Mechanotransduction and Stiffness-Sensing: Mechanisms and Opportunities to Control Multiple Molecular Aspects of Cell Phenotype as a Design Cornerstone of Cell-Instructive Biomaterials for Articular Cartilage Repair


Abstract
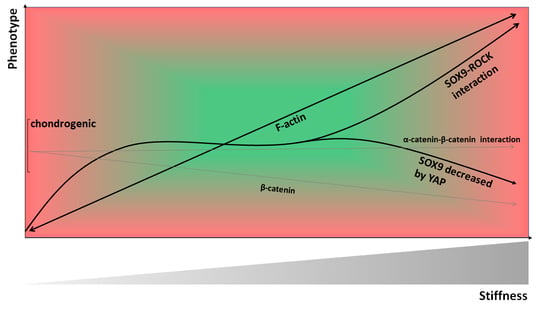
:
1. Introduction
2. Clinical Use of CHs and MSCs in AC Repair Procedures
3. Overview: How Do Cells Sense Their Environment?
4. Stiffness Sensing
5. Proteins and Structures Involved in Stiffness Sensing
5.1. Focal Adhesions
5.2. FA Focal Complex
5.3. Integrins
5.4. Focal Adhesion Kinase
5.5. Rho GTPases
5.6. Stress Fibers
6. Material Stiffness-Regulated Cell Proliferation
7. Material Stiffness-Regulated Cell Migration
8. Material Stiffness-Modulated MSC Shape and Lineage Determination
9. Material Stiffness-Modulated CH Shape, Cytoskeleton, and Phenotype
10. Material Stiffness Changes Modulate Nuclear Shape and Nuclear Lamina and Inner Membrane Composition for Controlling mRNA Expression and MSC Differentiation
11. TGF-β1-Induced Lineage Determination of MSCs is Modulated by Material Stiffness
12. TGF-β1- and IL-1 β-Induced Changes in CH Stiffness and Traction Force are Material-Stiffness Dependent
13. Substrate Stiffness-Modulated Cell Surface Growth Factor Receptor Composition
14. Rho GTPases in Substrate Stiffness-Modulated MSC Differentiation and CH Phenotype
15. Substrate Stiffness-Modulated Integrin Subunit Expression of MSCs and CHs
16. Differential MSC Behavior in 2D vs. 3D
17. Immuno-Modulative and Angiogenic Role of Material Stiffness in MSCs
18. The Role of Material Stiffness in Inducing Re-Differentiation of CHs after Serial Expansion-Induced De-Differentiation
19. The Role of Material Stiffness-Dependent β-Catenin Signaling in CH De-Differentiation
20. Collagen Type II Fragment Production and Subsequent Catabolic Effects are Modulated by Rho/ROCK Activation in CHs
21. Biomaterials Used for Clinically Inducing Human AC Repair
22. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AC | articular cartilage |
| ACAN | aggrecan |
| ACI | autologous chondrocyte implantation |
| ALP | alkaline phosphatase |
| AMICTM | autologous matrix-induced chondrogenesis |
| ATF2 | activation transcription factor 2 |
| Arp2/3 | actin-related protein 2/3 |
| BMP | bone morphogenetic protein |
| CHs | chondrocytes |
| cCHs | chicken chondrocytes |
| COL1A2 | collagen type I alpha II chain |
| COL2A1 | collagen type II alpha I chain |
| Col10A1 | collagen type 10 alpha I chain |
| CREB | cAMP response element-binding protein |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| ERKs | extracellular signal-regulated kinases |
| FAK | focal adhesion kinase |
| FAs | focal adhesions |
| GAGs | glycosaminoglycans |
| gCHs | goat chondrocytes |
| GEF | guanine-exchange factor |
| hASCs | human adipose-derived stem cells |
| hMSCs | human mesenchymal stem cells |
| IL-1β | Interleukin 1 β |
| IRAK | Interleukin-1 receptor associated kinase |
| JNK | Jun NH2-terminal kinase |
| LINC | Linker of nucleoskeleton to cytoskeleton |
| LOX | lysil oxidase |
| LPL | Lipoprotein lipase |
| µRB | microribbon |
| MMP-13 | matrix metalloproteinase 13 |
| MSCs | mesenchymal stem cells |
| mCHs | murine chondrocytes |
| MLCK | myosin light chain kinase |
| myosin II | myosin phosphatase II |
| NSCs | neural stem cells |
| OA | osteoarthritis |
| PAA | polyacrylamide |
| pCHs | porcine chondrocytes |
| PEGDA | poly(ethylene)glycol diacrylate |
| rabCHs | rabbit chondrocytes |
| rCHs | rat chondrocytes |
| rMSCs | rat mesenchymal stem cells |
| rNSCs | rat neural stem cells |
| ROCK | Rho associated protein kinase |
| ROS | Reactive oxygen species |
| RUNX2 | runt related transcription factor 2 |
