicity
4.3. Difficulties in Characterization
5. Does the Pharma Industry Work with Liquids?
6. Are API-ILs Arriving in Pharma? The case of Glaxo Smith Kline GSK2838232
7. Other Antiviral API-ILs Already Exist: The Case of Acyclovir
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| U.S. FDA | U.S. Food and Drug Administration |
| [Cho][Acy] | Choline Acyclovir |
| [H2Acy][Doc] | Acyclovir Docusate |
| [H2Acy]Cl | Acyclovir Chloride |
| [N1,1,1,16][Acy] | N-hexadecyl-N,N,N-Trimethylammonium Acyclovir |
| [N1,4,4,4][Acy] | N,N,N-Tributyl-N-methylammonium Acyclovir |
| [N4,4,4,4][Acy] | N,N,N,N-Tetrabutylammonium Acyclovir |
| [P4,4,4,4][Acy] | Tetrabutylphosphonium Acyclovir |
| [P4,4,4,4][Ibu] | Tetrabutylphosphonium Ibuprofenate |
| API | Active Pharmaceutical Ingredients |
| API-IL | Active Pharmaceutical Ingredient—Ionic Liquid |
| BCS | Biopharmaceutical Classification System |
| COVID-19 | Coronavirus Disease 2019 |
| DLS | Dynamic Light Scattering |
| EUA | Emergency Use Authorization |
| FT-IR | Fourier-Transform Infrared |
| GMP | Good Manufacturing Practice |
| GSK | GlaxoSmithKline |
| HPLC | High-Performance Liquid Chromatography |
| HR-MAS | High-Resolution Magic Angle Spinning |
| IL | Ionic Liquid |
| NMR | Nuclear Magnetic Resonance |
| PBS | Phosphate Buffer Saline |
| PFG-NMR | Pulse-Field-Gradient |
| SGF | Simulated Gastric Fluid |
| SIF | Simulated Intestinal Fluid |
References
- Potential Antiviral Drugs under Evaluation for the Treatment of COVID-19. Available online: https://www.covid19treatmentguidelines.nih.gov/antiviral-therapy/ (accessed on 29 June 2020).
- COVID-19: Prevention & Investigational Treatments. Available online: https://www.drugs.com/condition/covid-19.htmlhttps://www.covid19treatmentguidelines.nih.gov/antiviral-therapy/ (accessed on 29 June 2020).
- De Savi, C.; Hughes, D.L.; Kvaerno, L. Quest for a COVID-19 Cure by repurposing small-molecule drugs: Mechanism of action, clinical development, synthesis at scale, and outlook for supply. Org. Process Res. Dev. 2020, 24, 940–976. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Brittain, H.G.; Suryanarayanan, R. Thermoanalytical and crystallographic methods. In Polymorphism in Pharmaceutical Solids. Drugs and the Pharmaceutical Sciences Series, 2nd ed.; Brittain, H.G., Ed.; Informa Healthcare: New York, NY, USA, 2009; Volume 192, pp. 318–346. [Google Scholar]
- Characteristics of Potential Antiviral Agents under Evaluation for Treatment of COVID-19. Available online: https://www.covid19treatmentguidelines.nih.gov/tables/table-2b/ (accessed on 29 June 2020).
- Shamshina, J.L.; Kelley, S.P.; Gurau, G.; Rogers, R.D. Chemistry: Develop ionic liquid drugs. Nature 2015, 528, 188–189. [Google Scholar] [CrossRef] [Green Version]
- Pernak, J.; Skrzypczak, A.; Lota, G.; Frackowiak, E. Synthesis and properties of trigeminal tricationic ionic liquids. Chem Eur. J. 2007, 13, 3106–3112. [Google Scholar] [CrossRef] [PubMed]
- Shamshina, J.L.; Berton, P.; Wang, H.; Zhou, X.; Gurau, G.; Rogers, R.D. Ionic liquids in the pharmaceutical industry. In Green Techniques for Organic Synthesis and Medicinal Chemistry, 2nd ed.; Zhang, W., Cue, B., Eds.; John Wiley and Sons: Singapore, 2018; pp. 541–577. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals, IUCR Monographs on Crystallography 14; Oxford Science Publications: Oxford, UK, 2002. [Google Scholar]
- Morissette, S.L.; Soukasene, S.; Levinson, D.; Cima, M.J.; Almarsson, O. Elucidation of crystal form diversity of the HIV protease inhibitor ritonavir by high-throughput crystallization. Proc. Natl. Acad. Sci. USA 2003, 100, 2180–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandi, P.P.R.; Kura, R.R.; Rapoli, R.R.; Dasari, M.R.; Kesireddy, S.C.R. Novel polymorphs of Lopinair. U.S. Patent 13,121,505, 6 February 2009. [Google Scholar]
- Jensen, A.F.; Junager, F.; Laustsen, S.B.; Rasmussen, P.; Jessen, C.U.; Kornø, H.T.; Bondo, J. Two polymorphs—Which one is stable at ambient conditions? Acta Cryst. 2005, A61, c292–c293. [Google Scholar] [CrossRef] [Green Version]
- Odendaal, R.W. Amorphism and Polymorphism of Azithromycin. Ph.D. Thesis, North West University, Potchefstroom, South Africa, 2012. Available online: https://repository.nwu.ac.za/handle/10394/9528 (accessed on 29 June 2020).
