Endocytosis of Connexin 36 is Mediated by Interaction with Caveolin-1


Abstract
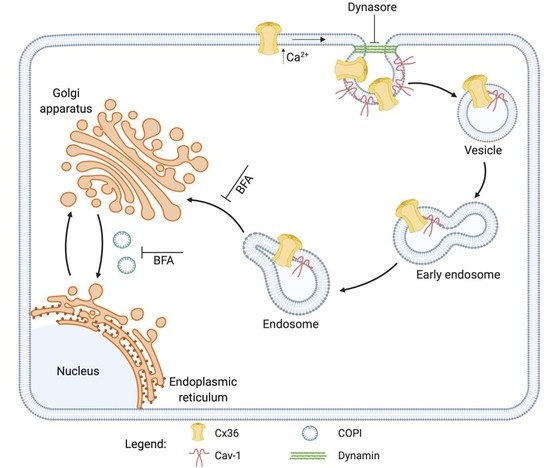
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Cx36 Co-Localizes With and Is in Close Proximity to Cav-1 in Neuro 2a Cells
2.2. Calcium Enhances the Interaction Between Cx36 and Cav-1
2.3. Cx36 and Cav-1 Co-Localize More With Golgi Than the ER and Their Interaction is Reduced With BFA Treatment
2.4. Cav-1 Affects Both Vesicular and Membrane Transport of Cx36
2.5. Cav-1 Depletes Levels of Cx36 From the Membrane via Endocytosis
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction and Site-Directed Mutagenesis
4.2. Cell Culture and Transient Transfection
4.3. Pharmacology
4.4. Western Blot
4.5. Coimmunoprecipitation (CoIP)
4.6. Cell Surface Biotinylation Assay
4.7. Confocal Microscopy, Co-Localization, and Immunofluorescence
4.8. Förster Resonance Energy Transfer Analysis (FRET)
4.9. Fluorescence Recovery After Photobleaching (FRAP)
4.10. Total Internal Reflection Fluorescence (TIRF)
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Laird, D.W. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010, 20, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Thévenin, A.F.; Kowal, T.J.; Fong, J.T.; Kells, R.M.; Fisher, C.G.; Falk, M.M. Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology 2013, 28, 93–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, J.I.; Rash, J.E. Cx36, Cx43 and Cx45 in mouse and rat cerebellar cortex: Species-specific expression, compensation in Cx36 null mice and co-localization in neurons vs. glia. Eur. J. Neurosci. 2017, 46, 1790–1804. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Lynn, B.D.; Senecal, J.M.M.; Stecina, K. Connexin36 Expression in Primary Afferent Neurons in Relation to the Axon Reflex and Modality Coding of Somatic Sensation. Neuroscience 2018, 383, 216–234. [Google Scholar] [CrossRef]
- Nagy, J.I.; Pereda, A.E.; Rash, J.E. On the occurrence and enigmatic functions of mixed (chemical plus electrical) synapses in the mammalian CNS. Neurosci. Lett. 2019, 695, 53–64. [Google Scholar] [CrossRef]
- Alev, C.; Urscheld, S.; Sonntag, S.; Zoidl, G.; Fort, A.G.; Höher, T.; Matsubara, M.; Willecke, K.; Spray, D.C.; Dermietzel, R. The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 20964–20969. [Google Scholar] [CrossRef] [Green Version]
- Del Corsso, C.; Iglesias, R.; Zoidl, G.; Dermietzel, R.; Spray, D.C. Calmodulin dependent protein kinase increases conductance at gap junctions formed by the neuronal gap junction protein connexin36. Brain Res. 2012, 1487, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Burr, G.S.; Mitchell, C.K.; Keflemariam, Y.J.; Heidelberger, R.; O’Brien, J. Calcium-dependent binding of calmodulin to neuronal gap junction proteins. Biochem. Biophys. Res. Commun. 2005, 335, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lynn, B.D.; Nagy, J.I. The effector and scaffolding proteins AF6 and MUPP1 interact with connexin36 and localize at gap junctions that form electrical synapses in rodent brain. Eur. J. Neurosci. 2012, 35, 166–181. [Google Scholar] [CrossRef]
