Aquilariae Lignum Methylene Chloride Fraction Attenuates IL-1β-Driven Neuroinflammation in BV2 Microglial Cells


Abstract
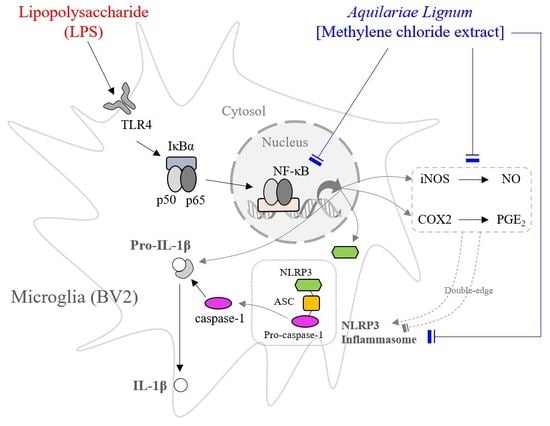
:
1. Introduction
2. Results
2.1. Fingerprinting of ALF
2.2. Effects on Cell Viability and Nitric Oxide (NO) Production
2.3. Effects on the Molecules Associated with Inflammation
2.4. Effects on Nuclear Factor-Kappa B (NF-κB) Translocation
2.5. Effects on Inflammatory Cytokines
2.6. Effects on the NLRP3 Inflammasome Pathway
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of the Aquilariae Lignum Methylene Chloride Fraction (ALF)
4.3. Fingerprinting Analysis of ALF
4.4. Cell Culture and Cell Viability
4.5. NO Assay
4.6. PGE2 Assay
4.7. Proinflammatory Cytokine Activity
4.8. Caspase-1 Assay
4.9. Western Blot Analysis
4.10. RNA Isolation and Quantitative Real-Time PCR
4.11. Immunofluorescence Staining
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.L.; Bowers, W.J. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J. Neuroimmune Pharmacol. 2012, 7, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in Alzheimer’s Disease: Risk Factors and Inflammation. Front. Neurol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Qi, Y.; Klyubin, I.; Cuello, A.C.; Rowan, M.J. NLRP3-dependent synaptic plasticity deficit in an Alzheimer’s disease amyloidosis model in vivo. Neurobiol. Dis. 2018, 114, 24–30. [Google Scholar] [CrossRef]
- Flores, J.; Noel, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.Z.; Cao, Q.; Liu, C. Targeting NLRP3 Inflammasome in the Treatment of CNS Diseases. Front. Mol. Neurosci. 2018, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.T.; Nguyen, V.D. Biodiversity of major bacterial groups in association with agarwood (Aquilaria Crassna) in Khanh Hoa province. J. Vietam. Environ. 2014, 6, 132–137. [Google Scholar]
- Lee, H.Y.; Lee, J.S.; Kim, H.G.; Kim, W.Y.; Lee, S.B.; Choi, Y.H.; Son, C.G. The ethanol extract of Aquilariae Lignum ameliorates hippocampal oxidative stress in a repeated restraint stress mouse model. BMC Complement. Altern. Med. 2017, 17, 397. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, W.Y.; Jeon, Y.J.; Lee, S.K.; Son, C.G. Aquilariae Lignum extract attenuates glutamate-induced neuroexcitotoxicity in HT22 hippocampal cells. Biomed. Pharmacother. 2018, 106, 1031–1038. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Singhal, G.; Baune, B.T. Microglia: An Interface between the Loss of Neuroplasticity and Depression. Front. Cell. Neurosci. 2017, 11, 270. [Google Scholar] [CrossRef] [Green Version]
- Erickson, M.A.; Dohi, K.; Banks, W.A. Neuroinflammation: A common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation 2012, 19, 121–130. [Google Scholar] [CrossRef] [Green Version]
- da Fonseca, A.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Gresa-Arribas, N.; Vieitez, C.; Dentesano, G.; Serratosa, J.; Saura, J.; Sola, C. Modelling neuroinflammation in vitro: A tool to test the potential neuroprotective effect of anti-inflammatory agents. PLoS ONE 2012, 7, e45227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, J.U.; Woodling, N.S.; Shi, J.; Andreasson, K.I. Inflammatory Cyclooxygenase Activity and PGE2 Signaling in Models of Alzheimer’s Disease. Curr. Immunol. Rev. 2015, 11, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Solfrizzi, V.; Panza, F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front. Aging Neurosci. 2010, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepanier, C.H.; Milgram, N.W. Neuroinflammation in Alzheimer’s disease: Are NSAIDs and selective COX-2 inhibitors the next line of therapy? J. Alzheimer’s. Dis. 2010, 21, 1089–1099. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Srinivasan, M.; Lahiri, D.K. Significance of NF-kappaB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef] [Green Version]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S.A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef]