| sCHs | sheep chondrocytes |
| SMAD | SMA- and MAD-related protein |
| SMCs | smooth muscle cells |
| sMSCs | synovium-derived mesenchymal stem cells |
| SOX9 | SRY-related HMG box-containing |
| TAZ | transcriptional co-activator with PDZ-binding motif |
| TGF-α | transforming growth factor α |
| TGF-β | transforming growth factor β |
| TGFR | transforming growth factor receptor |
| VEGF | vascular endothelial growth factor |
| YAP | Yes-associated protein |
References
- Park, J.; Kim, P.; Helen, W.; Engler, A.J.; Levchenko, A.; Kim, D.H. Control of stem cell fate and function by engineering physical microenvironments. Integr. Biol. 2012, 4, 1008–1018. [Google Scholar]
- Discher, D.E.; Janmey, P.; Wang, Y.-L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Seog, J.; Dean, D.; Rolauffs, B.; Wu, T.; Genzer, J.; Plaas, A.H.; Grodzinsky, A.J.; Ortiz, C. Nanomechanics of opposing glycosaminoglycan macromolecules. J. Biomech. 2005, 38, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.; Sheetz, M. Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Petridou, N.I.; Spiró, Z.; Heisenberg, C.-P. Multiscale force sensing in development. Nat. Cell Biol. 2017, 19, 581–588. [Google Scholar] [CrossRef]
- Kuettner, K.E.; Aydelotte, M.B.; Thonar, E.J. Articular cartilage matrix and structure: A minireview. J. Rheumatol. Suppl. 1991, 27, 46–48. [Google Scholar] [PubMed]
- Woo, S.L.Y.; Buckwalter, J.A. Injury and repair of the musculoskeletal soft tissues. Savannah, Georgia, June 18–20, 1987. J. Orthop. Res. 1988, 6, 907–931. [Google Scholar] [CrossRef]
- Buckwalter, J.A. Articular cartilage: Injuries and potential for healing. J. Orthop. Sports Phys. Ther. 1998, 28, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Aicher, W.K.; Rolauffs, B. The spatial organisation of joint surface chondrocytes: Review of its potential roles in tissue functioning, disease and early, preclinical diagnosis of osteoarthritis. Ann. Rheum. Dis. 2014, 73, 645–653. [Google Scholar] [CrossRef]
- Felka, T.; Rothdiener, M.; Bast, S.; Uynuk-Ool, T.; Zouhair, S.; Ochs, B.G.; De Zwart, P.; Stoeckle, U.; Aicher, W.K.; Hart, M.L.; et al. Loss of spatial organization and destruction of the pericellular matrix in early osteoarthritis in vivo and in a novel in vitro methodology. Osteoarthr. Cartil. 2016, 24, 1200–1209. [Google Scholar] [CrossRef] [Green Version]
- Rolauffs, B.; Williams, J.M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Distinct horizontal patterns in the spatial organization of superficial zone chondrocytes of human joints. J. Struct. Biol. 2008, 162, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolauffs, B.; Rothdiener, M.; Bahrs, C.; Badke, A.; Weise, K.; Kuettner, K.E.; Kurz, B.; Aurich, M.; Grodzinsky, A.; Aicher, W. Onset of preclinical osteoarthritis: The angular spatial organization permits early diagnosis. Arthritis Rheum. 2011, 63, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Rolauffs, B.; Williams, J.M.; Aurich, M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Proliferative remodeling of the spatial organization of human superficial chondrocytes distant from focal early osteoarthritis. Arthritis Rheum. 2010, 62, 489–498. [Google Scholar] [PubMed] [Green Version]
- Li, Y.; Wei, X.; Zhou, J.; Wei, L. The age-related changes in cartilage and osteoarthritis. BioMed Res. Int. 2013, 2013, 916530. [Google Scholar] [CrossRef]