- Li, X.; Lu, S.; Nagy, J.I. Direct association of connexin36 with zonula occludens-2 and zonula occludens-3. Neurochem. Int. 2009, 54, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Olson, C.; Lu, S.; Nagy, J.I. Association of connexin36 with zonula occludens-1 in HeLa cells, βtC-3 cells, pancreas, and adrenal gland. Histochem. Cell Biol. 2000, 122, 485–498. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lin, Y.P.; Mitchell, C.K.; Ram, S.; O’Brien, J. Two-color fluorescent analysis of connexin 36 turnover: Relationship to functional plasticity. J. Cell Sci. 2015, 128, 3888–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzigaluppi, P.; Isenia, S.C.; Haasdijk, E.D.; Elgersma, Y.; De Zeeuw, C.I.; van der Giessen, R.S.; de Jeu, M.T.G. Modulation of murine olivary connexin 36 gap junctions by PKA and CaMKII. Front. Cell. Neurosci. 2017, 11, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.A.; Del Corsso, C.; Zoidl, C.; Donaldson, L.W.; Spray, D.C.; Zoidl, G. Tubulin-Dependent Transport of Connexin-36 Potentiates the Size and Strength of Electrical Synapses. Cells 2019, 8, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, A.L.; Schubert, W.; Spray, D.C.; Lisanti, M.P. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry 2002, 41, 5754–5764. [Google Scholar] [CrossRef]
- Langlois, S.; Cowan, K.N.; Shao, Q.; Cowan, B.J.; Laird, D.W. Caveolin-1 and -2 Interact with Connexin43 and Regulate Gap Junctional Intercellular Communication in Keratinocytes. Mol. Biol. Cell 2008, 19, 912–928. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.W.; Hnasko, R.; Schubert, W.; Lisanti, M.P. Role of caveolae and caveolins in health and disease. Physiol. Rev. 2004, 84, 1341–1379. [Google Scholar] [CrossRef]
- Head, B.P.; Insel, P.A. Do caveolins regulate cells by actions outside of caveolae? Trends Cell Biol. 2007, 17, 51–57. [Google Scholar] [CrossRef]
- Galbiati, F.; Volonté, D.; Gil, O.; Zanazzi, G.; Salzer, J.L.; Sargiacomo, M.; Scherer, P.E.; Engelman, J.A.; Schlegel, A.; Parenti, M.; et al. Expression of caveolin-1 and -2 in differentiating PC 12 cells and dorsal root ganglion neurons: Caveolin-2 is up-regulated in response to cell injury. Proc. Natl. Acad. Sci. USA 1998, 95, 10257–10262. [Google Scholar] [CrossRef] [Green Version]
- Boulware, M.I.; Kordasiewicz, H.; Mermelstein, P.G. Caveolin proteins are essential for distinct effects of membrane estrogen receptors in neurons. J. Neurosci. 2007, 27, 9941–9950. [Google Scholar] [CrossRef]
- Condorelli, D.F.; Parenti, R.; Spinella, F.; Salinaro, A.T.; Belluardo, N.; Cardile, V.; Cicirata, F. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur. J. Neurosci. 1998, 10, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Bu, J.; Bruckner, S.R.; Sengoku, T.; Geddes, J.W.; Estus, S. Glutamate regulates caveolin expression in rat hippocampal neurons. J. Neurosci. Res. 2003, 72, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.E.A.; Madison, D.V. A novel SNAP25-caveolin complex correlates with the onset of persistent synaptic potentiation. J. Neurosci. 2000, 20, 5997–6006. [Google Scholar] [CrossRef]
- Gaudreault, S.B.; Blain, J.F.; Gratton, J.P.; Poirier, J. A role for caveolin-1 in post-injury reactive neuronal plasticity. J. Neurochem. 2005, 92, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Pun, S.; Stallmach, L.; Sadasivam, G.; Santos, A.F.; Caroni, P.; Fuhrer, C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006, 25, 4050–4060. [Google Scholar] [CrossRef] [Green Version]
- Mauch, D.H.; Nägier, K.; Schumacher, S.; Göritz, C.; Müller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Head, B.P.; Patel, H.H.; Tsutsumi, Y.M.; Hu, Y.; Mejia, T.; Mora, R.C.; Insel, P.A.; Roth, D.M.; Drummond, J.C.; Patel, P.M. Caveolin-1 expression is essential for N -methyl- D -aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008, 22, 828–840. [Google Scholar] [CrossRef] [Green Version]
