Evolution and Expression of the Membrane Attack Complex and Perforin Gene Family in the Poaceae

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Identification of MACPF Genes in Poaceae Genomes
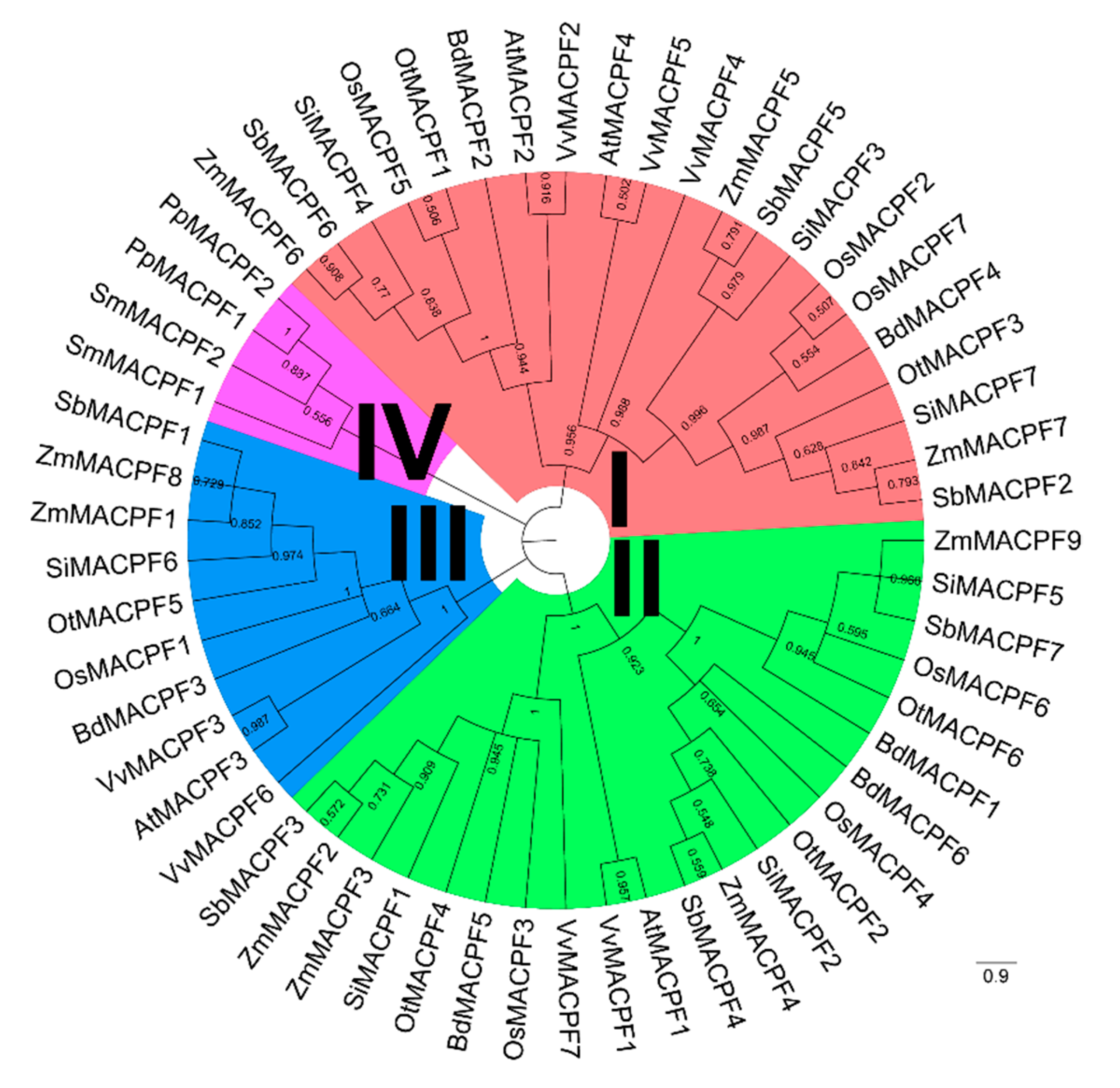
2.2. Phylogenetic and Structure Analysis
2.3. MACPF Gene Localization and Gene Duplication
2.4. Synteny Analysis of the MACPF Gene Family in Poaceae
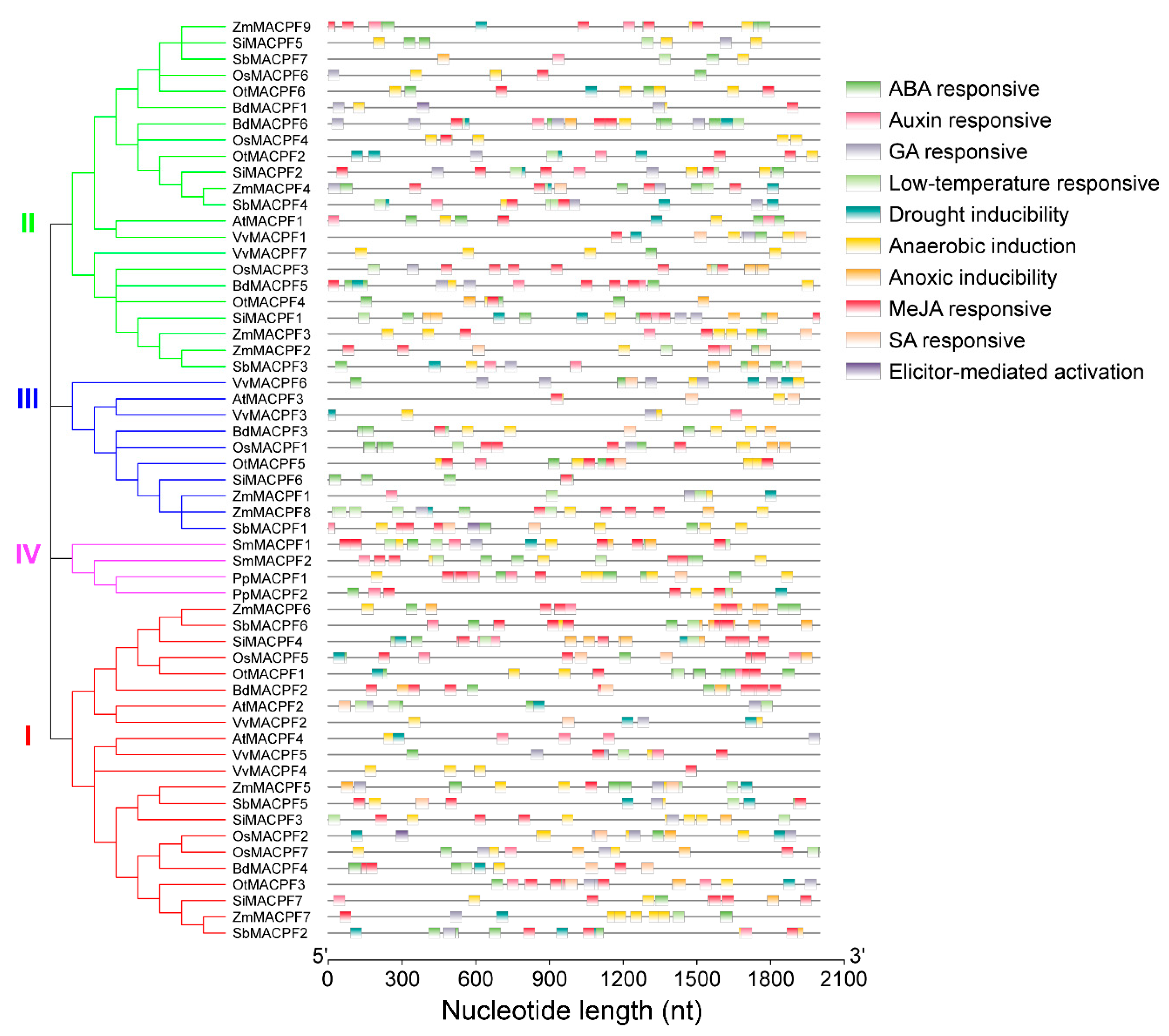
2.5. Analysis of Stress-Responsive cis-Regulatory Elements in MACPF Promoters
2.6. Expression Profile of MACPF Genes in Poaceae
2.7. Involvement of MACPF Genes in Response to Stresses
2.8. Expression of Rice MACPF Genes in Response to Jasmonic Acid Treatment
3. Discussion
3.1. The MACPF Gene Family is Conserved across Land Plants
3.2. MACPF Duplications are an Important Feature Across the Poaceae
3.3. MACPF Function and Gene Expression
4. Materials and Methods
4.1. Data Retrieval of MACPF Genes
4.2. Gene Exon–Intron Structure Predictions
4.3. Analysis of Domain Combinations and Architecture of MACPF Promoters and MACPF Proteins
4.4. Phylogenetic Analysis
4.5. Duplication Events and Synteny Analysis of MACPF Genes in the Poaceae
4.6. Gene Expression Analysis
4.7. RNA Extraction and Quantitative RT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| ABRE | ABA-responsive element |
| AS | Alternative splicing |
| Bd | Brachypodium distachyon |
| CAD1 | Constitutively activated cell death 1 |
| GA | Gibberellin |
| GSDS | Gene Structure Display Server |
| HMM | Hidden Markov model |
| Ka | Non-synonymous distance |
| Ks | Synonymous distance |
| LTRE | Low temperature responsive element |
| MAC | Membrane attack complex |
| MACPF | Membrane Attack Complex and Perforin |
| MBS | MYB binding site |
| MeJA | Methyl jasmonate |
| MEME | Multiple EM for motif elicitation |
| MW | Molecular weight |
| NSL1 | Necrotic spotted lesions 1 |
| NSL2 | Necrotic spotted lesions 2 |
| Os | Oryza sativa |
| Ot | Oropetium thomaeum |
| pI | Isoelectric point |
| Pp | Physcomitrium (Physcomitrella) patens |
| qRT-PCR | Quantitative reverse transcription PCR |
| SA | Salicylic acid |
| Sb | Sorghum bicolor |
| Si | Setaria italica |
| Sm | Selaginella moellendorffii |
| Zm | Zea mays |
References
- Tschopp, J.; Masson, D.; Stanley, K.K. Structural/functional similarity between proteins involved in complement- and cytotoxic T-lymphocyte-mediated cytolysis. Nature 1986, 322, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Rosado, C.J.; Buckle, A.M.; Law, R.H.; Butcher, R.E.; Kan, W.T.; Bird, C.H.; Ung, K.; Browne, K.A.; Baran, K.; Bashtannyk-Puhalovich, T.A.; et al. A common fold mediates vertebrate defense and bacterial attack. Science 2007, 317, 1548–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Hagelsieb, G.; Vitug, B.; Medrano-Soto, A.; Saier, M.H., Jr. The Membrane Attack Complex/Perforin Superfamily. J. Mol. Microb. Biotech. 