- Sarbajna, R.M.; Preetam, A.; Sivalakshmi, A.; Devi, A.S.; Suryanarayana, M.V.; Sethi, M.; Dutta, D. Studies on crystal modifications of Ganciclovir. Mol. Cryst. Liquid Cryst. 2011, 537, 141–154. [Google Scholar] [CrossRef]
- Roque-Flores, R.L.; Guzei, I.A.; Matos, J.R.; Yu, L. Polymorphs of the antiviral drug ganciclovir. Acta Cryst. 2017, C73, 1116–1120. [Google Scholar] [CrossRef]
- Lutker, K.M.; Quiñones, R.; Xu, J.; Ramamoorthy, A.; Matzger, A.J. Polymorphs and hydrates of Acyclovir. J. Pharm. Sci. 2011, 100, 949–963. [Google Scholar] [CrossRef] [Green Version]
- Noonan, T.J.; Mzondo, B.; Bournea, S.A.; Caira, M.R. Polymorphism of the antiviral agent clevudine. Cryst. Eng. Comm. 2016, 18, 8172–8181. [Google Scholar] [CrossRef]
- Karpinski, P.H. Polymorphism of active pharmaceutical ingredients. Chem. Eng. Technol. 2006, 29, 233–237. [Google Scholar] [CrossRef]
- Chawla, G.; Bansal, A.K. Challenges in polymorphism of pharmaceuticals. CRIPS 2004, 5, 9–12. [Google Scholar]
- Raza, K.; Kumar, P.; Ratan, S.; Malik, R.; Arora, S. Polymorphism: The phenomenon affecting the performance of drugs. SOJ Pharm. Pharm. Sci. 2014, 1, 10–20. [Google Scholar] [CrossRef] [Green Version]
- FDA Guidance for Industry. ANDAs: Pharmaceutical Solid Polymorphism. Chemistry, Manufacturing, and Controls Information. Available online: https://www.fda.gov/media/71375/download (accessed on 29 June 2020).