- Head, B.P.; Hu, Y.; Finley, J.C.; Saldana, M.D.; Bonds, J.A.; Miyanohara, A.; Niesman, I.R.; Ali, S.S.; Murray, F.; Insel, P.A.; et al. Neuron-targeted caveolin-1 protein enhances signaling and promotes arborization of primary neurons. J. Biol. Chem. 2011, 286, 33310–33321. [Google Scholar] [CrossRef] [Green Version]
- Bilderback, T.R.; Gazula, V.R.; Lisanti, M.P.; Dobrowsky, R.T. Caveolin interacts with Trk A and p75(NTR) and regulates neurotrophin signaling pathways. J. Biol. Chem. 1999, 274, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Björk, K.; Sjögren, B.; Svenningsson, P. Regulation of serotonin receptor function in the nervous system by lipid rafts and adaptor proteins. Exp. Cell Res. 2010, 316, 1351–1356. [Google Scholar] [CrossRef]
- Trouet, D.; Hermans, D.; Droogmans, G.; Nilius, B.; Eggermont, J. Inhibition of volume-regulated anion channels by dominant-negative caveolin-1. Biochem. Biophys. Res. Commun. 2001, 284, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Trouet, D.; Nilius, B.; Jacobs, A.; Remacle, C.; Droogmans, G.; Eggermont, J. Caveolin-1 modulates the activity of the volume-regulated chloride channel. J. Physiol. 1999, 520, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Toselli, M.; Biella, G.; Taglietti, V.; Cazzaniga, E.; Parenti, M. Caveolin-1 expression and membrane cholesterol content modulate N-type calcium channel activity in NG108-15 cells. Biophys. J. 2005, 89, 2443–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, D.; Liu, J.; Harris, A.L. Lipid rafts prepared by different methods contain different connexin channels, but gap junctions are not lipid rafts. Biochemistry 2005, 44, 13027–13042. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Misumi, Y.; Miki, K.; Takatsuki, A.; Tamura, G.; Ikehara, Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 1986, 261, 11398–11403. [Google Scholar]
- Kirchhausen, T.; Macia, E.; Pelish, H.E. Use of Dynasore, the Small Molecule Inhibitor of Dynamin, in the Regulation of Endocytosis. Methods Enzymol. 2008, 438, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Laird, D.W.; Puranam, K.L.; Revel, J. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 1991, 273, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Fallon, R.F.; Goodenough, D.A. Five-hour half-life of mouse liver gap-junction protein. J. Cell Biol. 1981, 90, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Gumpert, A.M.; Varco, J.S.; Baker, S.M.; Piehl, M.; Falk, M.M. Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 2008, 582, 2887–2892. [Google Scholar] [CrossRef] [Green Version]
- Fiorini, C.; Gilleron, J.; Carette, D.; Valette, A.; Tilloy, A.; Chevalier, S.; Segretain, D.; Pointis, G. Accelerated internalization of junctional membrane proteins (connexin 43, N-cadherin and ZO-1) within endocytic vacuoles: An early event of DDT carcinogenicity. Biochim. Biophys. Acta Biomembr. 2008, 1778, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Piehl, M.; Lehmann, C.; Gumpert, A.M.; Denizot, J.-P.; Segretain, D.; Falk, M.M. Internalization of Large Double-Membrane Intercellular Vesicles by a Clathrin-dependent Endocytic Process. Mol. Biol. Cell 2007, 18, 337–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, K.; Chodock, R.; Hand, A.R.; Laird, D.W. The origin of annular junctions: A mechanism of gap junction internalization. J. Cell Sci. 2001, 114, 763–773. [Google Scholar] [PubMed]