- Stolz, M.; Gottardi, R.; Raiteri, R.; Miot, S.; Martin, I.; Imer, R.; Staufer, U.; Raducanu, A.; Düggelin, M.; Baschong, W.; et al. Early detection of aging cartilage and osteoarthritis in mice and patient samples using atomic force microscopy. Nat. Nanotechnol. 2009, 4, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Lin, Y.H.; Lin, S.; Tsai-Wu, J.J.; Wu, C.H.; Jiang, C.C. Surface ultrastructure and mechanical property of human chondrocyte revealed by atomic force microscopy. Osteoarthr. Cartil. 2008, 16, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Lahm, A.; Mrosek, E.; Spank, H.; Erggelet, C.; Kasch, R.; Esser, J.; Merk, H. Changes in content and synthesis of collagen types and proteoglycans in osteoarthritis of the knee joint and comparison of quantitative analysis with Photoshop-based image analysis. Arch. Orthop. Trauma Surg. 2010, 130, 557–564. [Google Scholar] [CrossRef]
- Gottardi, R.; Hansen, U.; Raiteri, R.; Loparic, M.; Düggelin, M.; Mathys, D.; Friederich, N.F.; Bruckner, P.; Stolz, M. Supramolecular organization of collagen fibrils in healthy and osteoarthritic human knee and hip joint cartilage. PLoS ONE 2016, 11, e0163552. [Google Scholar] [CrossRef]
- DiMicco, M.A.; Patwari, P.; Siparsky, P.N.; Kumar, S.; Pratta, M.A.; Lark, M.W.; Kim, Y.-J.; Grodzinsky, A.J. Mechanisms and kinetics of glycosaminoglycan release following in vitro cartilage injury. Arthritis Rheum. 2004, 50, 840–848. [Google Scholar] [CrossRef]
- Williamson, A.K.; Chen, A.C.; Sah, R.L. Compressive properties and function-composition relationships of developing bovine articular cartilage. J. Orthop. Res. 2001, 19, 1113–1121. [Google Scholar] [CrossRef]
- Williamson, A.K.; Chen, A.C.; Masuda, K.; Thonar, E.J.M.; Sah, R.L. Tensile mechanical properties of bovine articular cartilage: Variations with growth and relationships to collagen network components. J. Orthop. Res. 2003, 21, 872–880. [Google Scholar] [CrossRef]
- Williamson, A.K.; Masuda, K.; Thonar, E.J.M.; Sah, R.L. Growth of immature articular cartilage in vitro: Correlated variation in tensile biomechanical and collagen network properties. Tissue Eng. 2003, 9, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Vunjak-Novakovic, G.; Martin, I.; Obradovic, B.; Treppo, S.; Grodzinsky, A.J.; Langer, R.; Freed, L.E. Bioreactor cultivation conditions modulate the composition and mechanical properties of tissue-engineered cartilage. J. Orthop. Res. 1999, 17, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Kempson, G.E.; Muir, H.; Pollard, C.; Tuke, M. The tensile properties of the cartilage of human femoral condyles related to the content of collagen and glycosaminoglycans. Biochim. Biophys. Acta 1973, 297, 456–472. [Google Scholar] [CrossRef]
- Kelly, D.J.; Crawford, A.; Dickinson, S.C.; Sims, T.J.; Mundy, J.; Hollander, A.P.; Prendergast, P.J.; Hatton, P.V. Biochemical markers of the mechanical quality of engineered hyaline cartilage. J. Mater. Sci. Mater. Med. 2007, 18, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Rolauffs, B.; Kurz, B.; Felka, T.; Rothdiener, M.; Uynuk-Ool, T.; Aurich, M.; Frank, E.; Bahrs, C.; Badke, A.; Stöckle, U.; et al. Stress-vs-time signals allow the prediction of structurally catastrophic events during fracturing of immature cartilage and predetermine the biomechanical, biochemical, and structural impairment. J. Struct. Biol. 2013, 183, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolauffs, B.; Muehleman, C.; Li, J.; Kurz, B.; Kuettner, K.E.; Frank, E.; Grodzinsky, A.J. Vulnerability of the superficial zone of immature articular cartilage to compressive injury. Arthritis Rheum. 2010, 62, 3016–3027. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.E.; Akhtar, R.; Comerford, E.J.; Bates, K.T. The effect of ageing and osteoarthritis on the mechanical properties of cartilage and bone in the human knee joint. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Kleemann, R.U.; Krocker, D.; Cedraro, A.; Tuischer, J.; Duda, G.N. Altered cartilage mechanics and histology in knee osteoarthritis: Relation to clinical assessment (ICRS Grade). Osteoarthr. Cartil. 2005, 13, 958–963. [Google Scholar] [CrossRef] [Green Version]