2017, 27, 252–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukoyanova, N.; Hoogenboom, B.W.; Saibil, H.R. The membrane attack complex, perforin and cholesterol-dependent cytolysin superfamily of pore-forming proteins. J. Cell Sci. 2016, 129, 2125–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosado, C.J.; Kondos, S.; Bull, T.E.; Kuiper, M.J.; Law, R.H.; Buckle, A.M.; Voskoboinik, I.; Bird, P.I.; Trapani, J.A.; Whisstock, J.C.; et al. The MACPF/CDC family of pore-forming toxins. Cell. Microbiol. 2008, 10, 1765–1774. [Google Scholar] [CrossRef] [Green Version]
- Zaitseva, J.; Vaknin, D.; Krebs, C.; Doroghazi, J.; Milam, S.L.; Balasubramanian, D.; Duck, N.B.; Freigang, J. Structure-function characterization of an insecticidal protein GNIP1Aa, a member of an MACPF and beta-tripod families. Proc. Natl. Acad. Sci. USA 2019, 116, 2897–2906. [Google Scholar] [CrossRef] [Green Version]
- Hadders, M.A.; Beringer, D.X.; Gros, P. Structure of C8α-MACPF reveals mechanism of membrane attack in complement immune defense. Science 2007, 317, 1552–1554. [Google Scholar] [CrossRef]
- Law, R.H.; Lukoyanova, N.; Voskoboinik, I.; Caradoc-Davies, T.T.; Baran, K.; Dunstone, M.A.; D’Angelo, M.E.; Orlova, E.V.; Coulibaly, F.; Verschoor, S.; et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 2010, 468, 447–451. [Google Scholar] [CrossRef]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef]
- Morita-Yamamuro, C.; Tsutsui, T.; Sato, M.; Yoshioka, H.; Tamaoki, M.; Ogawa, D.; Matsuura, H.; Yoshihara, T.; Ikeda, A.; Uyeda, I.; et al. The Arabidopsis gene CAD1 controls programmed cell death in the plant immune system and encodes a protein containing a MACPF domain. Plant Cell Physiol. 2005, 46, 902–912. [Google Scholar] [CrossRef] [Green Version]
- Esser, A.F. The membrane attack complex of complement. Assembly, structure and cytotoxic activity. Toxicology 1994, 87, 229–247. [Google Scholar] [CrossRef]
- Noutoshi, Y.; Kuromori, T.; Wada, T.; Hirayama, T.; Kamiya, A.; Imura, Y.; Yasuda, M.; Nakashita, H.; Shirasu, K.; Shinozaki, K. Loss of Necrotic Spotted Lesions 1 associates with cell death and defense responses in Arabidopsis thaliana. Plant Mol. Biol. 2006, 62, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, T.; Morita-Yamamuro, C.; Asada, Y.; Minami, E.; Shibuya, N.; Ikeda, A.; Yamaguchi, J. Salicylic acid and a chitin elicitor both control expression of the CAD1 gene involved in the plant immunity of Arabidopsis. Biosci. Biotech. Biochem. 