- Falk, M.M.; Baker, S.M.; Gumpert, A.M.; Segretain, D.; Buckheit, R.W. Gap Junction Turnover Is Achieved by the Internalization of Small Endocytic Double-Membrane Vesicles. Mol. Biol. Cell 2009, 20, 3342–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaietta, G.; Deerinck, T.J.; Adams, S.R.; Bouwer, J.; Tour, O.; Laird, D.W.; Sosinsky, G.E.; Tsien, R.Y.; Ellisman, M.H. Multicolor and electron microscopic imaging of connexin trafficking. Science 2002, 296, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Lauf, U.; Giepmans, B.N.G.; Lopez, P.; Braconnot, S.; Chen, S.C.; Falk, M.M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10446–10451. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Sottile, J. Caveolin-1-dependent β1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371. [Google Scholar] [CrossRef] [Green Version]
- Marchiando, A.M.; Shen, L.; Vallen Graham, W.; Weber, C.R.; Schwarz, B.T.; Austin, J.R.; Raleigh, D.R.; Guan, Y.; Watson, A.J.M.; Montrose, M.H.; et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 2010, 189, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liang, M.; Liu, L.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005, 67, 1844–1854. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Deviez, D.J.; Howes, M.T.; Laval, S.H.; Bushby, K.; Hancock, J.F.; Parton, R.G. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J. Biol. Chem. 2008, 283, 6476–6488. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, S.; Watson, R.T.; Khan, A.H.; Pessin, J.E. The adipocyte plasma membrane caveolin functional/structural organization is necessary for the efficient endocytosis of GLUT4. J. Biol. Chem. 2003, 278, 10683–10690. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.W.; Zu, X.Y.; Tuo, Q.H.; Chen, L.X.; Lei, X.Y.; Li, K.; Tang, C.K.; Liao, D.F. Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacol. Sin. 2010, 31, 1336–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, M.I.; Krizman-Genda, E.; Robinson, M.B. Caveolin-1 regulates the delivery and endocytosis of the glutamate transporter, excitatory amino acid carrier. J. Biol. Chem. 2007, 282, 29855–29865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyse, B.D.; Prior, I.A.; Qian, H.; Morrow, I.C.; Nixon, S.; Muncke, C.; Kurzchalia, T.V.; Thomas, W.G.; Parton, R.G.; Hancock, J.F. Caveolin interacts with the angiotensin II type I receptor during exocytic transport but not at the plasma membrane. J. Biol. Chem. 2003, 278, 23738–23746. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Deviez, D.J.; Martin, S.; Laval, S.H.; Lo, H.P.; Cooper, S.T.; North, K.N.; Bushby, K.; Parton, R.G. Aberrant dysferlin trafficking in cells lacking caveolin or expressing dystrophy mutants of caveolin-3. Hum. Mol. Genet. 2006, 15, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotgia, F.; Razani, B.; Bonuccelli, G.; Schubert, W.; Battista, M.; Lee, H.; Capozza, F.; Schubert, A.L.; Minetti, C.; Buckley, J.T.; et al. Intracellular Retention of Glycosylphosphatidyl Inositol-Linked Proteins in Caveolin-Deficient Cells. Mol. Cell. Biol. 2002, 22, 3905–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, P.U.; Guay, G.; Altschuler, Y.; Nabi, I.R. Caveolin-1 is a negative regulator of caveolae-mediated endocytosis to the endoplasmic reticulum. J. Biol. Chem. 2002, 277, 3371–3379. [Google Scholar] [CrossRef] [Green Version]
- Nabi, I.R.; Le, P.U. Caveolae/raft-dependent endocytosis. J. Cell Biol. 2003, 161, 673–677. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef]