- Florence, A.J. Approaches to high-throughput physical form screening and discovery. In Polymorphism in Pharmaceutical Solids; Brittain, H.G., Ed.; Informa Healthcare: New York, NY, USA, 2009; pp. 139–184. [Google Scholar]
- Barr, G.; Dong, W.; Gilmore, C.J. High-throughput powder diffraction. II. Applications of clustering methods and multivariate data analysis. J. Appl. Crystallography. 2004, 37, 243–252. [Google Scholar] [CrossRef]
- Schuster, D.; Laggner, C.; Langer, T. Why drugs fail—A study on side effects in new chemical entities. Curr. Pharm. Des. 2005, 11, 3545–3559. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowska, Z.; Paluch, M.A.; Grzybowski, K.; Adrjanowicz, K.; Grzybowska, K.; Kaminski, K.; Wlodarczyk, P.; Pionteck, J. Study of molecular dynamics of pharmaceutically important protic ionic liquid-verapamil hydrochloride. I. Test of thermodynamic scaling. J. Chem. Phys. 2009, 131, 104505/1–104505/14. [Google Scholar] [CrossRef]
- Schade, D.; Kotthaus, J.; Riebling, L.; Kotthaus, J.; Mueller-Fielitz, H.; Raasch, W.; Hoffmann, A.; Schmidtke, M.; Clement, B. Zanamivir amidoxime- and N-hydroxyguanidine-based prodrug approaches to tackle poor oral bioavailability. J. Pharm. Sci. 2015, 104, 3208–3219. [Google Scholar] [CrossRef]
- Nagappa, A.N.; Agarwal, G.; Chikkamath, V.; Agarwal, S.; Rani, R.; Karar, P.K. Acyclovir sodium potential as an antiviral drug is limited by its low oral bioavailability (20–30%) with short half-life (2–3 h) with poor plasma protein binding. Formulation and evaluation of Acyclovir sodium solid lipid microparticles. Am. J. Adv. Drug Deliv. 2016, 4, 78–84. [Google Scholar]
- Giesler, K.E.; Marengo, J.; Liotta, D.C. Reduction sensitive lipid conjugates of tenofovir: Synthesis, stability, and antiviral activity. J. Med. Chem. 2016, 59, 7097–7110. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H. Biological assay challenges from compound solubility: Strategies for bioassay optimization (Review). Drug Discov. Today 2006, 11, 446–451. [Google Scholar] [CrossRef]
- Paulekuhn, G.S.; Dressman, J.B.; Saal, C. Trends in active pharmaceutical ingredient salt selection based on analysis of the orange book database. J. Med. Chem. 2007, 50, 6665–6672. [Google Scholar] [CrossRef]
- Prohotsky, D.L.; Zhao, F. A survey of top 200 drugs—Inconsistent practice of drug strength expression for drugs containing salt forms. J. Pharm. Sci. 2012, 101, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Crystal and co-crystal. CrystEngComm 2003, 5, 466–467. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Banerjee, R.; Bhatt, P.M.; Ravindra, N.V.; Desiraju, G.R. Saccharin salts of active pharmaceutical ingredients, their crystal structures, and increased water solubilities. Cryst. Growth Des. 2005, 5, 2299–2309. [Google Scholar] [CrossRef]
- Sopyan, I.; Alfauziah, T.O.; Gozali, D. Better in solubility enhancement: Salt or cocrystal? Int. J. Res. Pharm. Sci. 2019, 10, 3013–3025. [Google Scholar] [CrossRef]
- Guidance for Industry on Regulatory Classification of Pharmaceutical Co-Crystals. Available online: https://www.federalregister.gov/documents/2013/04/26/2013-09872/guidance-for-industry-on-regulatory-classification-of-pharmaceutical-co-crystals-availability (accessed on 13 July 2020).
- Regulatory Classification of Pharmaceutical Co-Crystals Guidance for Industry. Available online: https://www.fda.gov/media/81824/download (accessed on 13 July 2020).
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, salts, and cocrystals: What’s in a name? Crystal Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Kelley, S.P.; Narita, A.; Holbrey, J.D.; Green, K.D.; Reichert, W.M.; Rogers, R.D. Understanding the effects of ionicity in salts, solvates, co-crystals, Ionic co-crystals, and ionic liquids, rather than nomenclature, is critical to understanding their behavior. Crystal Growth Des. 2013, 13, 965–975. [Google Scholar] [CrossRef]