2006, 70, 2042–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, S.; Sogame, M.; Hata, M.; Singkaravanit-Ogawa, S.; Pislewska-Bednarek, M.; Onozawa-Komori, M.; Nishiuchi, T.; Hiruma, K.; Saitoh, H.; Terauchi, R.; et al. Dysfunction of Arabidopsis MACPF domain protein activates programmed cell death via tryptophan metabolism in MAMP-triggered immunity. Plant J. 2017, 89, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Nomura, K.; Wang, X.; Sohrabi, R.; Xu, J.; Yao, L.; Paasch, B.C.; Ma, L.; Kremer, J.; Cheng, Y.; et al. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 2020, 580, 653–657. [Google Scholar] [CrossRef]
- Asada, Y.; Yamamoto, M.; Tsutsui, T.; Yamaguchi, J. The Arabidopsis NSL2 negatively controls systemic acquired resistance via hypersensitive response. Plant Biotechnol. 2011, 28, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Project, International Rice Genome Sequencing. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J.; et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanBuren, R.; Bryant, D.; Edger, P.P.; Tang, H.; Burgess, D.; Challabathula, D.; Spittle, K.; Hall, R.; Gu, J.; Lyons, E.; et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 2015, 527, 508–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauser, L.; Wieloch, W.; Stougaard, J. Evolution of NIN-Like proteins in Arabidopsis, rice, and Lotus japonicus. J. Mol. Evol. 2005, 60, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Akhunov, E.D.; Sehgal, S.; Liang, H.; Wang, S.; Akhunova, A.R.; Kaur, G.; Li, W.; Forrest, K.L.; See, D.; Simkova, H.; et al. Comparative analysis of syntenic genes in grass genomes reveals accelerated rates of gene structure and coding sequence evolution in polyploid wheat. Plant Physiol. 2013, 161, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. TRENDS Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Nei, M. The new mutation theory of phenotypic evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 12235–12242. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools—An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020. [Google Scholar] [CrossRef]
- Wang, W.; Chen, Q.; Xu, S.; Liu, W.C.; Zhu, X.; Song, C.P. Trehalose-6-phosphate phosphatase E modulates ABA-controlled root growth and stomatal movement in Arabidopsis. J. Integr. Plant Biol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.J.; Luo, Y.F.; Liao, B.; Xie, L.J.; Chen, L.; Xiao, S.; Li, J.T.; Hu, S.N.; Shu, W.S. Comparative transcriptome analysis of transporters, phytohormone and lipid metabolism pathways in response to arsenic stress in rice (Oryza sativa). New Phytol. 2012, 195, 97–112. [Google Scholar] [CrossRef]