- Scherer, P.E.; Lewis, Y.; Volonte, D.; Engelman, J.A.; Galbiati, F.; Couet, J.; Kohtz, D.S.; Van Donselaar, E.; Peters, P.; Lisanti, M.P. Cell-type and Tissue-specific Expression of Caveolin-2. J. Biol. Chem. 1997, 272, 29337–29346. [Google Scholar] [CrossRef] [Green Version]
- Gorodinsky, A.; Harris, D.A. Glycolipid-anchored proteins in neuroblastoma cells form detergent- resistant complexes without caveolin. J. Cell Biol. 1995, 129, 619–627. [Google Scholar] [CrossRef]
- Peyroche, A.; Antonny, B.; Robineau, S.; Acker, J.; Cherfils, J.; Jackson, C.L. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: Involvement of specific residues of the Sec7 domain. Mol. Cell 1999, 3, 275–285. [Google Scholar] [CrossRef]
- Springer, S.; Spang, A.; Schekman, R. A primer on vesicle budding. Cell 1999, 97, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Lippincott-Schwartz, J.; Yuan, L.; Tipper, C.; Amherdt, M.; Orci, L.; Klausner, R.D. Brefeldin A’s effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 1991, 67, 601–616. [Google Scholar] [CrossRef]
- Morgan, A.J.; Jacob, R. Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochem. J. 1994, 300, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamble, E.; Koch, C. The dynamics of free calcium in dendritic spines in response to repetitive synaptic input. Science 1987, 236, 1311–1315. [Google Scholar] [CrossRef]
- Siu, R.C.F.; Smirnova, E.; Brown, C.A.; Zoidl, C.; Spray, D.C.; Donaldson, L.W.; Zoidl, G. Structural and functional consequences of connexin 36 (Cx36) interaction with calmodulin. Front. Mol. Neurosci. 2016, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaranarayanan, S.; Ryan, T.A. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat. Neurosci. 2001, 4, 129–136. [Google Scholar] [CrossRef]
- Han, Y.; Massey, S.C. Electrical synapses in retinal ON cone bipolar cells: Subtype-specific expression of connexins. PNAS 2005, 102, 13313–13318. [Google Scholar] [CrossRef] [Green Version]
- Neves, G.; Gomis, A.; Lagnado, L. Calcium influx selects the fast mode of endocytosis in the synaptic terminal of retinal bipolar cells. Proc. Natl. Acad. Sci. USA 2001, 98, 15282–15287. [Google Scholar] [CrossRef] [Green Version]
- Daly, C.; Sugimori, M.; Moreira, J.E.; Ziff, E.B.; Llina, R. Synaptophysin regulates clathrin-independent endocytosis of synaptic vesicles. PNAS 2000, 97, 6120–6125. [Google Scholar] [CrossRef] [Green Version]
- Timonina, K.; Kotova, A.; Zoidl, G. Role of an aromatic–aromatic interaction in the assembly and trafficking of the zebrafish panx1a membrane channel. Biomolecules 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.M.; Galliardt, H.; Schneider, J.; Barisas, B.G.; Seidel, T. Quantification of Förster resonance energy transfer by monitoring sensitized emission in living plant cells. Front. Plant. Sci. 2013, 4, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotova, A.; Timonina, K.; Zoidl, G.R. Endocytosis of Connexin 36 is Mediated by Interaction with Caveolin-1. Int. J. Mol. Sci. 2020, 21, 5401. https://doi.org/10.3390/ijms21155401
Kotova A, Timonina K, Zoidl GR. Endocytosis of Connexin 36 is Mediated by Interaction with Caveolin-1. International Journal of Molecular Sciences. 2020; 21(15):5401. https://doi.org/10.3390/ijms21155401
Chicago/Turabian StyleKotova, Anna, Ksenia Timonina, and Georg R. Zoidl. 2020. "Endocytosis of Connexin 36 is Mediated by Interaction with Caveolin-1" International Journal of Molecular Sciences 21, no. 15: 5401. https://doi.org/10.3390/ijms21155401