- Makary, P. Principles of salt formation. UK J. Pharm. Biosci. 2014, 2, 1–4. [Google Scholar] [CrossRef]
- Hilfiker, R. Polymorphism in the pharmaceutical industry. Chem Today 2006, 21, 41–44. [Google Scholar]
- Gould, P.L. Salt selection for basic drugs. Int. J. Pharm. 1986, 33, 201–217. [Google Scholar] [CrossRef]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Varghese Gupta, S. Salts of therapeutic agents: Chemical, physicochemical, and biological considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef] [Green Version]
- Hough, W.L.; Smiglak, M.; Rodriguez, H.; Swatloski, R.P.; Spear, S.K.; Daly, D.T.; Pernak, J.; Grisel, J.E.; Carliss, R.D.; Soutullo, M.D.; et al. The third evolution of ionic liquids: Active pharmaceutical ingredients. New J. Chem. 2007, 31, 1429–1436. [Google Scholar] [CrossRef]
- Welton, T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Shadid, M.; Gurau, G.; Shamshina, J.L.; Chuang, B.C.; Hailu, S.; Guan, E.; Chowdhury, S.K.; Wu, J.T.; Rizvi, S.A.A.; Griffin, R.J. Sulfasalazine in ionic liquid form with improved solubility and exposure. Med. Chem. Comm. 2015, 6, 1837–1841. [Google Scholar] [CrossRef]
- Shamshina, J.L.; Cojocaru, O.A.; Kelley, S.P.; Bica, K.; Wallace, S.P.; Gurau, G.; Rogers, R.D. Acyclovir as an ionic liquid cation or anion can improve aqueous solubility. Acs Omega 2017, 2, 3483–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, J.M.M.; Florindo, C.; Pereiro, A.B.; Vieira, N.S.M.; Matias, A.A.; Duarte, C.M.M.; Rebelo, L.P.N.; Marrucho, I.M. Cholinium-based ionic liquids with pharmaceutically active anions. RSC Adv. 2014, 4, 28126–28132. [Google Scholar] [CrossRef]
- Egorova, K.S.; Gordeev, E.G.; Ananikov, V.P. Biological activity of ionic liquids and their application in pharmaceutics and medicine. Chem. Rev. 2017, 117, 7132–7189. [Google Scholar] [CrossRef]
- Shamshina, J.L.; Barber, P.S.; Rogers, R.D. Ionic liquids in drug delivery. Expert Opin. Drug Deliv. 2013, 10, 1367–1381. [Google Scholar] [CrossRef]
- Wang, H.; Gurau, G.; Shamshina, J.L.; Cojocaru, O.A.; Janikowski, J.; MacFarlane, D.R.; Davis, J.H., Jr.; Rogers, R.D. Simultaneous membrane transport of two active pharmaceutical ingredients by charge assisted hydrogen bond complex formation. Chem. Sci. 2014, 5, 3449–3456. [Google Scholar] [CrossRef]
- Miwa, Y.; Hamamoto, H.; Ishida, T. Lidocaine self-sacrificially improves the skin permeation of the acidic and poorly water-soluble drug Etodolac via its transformation into an ionic liquid. Eur. J. Pharm. Biopharm. 2016, 102, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Clinical trials. Etodolac-Lidocaine Patch in Subjects Experiencing Acute Delayed Onset Muscle Soreness. Available online: https://clinicaltrials.gov/ct2/show/NCT02695381 (accessed on 20 July 2020).
- Topline Phase III Clinical Trial Results for ETOREAT® in the United States. Available online: http://pdf.irpocket.com/C4586/xAmX/x8qh/SVn5.pdf (accessed on 20 July 2020).
- Ostadjoo, S.; Berton, P.; Shamshina, J.L.; Rogers, R.D. Scaling-up ionic liquid-based technologies: How much do we care about their toxicity? Prima facie information on 1-ethyl-3-methylimidazolium acetate. Toxicol. Sci. 2018, 161, 249–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peplow, M. Warning shot for green chemistry. Nature 2005. [Google Scholar] [CrossRef]
- Pham, T.P.T.; Cho, C.-W.; Yun, Y.-S. Environmental fate and toxicity of ionic liquids: A review. Water Res. 2010, 44, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Balk, A.; Holzgrabe, U.; Meinel, L. ‘Pro et contra’ ionic liquid drugs—Challenges and opportunities for pharmaceutical translation. Eur. J. Pharm. Biopharm. 2015, 94, 291–304. [Google Scholar] [CrossRef] [PubMed]
- FDA Guidance Document “Oral Solutions and Suspensions (8/94)”. Available online: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-guides/oral-solutions-and-suspensions-894 (accessed on 30 June 2020).