- Jain, M.; Nijhawan, A.; Arora, R.; Agarwal, P.; Ray, S.; Sharma, P.; Kapoor, S.; Tyagi, A.K.; Khurana, J.P. F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 2007, 143, 1467–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, A.; Klein, R.R.; Dugas, D.V.; Lu, Z.Y.; Regulski, M.; Klein, P.E.; Ware, D. Expanding and Vetting Sorghum bicolor Gene Annotations through Transcriptome and Methylome Sequencing. Plant Genome 2014, 7. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoopes, G.M.; Hamilton, J.P.; Wood, J.C.; Esteban, E.; Pasha, A.; Vaillancourt, B.; Provart, N.J.; Buell, C.R. An updated gene atlas for maize reveals organ-specific and stress-induced genes. Plant J. 2019, 97, 1154–1167. [Google Scholar] [CrossRef] [Green Version]
- Sibout, R.; Proost, S.; Hansen, B.O.; Vaid, N.; Giorgi, F.M.; Ho-Yue-Kuang, S.; Legee, F.; Cezart, L.; Bouchabke-Coussa, O.; Soulhat, C.; et al. Expression atlas and comparative coexpression network analyses reveal important genes involved in the formation of lignified cell wall in Brachypodium distachyon. New Phytol. 2017, 215, 1009–1025. [Google Scholar] [CrossRef] [Green Version]
- Ota, K.; Butala, M.; Viero, G.; Dalla Serra, M.; Sepcic, K.; Macek, P. Fungal MACPF-like proteins and aegerolysins: Bi-component pore-forming proteins? Subcell. Biochem. 2014, 80, 271–291. [Google Scholar] [CrossRef]
- Kondos, S.C.; Hatfaludi, T.; Voskoboinik, I.; Trapani, J.A.; Law, R.H.; Whisstock, J.C.; Dunstone, M.A. The structure and function of mammalian membrane-attack complex/perforin-like proteins. Tissue Antigens 2010, 76, 341–351. [Google Scholar] [CrossRef]
- Wade, K.R.; Tweten, R.K. The Apicomplexan CDC/MACPF-like pore-forming proteins. Curr. Opin. Microbiol. 2015, 26, 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swigonova, Z.; Lai, J.; Ma, J.; Ramakrishna, W.; Llaca, V.; Bennetzen, J.L.; Messing, J. Close split of sorghum and maize genome progenitors. Genome Res. 2004, 14, 1916–1923. [Google Scholar] [CrossRef] [Green Version]
- Staiger, D.; Brown, J.W.S. Alternative Splicing at the Intersection of Biological Timing, Development, and Stress Responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.N.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the Alternative Splicing Landscape in Plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashirullah, A.; Cooperstock, R.L.; Lipshitz, H.D. Spatial and temporal control of RNA stability. Proc. Natl. Acad. Sci. USA 2001, 98, 7025–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Sun, F.; Yan, L.; Li, R.; Bai, J.; Caetano-Anolles, G. Genome-Wide Identification and Characterization of the Vacuolar H+-ATPase Subunit H Gene Family in Crop Plants. Int. J. Mol. Sci. 2019, 20, 5125. [Google Scholar] [CrossRef] [Green Version]