- Hunziker, E.B.; Lippuner, K.; Keel MJ, B.; Shintani, N. An educational review of cartilage repair: Precepts & practice–myths & misconceptions–progress & prospects. Osteoarthr. Cartil. 2015, 23, 334–350. [Google Scholar]
- Niemeyer, P.; Albrecht, D.; Andereya, S.; Angele, P.; Ateschrang, A.; Aurich, M.; Baumann, M.; Bosch, U.; Erggelet, C.; Fickert, S.; et al. Autologous chondrocyte implantation (ACI) for cartilage defects of the knee: A guideline by the working group “Clinical Tissue Regeneration” of the German Society of Orthopaedics and Trauma (DGOU). Knee 2016, 23, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Aurich, M.; Bedi, H.S.; Smith, P.J.; Rolauffs, B.; Mückley, T.; Clayton, J.; Blackney, M. Arthroscopic treatment of osteochondral lesions of the ankle with matrix-associated chondrocyte implantation: Early clinical and magnetic resonance imaging results. Am. J. Sports Med. 2011, 39, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ochs, B.G.; Müller-Horvat, C.; Albrecht, D.; Schewe, B.; Weise, K.; Aicher, W.K.; Rolauffs, B. Remodeling of articular cartilage and subchondral bone after bone grafting and matrix-associated autologous chondrocyte implantation for osteochondritis dissecans of the knee. Am. J. Sports Med. 2011, 39, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Ochs, B.G.; Müller-Horvat, C.; Rolauffs, B.; Fritz, J.; Weise, K.; Schewe, B. Treatment of osteochondritis dissecans of the knee: One-step procedure with bone grafting and matrix-supported autologous chondrocyte transplantation. Z. Orthop. Unfall. 2007, 145, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Steadman, J.R.; Rodkey, W.G.; Briggs, K.K. Microfracture to treat full-thickness chondral defects: Surgical technique, rehabilitation, and outcomes. J. Knee Surg. 2002, 15, 170–176. [Google Scholar] [PubMed]
- Gao, L.; Orth, P.; Cucchiarini, M.; Madry, H. Autologous Matrix-Induced Chondrogenesis: A Systematic Review of the Clinical Evidence. Am. J. Sports Med. 2019, 47, 222–231. [Google Scholar] [CrossRef]
- Steinwachs, M.R.; Gille, J.; Volz, M.; Anders, S.; Jakob, R.; De Girolamo, L.; Volpi, P.; Schiavone-Panni, A.; Scheffler, S.; Reiss, E.; et al. Systematic Review and Meta-Analysis of the Clinical Evidence on the Use of Autologous Matrix-Induced Chondrogenesis in the Knee. Cartilage 2019, 1947603519870846. [Google Scholar] [CrossRef] [Green Version]
- Sreenivasappa, H.; Chaki, S.P.; Lim, S.M.; Trzeciakowski, J.P.; Davidson, M.W.; Rivera, G.M.; Trache, A. Selective regulation of cytoskeletal tension and cell–matrix adhesion by RhoA and Src. Integr. Biol. 2014, 6, 743–754. [Google Scholar] [CrossRef]
- Bae, Y.H.; Mui, K.L.; Hsu, B.Y.; Liu, S.L.; Cretu, A.; Razinia, Z.; Xu, T.; Puré, E.; Assoian, R.K. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 2014, 7, ra57. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Placone, J.K.; Engler, A.J. Understanding the extracellular forces that determine cell fate and maintenance. Development 2017, 144, 4261–4270. [Google Scholar] [CrossRef] [Green Version]
- Van Helvert, S.; Storm, C.; Friedl, P. Mechanoreciprocity in cell migration. Nat. Cell Biol. 2018, 20, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Gefen, A.; Margulies, S.S. Are in vivo and in situ brain tissues mechanically similar? J. Biomech. 2004, 37, 1339–1352. [Google Scholar] [CrossRef]