- Cascella, M.; Bimonte, S.; Muzio, M.R.; Schiavone, V.; Cuomo, A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agent. Cancer 2017, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.C.; Robbins, M.E. PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic. Biol. Med. 2008, 45, 1695–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, R.M. Role of caspase 1 in neurologic disease. Arch. Neurol. 2000, 57, 1273–1276. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Petrin, D.; Zhang, Y.; Bergeron, C.; Goodyer, C.G.; LeBlanc, A.C. Caspase-1 activation of caspase-6 in human apoptotic neurons. Cell Death Differ. 2006, 13, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Takeuchi, H. Neurotoxicity by microglia: Mechanisms and potential therapeutic strategy. Clin. Exp. Neuroimmunol. 2010, 1, 12–21. [Google Scholar] [CrossRef]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Rathinam, V.A.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat. Immunol. 2013, 14, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Sokolowska, M.; Chen, L.Y.; Liu, Y.; Martinez-Anton, A.; Qi, H.Y.; Logun, C.; Alsaaty, S.; Park, Y.H.; Kastner, D.L.; Chae, J.J.; et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J. Immunol. 2015, 194, 5472–5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tao, J.; Yao, Y. Prostaglandin E2 Activates NLRP3 Inflammasome in Endothelial Cells to Promote Diabetic Retinopathy. Horm. Metab. Res. 2018, 50, 704–710. [Google Scholar] [CrossRef]
- Sheppe, A.E.F.; Kummari, E.; Walker, A.; Richards, A.; Hui, W.W.; Lee, J.H.; Mangum, L.; Borazjani, A.; Ross, M.K.; Edelmann, M.J. PGE2 Augments Inflammasome Activation and M1 Polarization in Macrophages Infected with Salmonella Typhimurium and Yersinia enterocolitica. Front. Microbiol. 2018, 9, 2447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, W.; Zhang, J. Effects of novel anxiolytic 4-butyl-alpha-agarofuran on levels of monoamine neurotransmitters in rats. Eur. J. Pharmacol. 2004, 504, 39–44. [Google Scholar] [CrossRef]
- Thangnipon, W.; Suwanna, N.; Kitiyanant, N.; Soi-Ampornkul, R.; Tuchinda, P.; Munyoo, B.; Nobsathian, S. Protective role of N-trans-feruloyltyramine against beta-amyloid peptide-induced neurotoxicity in rat cultured cortical neurons. Neurosci. Lett. 2012, 513, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.X.; Zhu, Z.X.; Pang, D.R.; Li, Y.T.; Huang, Z.; Shi, S.P.; Zheng, J.; Zhang, Q.; Zhao, Y.F.; Tu, P.F.; et al. Anti-neuroinflammatory sesquiterpenes from Chinese eaglewood. Fitoterapia 2015, 106, 115–121. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Primer Sequence (5′→ 3′) |
|---|---|---|
| iNOS | Forward | GGC AGC CTG TGA GAC CTT TG TGC ATT GGA AGT GAA GCG TTT |
| Reverse | ||
| COX-2 | Forward | CAG CAA CTC CTT GCT GTT CC TGG GCA AAG AAT GCA AAC ATC |
| Reverse | ||
| TNF-α | Forward | CTC CCA GGT TCT CTT CAA GG TGG AAG ACT CCT CCC AGG TA |
| Reverse | ||
| IL-1β | Forward | AAG TTG ACG GAC CCC AAA AGA TTG ATG TGC TGC GAG AT |
| Reverse | ||
| GAPDH | Forward | CAT GGC CTT CCG TGT TCC T CCT GCT TCA CCA CCT TCT TGA |
| Reverse |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-S.; Jeon, Y.-J.; Kang, J.-Y.; Lee, S.-K.; Lee, H.-D.; Son, C.-G. Aquilariae Lignum Methylene Chloride Fraction Attenuates IL-1β-Driven Neuroinflammation in BV2 Microglial Cells. Int. J. Mol. Sci. 2020, 21, 5465. https://doi.org/10.3390/ijms21155465
Lee J-S, Jeon Y-J, Kang J-Y, Lee S-K, Lee H-D, Son C-G. Aquilariae Lignum Methylene Chloride Fraction Attenuates IL-1β-Driven Neuroinflammation in BV2 Microglial Cells. International Journal of Molecular Sciences. 2020; 21(15):5465. https://doi.org/10.3390/ijms21155465
Chicago/Turabian StyleLee, Jin-Seok, Yoo-Jin Jeon, Ji-Yun Kang, Sam-Keun Lee, Hwa-Dong Lee, and Chang-Gue Son. 2020. "Aquilariae Lignum Methylene Chloride Fraction Attenuates IL-1β-Driven Neuroinflammation in BV2 Microglial Cells" International Journal of Molecular Sciences 21, no. 15: 5465. https://doi.org/10.3390/ijms21155465