- Vandepoele, K.; Simillion, C.; Van de Peer, Y. Evidence that rice and other cereals are ancient aneuploids. Plant Cell 2003, 15, 2192–2202. [Google Scholar] [CrossRef]
- Yu, J.; Wang, J.; Lin, W.; Li, S.; Li, H.; Zhou, J.; Ni, P.; Dong, W.; Hu, S.; Zeng, C.; et al. The Genomes of Oryza sativa: A history of duplications. PLoS Biol. 2005, 3, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.; Zhang, Y.; Deng, X.; Li, S.; Zhang, C.; Li, Y. Comparative Genomic and Transcriptomic Analysis Suggests the Evolutionary Dynamic of GH3 Genes in Gramineae Crops. Front. Plant Sci. 2019, 10, 1297. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.S.; Raes, J. Duplication and divergence: The evolution of new genes and old ideas. Annu. Rev. Genet. 2004, 38, 615–643. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, T.; Shinozaki, K. Research on plant abiotic stress responses in the post-genome era: Past, present and future. Plant J. 2010, 61, 1041–1052. [Google Scholar] [CrossRef]
- Blazquez, M.A.; Nelson, D.C.; Weijers, D. Evolution of Plant Hormone Response Pathways. Annu. Rev. Plant Biol. 2020, 71, 327–353. [Google Scholar] [CrossRef]
- Palenik, B.; Grimwood, J.; Aerts, A.; Rouze, P.; Salamov, A.; Putnam, N.; Dupont, C.; Jorgensen, R.; Derelle, E.; Rombauts, S.; et al. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. USA 2007, 104, 7705–7710. [Google Scholar] [CrossRef] [Green Version]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochnik, S.E.; Umen, J.; Nedelcu, A.M.; Hallmann, A.; Miller, S.M.; Nishii, I.; Ferris, P.; Kuo, A.; Mitros, T.; Fritz-Laylin, L.K.; et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 2010, 329, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A.; Salamov, A.; Shapiro, H.; Nishiyama, T.; Perroud, P.F.; Lindquist, E.A.; Kamisugi, Y.; et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; dePamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef]
- Finn, R.D.; Mistry, J.; Schuster-Bockler, B.; Griffiths-Jones, S.; Hollich, V.; Lassmann, T.; Moxon, S.; Marshall, M.; Khanna, A.; Durbin, R.; et al. Pfam: Clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. [Google Scholar] [CrossRef] [Green Version]
- Kersey, P.J.; Allen, J.E.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; Hughes, D.S.; Humphrey, J.; Kerhornou, A.; Khobova, J.; et al. Ensembl Genomes 2013: Scaling up access to genome-wide data. Nucleic Acids Res. 2014, 42, D546–D552. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.J.; Feng, D.R.; Li, W.Y.; Wang, H.B.; Wang, J.F.; Liu, B. Comprehensive and evolutionary analysis of protein tyrosine phosphatases (PTP) in the green plants. Plant Omics 2013, 6, 215–223. [Google Scholar]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.J.; Chen, F.; Peng, Y.J.; Xie, L.J.; Liu, D.; Han, M.Q.; Chen, F.; Xiao, S.; Huang, J.C.; Li, J. Arabidopsis thaliana Plants Engineered to Produce Astaxanthin Show Enhanced Oxidative Stress Tolerance and Bacterial Pathogen Resistance. J. Agric. Food Chem. 2019, 67, 12590–12598. [Google Scholar] [CrossRef]