- Engler, A.J.; Griffin, M.A.; Sen, S.; Boönnemann, C.G.; Sweeney, H.L.; Discher, D.E. Myotubes differentiate optimally on substrates with tissue-like stiffness: Pathological implications for soft or stiff microenvironments. J. Cell Biol. 2004, 166, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, C.S.; Fu, J. Forcing stem cells to behave: A biophysical perspective of the cellular microenvironment. Annu. Rev. Biophys. 2012, 41, 519–542. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Doss, B.; Lim, C.T.; Voituriez, R.; Ladoux, B. Single cell rigidity sensing: A complex relationship between focal adhesion dynamics and large-scale actin cytoskeleton remodeling. Cell Adh. Migr. 2016, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Ouyang, M.; Van den Dries, K.; McGhee, E.J.; Tanaka, K.; Anderson, M.D.; Groisman, A.; Goult, B.T.; Anderson, K.I.; Schwartz, M.A. Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J. Cell Biol. 2016, 213, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O.; Destree, A.T. Relationships between fibronectin (LETS protein) and actin. Cell 1978, 15, 875–886. [Google Scholar] [CrossRef]
- Singer, I.I. Association of fibronectin and vinculin with focal contacts and stress fibers in stationary hamster fibroblasts. J. Cell Biol. 1982, 92, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Tamkun, J.W.; DeSimone, D.W.; Fonda, D.; Patel, R.S.; Buck, C.; Horwitz, A.F.; Hynes, R.O. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 1986, 46, 271–282. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.-C.; Han, X.; Hsiao, C.T.; Yates, J.R., III; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–519. [Google Scholar] [CrossRef] [Green Version]
- Zaidel-Bar, R.; Cohen, M.; Addadi, L.; Geiger, B. Hierarchical Assembly of Cell–Matrix Adhesion Complexes; Portland Press Ltd.: London, UK, 2004. [Google Scholar]
- Pelham, R.J.; Wang, Y.-L. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelham, R.J., Jr.; Wang, Y.-L. Cell locomotion and focal adhesions are regulated by the mechanical properties of the substrate. Biol. Bull. 1998, 194, 348–350. [Google Scholar] [CrossRef] [Green Version]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale architecture of integrin-based cell adhesions. Nature 2010, 468, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Case, L.B.; Baird, M.A.; Shtengel, G.; Campbell, S.L.; Hess, H.F.; Davidson, M.W.; Waterman, C.M. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 2015, 17, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.E. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2000, 2, E231–E236. [Google Scholar] [CrossRef]
- Schaller, M.D. Paxillin: A focal adhesion-associated adaptor protein. Oncogene 2001, 20, 6459–6472. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.S.; Baumann, F.; Daday, C.; Redondo, P.; Durner, E.; Jobst, M.A.; Milles, L.F.; Mercadante, D.; Pippig, D.A.; Gaub, H.E.; et al. Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 6766–6774. [Google Scholar] [CrossRef] [Green Version]
- Salgado-Lucio, M.L.; Ramírez-Ramírez, D.; Jorge-Cruz, C.Y.; Roa-Espitia, A.L.; Hernández-González, E.O. FAK regulates actin polymerization during capacitation via the ERK2/GEF- H1/RhoA signaling pathway. J. Cell Sci. 2020, 133, jcs239186. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Jones, G.E.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Case, L.B.; Waterman, C.M. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 2015, 17, 955–963. [Google Scholar] [CrossRef]
- Hoffman, B.D.; Grashoff, C.; Schwartz, M.A. Dynamic molecular processes mediate cellular mechanotransduction. Nature 2011, 475, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Klapholz, B.; Brown, N.H. Talin—The master of integrin adhesions. J. Cell Sci. 2017, 130, 2435–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.M.; Kutscher, B.; Chen, H.; Murphy, D.B.; Craig, S.W. A conformational switch in vinculin drives formation and dynamics of a talin-vinculin complex at focal adhesions. J. Biol. Chem. 2006, 281, 16006–16015. [Google Scholar] [CrossRef] [Green Version]