- Paduszyński, K.; Domańska, U. Viscosity of ionic liquids: An extensive database and a new group contribution model based on a feed-forward artificial neural network. J. Chem. Inf. Model. 2014, 54, 1311–1324. [Google Scholar] [CrossRef]
- Koi, Z.K.; Yahya, V.Z.N.; Talipa, R.A.A.; Kurnia, K. Prediction of the viscosity of imidazolium-based ionic liquids at different temperatures using the quantitative structure property relationship approach. New J. Chem. 2019, 43, 16207–16217. [Google Scholar] [CrossRef]
- Chatel, G.; Pereira, J.F.B.; Debbeti, V.; Wang, H.; Rogers, R.D. Mixing ionic liquids —“Simple mixtures” or “double salts”? Green Chem. 2014, 16, 2051–2083. [Google Scholar] [CrossRef] [Green Version]
- McCrary, P.D.; Beasley, P.A.; Gurau, G.; Narita, A.; Barber, P.S.; Cojocaru, O.A.; Rogers, R.D. Drug specific tuning of an ionic liquid’s hydrophilic-lipophilic balance to improve water solubility of poorly soluble active pharmaceutical ingredients. New J. Chem. 2013, 37, 2196–2202. [Google Scholar] [CrossRef]
- Guidance for Industry; Guidance on Abbreviated New Drug Applications: Stability Testing of Drug Substances and Products; Availability. Available online: https://www.federalregister.gov/documents/2013/06/20/2013-14674/guidance-for-industry-guidance-on-abbreviated-new-drug-applications-stability-testing-of-drug (accessed on 15 July 2020).
- Frizzo, C.P.; Gindri, I.M.; Tier, A.Z.; Buriol, L.; Moreira, D.N.; Martins, M.A.P. Pharmaceutical salts: Solids to liquids by using ionic liquid design. In Ionic Liquids—New Aspects for the Future; Kadokawa, J.-I., Ed.; InTechOpen: London, UK, 2013; pp. 557–579. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, R.; Branco, L.C.; Marrucho, I.M.; Araújo, J.M.M.; Rebelo, L.P.N.M.; da Ponte, M.N.; Prudêncio, C.; Noronha, J.P.; Petrovski, Z. Development of novel ionic liquids based on ampicillin. Med. Chem. Comm. 2012, 3, 494–497. [Google Scholar] [CrossRef] [Green Version]
- Hough-Troutman, W.L.; Smiglak, M.; Griffin, S.; Reichert, W.M.; Mirska, I.; Jodynis-Liebert, J.; Adamska, T.; Nawrot, J.; Stasiewicz, M.; Rogers, R.D.; et al. Ionic liquids with dual biological function: Sweet and anti-microbial, hydrophobic quaternary ammonium-based salts. New J. Chem. 2009, 33, 26–33. [Google Scholar] [CrossRef]
- Bica, K.; Rijksen, C.; Nieuwenhuyzen, M.; Rogers, R.D. In search of pure liquid salt forms of aspirin: Ionic liquid approaches with acetylsalicylic acid and salicylic acid. Phys. Chem. Chem. Phys. 2012, 12, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Ananikov, V.P. Biological activity of ionic liquids involving ionic and covalent binding: Tunable drug development platform. In Encyclopedia of Ionic Liquids; Zhang, S., Ed.; Springer: Singapore, 2019; pp. 1–8. [Google Scholar] [CrossRef]
- Smith, D.J.; Shah, J.K.; Maginn, E.J. Molecular dynamics simulation study of the association of lidocainium docusate and its derivatives in aqueous solution. Mol. Pharm. 2015, 12, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Stoimenovski, J.; MacFarlane, D.R. Enhanced membrane transport of pharmaceutically active protic ionic liquids. Chem. Commun. 2011, 47, 11429–11431. [Google Scholar] [CrossRef] [PubMed]
- Megwa, S.A.; Cross, S.E.; Benson, H.A.E.; Roberts, M.S. Ion-pair formation as a strategy to enhance topical delivery of salicylic acid. J. Pharm. Pharmacol. 2010, 52, 919–928. [Google Scholar] [CrossRef] [PubMed]
- U.S. FDA Current Good Manufacturing Practice (CGMP) Regulations. Available online: https://www.fda.gov/drugs/pharmaceutical-quality-resources/current-good-manufacturing-practice-cgmp-regulations (accessed on 15 July 2020).