- Zhou, Y.; Tan, W.J.; Xie, L.J.; Qi, H.; Yang, Y.C.; Huang, L.P.; Lai, Y.X.; Tan, Y.F.; Zhou, D.M.; Yu, L.J.; et al. Polyunsaturated linolenoyl-CoA modulates ERF-VII-mediated hypoxia signaling in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 330–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant | Clade | Genome Size (Mb) | Total | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|---|---|---|
| Green alga (Ostreococcus tauri) | Chlorophytes | 12.56 | 0 | 0 | 0 | 0 | 0 |
| Green alga (Ostreococcus lucimarinus) | Chlorophytes | 13.2 | 0 | 0 | 0 | 0 | 0 |
| Green alga (Chlamydomonas reinhardtii) | Chlorophytes | 120 | 0 | 0 | 0 | 0 | 0 |
| Green alga (Volvox carteri) | Chlorophytes | 138 | 0 | 0 | 0 | 0 | 0 |
| Moss (Physcomitrium [Physcomitrella] patens) | Bryophytes | 472 | 2 | 0 | 0 | 0 | 2 |
| Spikemoss (Selaginella moellendorffii) | Lycophytes | 100 | 2 | 0 | 0 | 0 | 2 |
| Eudicot (Arabidopsis thaliana) | Eudicots | 135 | 4 | 2 | 1 | 1 | 0 |
| Eudicot (Vitis vinifera) | Eudicots | 500 | 7 | 3 | 2 | 2 | 0 |
| Monocot (Brachypodium distachyon) | Monocots | 355 | 6 | 2 | 3 | 1 | 0 |
| Monocot (Oryza sativa) | Monocots | 370 | 7 | 3 | 3 | 1 | 0 |
| Monocot (Zea mays) | Monocots | 2400 | 9 | 3 | 4 | 2 | 0 |
| Monocot (Sorghum bicolor) | Monocots | 730 | 7 | 3 | 3 | 1 | 0 |
| Monocot (Setaria italica) | Monocots | 490 | 7 | 3 | 3 | 1 | 0 |
| Monocot (Oropetium thomaeum) | Monocots | 245 | 6 | 2 | 3 | 1 | 0 |
| Total | - | - | 57 | 21 | 22 | 10 | 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Liu, D.; Chen, S.; Dai, Y.; Guo, W.; Zhang, X.; Wang, L.; Ma, S.; Xiao, M.; Qi, H.; et al. Evolution and Expression of the Membrane Attack Complex and Perforin Gene Family in the Poaceae. Int. J. Mol. Sci. 2020, 21, 5736. https://doi.org/10.3390/ijms21165736
Yu L, Liu D, Chen S, Dai Y, Guo W, Zhang X, Wang L, Ma S, Xiao M, Qi H, et al. Evolution and Expression of the Membrane Attack Complex and Perforin Gene Family in the Poaceae. International Journal of Molecular Sciences. 2020; 21(16):5736. https://doi.org/10.3390/ijms21165736
Chicago/Turabian StyleYu, Lujun, Di Liu, Shiyi Chen, Yangshuo Dai, Wuxiu Guo, Xue Zhang, Linna Wang, Sirui Ma, Ming Xiao, Hua Qi, and et al. 2020. "Evolution and Expression of the Membrane Attack Complex and Perforin Gene Family in the Poaceae" International Journal of Molecular Sciences 21, no. 16: 5736. https://doi.org/10.3390/ijms21165736