- Florindo, C.; Araujo, J.M.; Alves, F.; Matos, C.; Ferraz, R.; Prudencio, C.; Noronha, J.P.; Petrovski, Z.; Branco, L.; Rebelo, L.P.N.; et al. Evaluation of solubility and partition properties of ampicillin-based ionic liquids. Int. J. Pharm. 2013, 456, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, M.R.; Li, M.; El-Zahab, B.; Janes, M.E.; Hayes, D.; Warner, I.M. Design, synthesis, and biological evaluation of beta-lactam antibiotic-based imidazolium- and pyridinium-type ionic liquids. Chem. Biol. Drug Des. 2011, 78, 33–41. [Google Scholar] [CrossRef]
- Alves, F.; Oliveira, F.S.; Schroder, B.; Matos, C.; Marrucho, I.M. Synthesis, characterization, and liposome partition of a novel tetracycline derivative using the ionic liquids framework. J. Pharm. Sci. 2013, 102, 1504–1512. [Google Scholar] [CrossRef]
- Stoimenovski, J.; MacFarlane, D.R.; Bica, K.; Rogers, R.D. Crystalline vs. ionic liquid salt forms of active pharmaceutical ingredients: A position paper. Pharm. Res. 2010, 27, 521–526. [Google Scholar] [CrossRef]
- Wiedemann, C.; Hempel, G.; Bordusa, F. Reorientation dynamics and ion diffusivity of neat dimethylimidazolium dimethylphosphate probed by NMR spectroscopy. RSC Adv. 2019, 9, 35735–35750. [Google Scholar] [CrossRef] [Green Version]
- Mrestani-Klaus, C.; Richardt, A.; Wespe, C.; Stark, A.; Humpfer, E.; Bordusa, F. Structural studies on ionic liquid/water/peptide systems by HR-MAS NMR spectroscopy. ChemPhysChem 2012, 13, 1836–1844. [Google Scholar] [CrossRef]
- Daniel, C.I.; Vaca Chávez, F.; Feio, G.; Portugal, C.A.M.; Crespo, J.G.; Sebastião, P.J. 1H NMR relaxometry, viscometry, and PFG NMR studies of magnetic and nonmagnetic ionic liquids. J. Phys. Chem. B 2013, 117, 11877–11884. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Xu, W.; Angell, C.A. Ionic liquids by proton transfer: vapor pressure, conductivity, and the relevance of ΔpKa from aqueous solutions. J. Am. Chem. Soc. 2003, 125, 15411–15419. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, I.; Szabo, Y.; Desert, A.; Trefalt, G.; Oncsik, T.; Borkovec, M. Particle aggregation mechanisms in ionic liquids. Phys. Chem. Chem. Phys. 2014, 16, 9515–9524. [Google Scholar] [CrossRef] [Green Version]
- Cutaia, K.; Chablani, L.; Zhao, F. Basics of compounding: Vehicles for compounded oral liquid medications: A review. Int. J. Pharm. Comp. 2018, 22, 480–489. [Google Scholar]
- Suitthimeathegorn, O.; Jaitely, V.; Florence, A.T. Novel anhydrous emulsions: Formulation as controlled release vehicles. Int. J. Pharm. 2005, 298, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.K.; Raw, A.; Lionberger, R.; Yu, L. Generic development of topical dermatologic products: Formulation development, process development, and testing of topical dermatologic products. Aaps J. 2013, 15, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Gala, U.; Pham, H.; Chauhan, H. Pharmaceutical applications of eutectic mixtures. J. Dev. Drugs 2013, 2, e130. [Google Scholar] [CrossRef]
- Pitha, J.; Irie, T.; Sklar, P.B.; Nye, J.S. Drug solubilizers to aid pharmacologists: Amorphous cyclodextrin derivatives. Life Sci. 1988, 43, 493–502. [Google Scholar] [CrossRef]
- Riebesehl, B.U. Drug delivery with organic solvents or colloidal dispersed systems. In The Practice of Medicinal Chemistry, 4th ed.; Academic Press: Oxford, UK, 2015; pp. 699–722. [Google Scholar]
- Mwesigwa, E.; Basit, A.W. An investigation into moisture barrier film coating efficacy and its relevance to drug stability in solid dosage forms. Int. J. Pharm. 2016, 497, 70–77. [Google Scholar] [CrossRef]
- Ahlneck, C.; Zografi, G. The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state. Int. J. Pharm. 1990, 62, 87–95. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physiochemical and physiological issues and lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Riisager, A.; Fehrmann, R.; Rodriguez, H.; Bica, K.; Rogers, R.D.; Daly, D.T.; Gurau, G. Supported biologically active compounds. WIPO (PCT) Patent WO2011110662A1, 15 September 2011. [Google Scholar]
- Bica, K.; Rodríguez, H.; Gurau, G.; Cojocaru, O.A.; Riisager, A.; Fehrmann, R.; Rogers, R.D. Pharmaceutically active ionic liquids with solids handling, enhanced thermal stability, and fast release. Chem. Commun. 2012, 48, 5422–5424. [Google Scholar] [CrossRef] [PubMed]
- All about Drugs: GSK 2838232. Available online: https://www.allfordrugs.com/2016/06/13/gsk-2838232/ (accessed on 15 July 2020).
- Hatcher, M.A.; Johns, B.A.; Martin, M.T.; Tabet, E.A.; Tang, J. Preparation of betulin derivatives for the treatment of HIV. WIPO (PCT) Patent WO2013090664A1, 20 June 2013. [Google Scholar]
- Johns, B.A. Derivatives of betulin. U.S. Patent US 9,102,685 B2, 11 August 2015. [Google Scholar]
- DeJesus, E.; Harward, S.; Jewell, R.C.; Johnson, M.; Dumont, E.; Wilches, V.; Halliday, F.; Talarico, C.L.; Jeffrey, J.; Gan, J.; et al. A Phase IIa study evaluating safety, pharmacokinetics, and antiviral activity of GSK2838232, a novel, second-generation maturation inhibitor, in participants with human immunodeficiency virus type 1 infection. Clin. Infect. Dis. 2019, Pii: ciz938. [Google Scholar] [CrossRef]
- Chemical Book GSK 2838232. Full IUPAC Chemical Name: 4-(((3R,5aR,5bR,7aR,9S,11aR,11bR,13aS)-3a-((R)-2-((4-chlorobenzy1)(2-(dimethylamino)ethyl)amino)-l-hydroxyethyl)-l-isopropyl-5a,5b,8,8,11a-pentamethyl-2-oxo-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,1a,11b,12,13,13a-octadecahydro-2H-cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid. Available online: https://www.chemicalbook.com/ProductChemicalPropertiesCB12664173_EN.htm (accessed on 18 August 2020).
- Johnson, M.; Jewell, R.C.; Gan, J.; Dumont, E.; Burns, O.; Johns, B.A. A Phase 1 study to assess the relative bioavailability, food effect, and safety of a tablet formulation of GSK2838232, a novel HIV maturation inhibitor in healthy participants after single and repeated doses. Clin. Pharmacol. Drug Dev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Safety and Efficacy Study of GSK2838232 in Human Immunodeficiency Virus (HIV)-1 Infected Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT03045861 (accessed on 15 July 2020).
- Burke, M.D. Transforming the Patient Experience through Long Acting Injectable/Implantable Formulations: New Opportunities and Technologies. In Proceedings of the American Association of Pharmaceutical Scientists (AAPS) PharmSci 360 Conference, Washington, DC, USA, 4–7 November 2018. [Google Scholar]
- Burke, M.D.; McQueen, L.; Rusk, S. Ionic Liquid salts of 4-4-(((3R,5aR,5bR,7aR,9S,11aR,11bR,13aS)-3a-((R)-2-((4-chlorobenzy1)(2-(dimethylamino)ethyl)amino)-l-hydroxyethyl)-l-isopropyl-5a,5b,8,8,11a-pentamethyl-2-oxo-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,1a,11b,12,13,13a-octadecahydro-2H-cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid. WIPO (PCT) Patent WO2019186342A1, 3 October 2019. [Google Scholar]
- Akhavein, N.; Velthuisen, E. Novel parenteral platforms for tunable long-acting administration of hydrophobic HIV compound. In Proceedings of the HIV Research for Prevention Conference, (PD09.02), Madrid, Spain, 21–25 October 2018. [Google Scholar]
- Bica, K.; Rogers, R.D. Confused ionic liquid ions—A “liquification” and dosage strategy for pharmaceutically active salts. Chem. Commun. 2010, 46, 1215–1217. [Google Scholar] [CrossRef]
- Rogers, R.D.; Daly, D.T.; MacFarlane, D.; Scott, J.L.; Seddon, K.R.; Gurau, G.; Bica, K.; Turanjanin, J.; Dean, P.M. Dual functioning ionic liquids and salts thereof. U.S. Patent 9,278,134, 29 December 2009. [Google Scholar]
- Pannu, J. Running ahead of Pandemics: Achieving in-advance Antiviral Drugs. Available online: https://www.mercatus.org/publications/covid-19-crisis-response/running-ahead-pandemics-achieving-advance-antiviral-drugs (accessed on 15 July 2020).
- Prescribing Information for Zovirax ® (Acyclovir). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/018828s031lbl.pdf (accessed on 15 July 2020).
- AHFS Drug Information American Society of Hospital Pharmacists (AHFS). Bethesda MD 765. Available online: http://www.ahfsdruginformation.com/ (accessed on 15 July 2020).
- Kristl, A.; Srcic, S.; Vrecer, F.; Sustar, B.; Vojnovic, D.D. Polymorphism and pseudopolymorphism: Influencing the dissolution properties of the guanine derivative Acyclovir. Int. J. Pharm. 1996, 139, 231–235. [Google Scholar] [CrossRef]
- Bergstrom, C.A.; Norinder, U.; Luthman, K.; Artursson, P. Experimental and computational screening models for prediction of aqueous drug solubility. Pharm. Res. 2002, 19, 182–188. [Google Scholar] [CrossRef]
- Nair, A.B.; Attimarad, M.; Al-Dhubiab, B.E.; Wadhwa, J.; Harsha, S.; Ahmed, M. Enhanced oral bioavailability of Acyclovir by inclusion complex using hydroxypropyl-β-cyclodextrin. Drug Deliv. 2014, 21, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Budavari, S.; O’Neil, M.; Smith, A.; Heckelman, P.; Obenchain, J. The Merck Index, 12th ed.; Taylor & Francis: Abingdon, UK; Merck & Co.: Whitehouse Station, NJ, USA, 1996; p. 27. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer or Water | mol/L | mg/mL | mg Active/ mL |
|---|---|---|---|
| Acyclovir (3Acy·2H2O), mp = 256.5 °C (dec) b | |||
| Water | 0.00699(1) | 1.574(2) | 1.495(2) |
| PBS | 0.00631(3) | 1.4(1) | 1.33(1) |
| SIF | 0.00444(1) | 1.0(1) | 0.95(1) |
| Na[Acy] a, mp = 256 °C | |||
| Water | 0.404(5) | 100.0 | 90.68 |
| [Cho][Acy] a, mp = ND c | |||
| Water | 2.68(5) | 880(20) | 600(10) |
| PBS | 2.92(5) | 960(20) | 660(10) |
| SIF | 2.83(9) | 930(30) | 650(20) |
| SGF | 3.8(1) | 1300(500) | 860(30) |
| [P4,4,4,4][Acy] a, mp = 95.1 °C d | |||
| Water | 1.66(3) | 800(10) | 373(7) |
| [N4,4,4,4][Acy] a, mp = 70.6 °C | |||
| Water | 0.952(8) | 444.7 | 213.7 |
| PBS | 0.9867(2) | 490(30) | 220(20) |
| SIF | 0.6394(2) | 300(20) | 140(10) |
| SGF | 0.7416(8) | 350 (2) | 170(20) |
| [N1,1,1,16][Acy] a, mp = 48.3 °C e | |||
| Water | 0.4496(25) | 229(1) | 101.3(5) |
| [H2Acy]Cl a, mp >290 °C | |||
| Water | 0.241(1) | 63.1(30) | 54.4(3) |
| PBS | 0.244(7) | 64(2) | 55(2) |
| SIF | 0.339(7) | 89(2) | 76(2) |
| SGF | 0.348(3) | 91.0(8) | 78.4(7) |
| [H2Acy][Doc] a, mp = ND c | |||
| Water | 0.01305(1) | 8.461(5) | 2.948(4) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shamshina, J.L.; Rogers, R.D. Are Myths and Preconceptions Preventing Us from Applying Ionic Liquid Forms of Antiviral Medicines to the Current Health Crisis? Int. J. Mol. Sci. 2020, 21, 6002. https://doi.org/10.3390/ijms21176002
Shamshina JL, Rogers RD. Are Myths and Preconceptions Preventing Us from Applying Ionic Liquid Forms of Antiviral Medicines to the Current Health Crisis? International Journal of Molecular Sciences. 2020; 21(17):6002. https://doi.org/10.3390/ijms21176002
Chicago/Turabian StyleShamshina, Julia L., and Robin D. Rogers. 2020. "Are Myths and Preconceptions Preventing Us from Applying Ionic Liquid Forms of Antiviral Medicines to the Current Health Crisis?" International Journal of Molecular Sciences 21, no. 17: 6002. https://doi.org/10.3390/ijms21176002