- Golji, J.; Mofrad, M.R. The interaction of vinculin with actin. PLoS Comput. Biol. 2013, 9, e1002995. [Google Scholar] [CrossRef]
- Kumar, A.; Anderson, K.L.; Swift, M.F.; Hanein, D.; Volkmann, N.; Schwartz, M.A. Local Tension on Talin in Focal Adhesions Correlates with F-Actin Alignment at the Nanometer Scale. Biophys. J. 2018, 115, 1569–1579. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Jiang, G.; Sutton, D.H.; Critchley, D.R.; Sheetz, M.P. Talin1 is critical for force-dependent reinforcement of initial integrin–cytoskeleton bonds but not tyrosine kinase activation. J. Cell Biol. 2003, 163, 409–419. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, G.; Cai, Y.; Monkley, S.J.; Critchley, D.R.; Sheetz, M.P. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 2008, 10, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Ng, W.P.; Kumar, S. Contributions of talin-1 to glioma cell-matrix tensional homeostasis. J. R. Soc. Interface 2012, 9, 1311–1317. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, H.; Tatsumi, H.; Sokabe, M. Mechanical forces facilitate actin polymerization at focal adhesions in a zyxin-dependent manner. J. Cell Sci. 2008, 121 Pt 17, 2795–2804. [Google Scholar] [CrossRef] [Green Version]
- Senger, F.; Pitaval, A.; Ennomani, H.; Kurzawa, L.; Blanchoin, L.; Théry, M. Spatial integration of mechanical forces by alpha-actinin establishes actin network symmetry. J. Cell Sci. 2019, 132, jcs236604. [Google Scholar] [CrossRef]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Riveline, D.; Zamir, E.; Balaban, N.Q.; Schwarz, U.S.; Ishizaki, T.; Narumiya, S.; Kam, Z.; Geiger, B.; Bershadsky, A.D. Focal contacts as mechanosensors: Externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J. Cell Biol. 2001, 153, 1175–1186. [Google Scholar] [CrossRef]
- Giannone, G.; Dubin-Thaler, B.J.; Rossier, O.; Cai, Y.; Chaga, O.; Jiang, G.; Beaver, W.; Döbereiner, H.-G.; Freund, Y.; Borisy, G.; et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 2007, 128, 561–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, S.; Svineng, G.; Wennerberg, K.; Armulik, A.; Lohikangas, L. Fibronectin-integrin interactions. Front. Biosci. 1997, 2, d126–d146. [Google Scholar] [CrossRef] [Green Version]
- Tulla, M.; Pentikäinen, O.T.; Viitasalo, T.; Käpylä, J.; Impola, U.; Nykvist, P.; Nissinen, L.; Johnson, M.S.; Heino, J. Selective binding of collagen subtypes by integrin α1I, α2I, and α10I domains. J. Biol. Chem. 2001, 276, 48206–48212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiuchi, R.; Takagi, J.; Hayashi, M.; Ido, H.; Yagi, Y.; Sanzen, N.; Tsuji, T.; Yamada, M.; Sekiguchi, K. Ligand-binding specificities of laminin-binding integrins: A comprehensive survey of laminin–integrin interactions using recombinant α3β1, α6β1, α7β1 and α6β4 integrins. Matrix Biol. 2006, 25, 189–197. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Zent, R.; Grant, R.; Rees DJ, G.; Hynes, R.O.; Ginsberg, M.H. The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 1999, 274, 28071–28074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca-Cusachs, P.; Del Rio, A.; Puklin-Faucher, E.; Gauthier, N.C.; Biais, N.; Sheetz, M.P. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. USA 2013, 110, E1361–E1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderwood, D.A.; Campbell, I.D.; Critchley, D.R. Talins and kindlins: Partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderwood, D.A.; Fujioka, Y.; de Pereda, J.M.; García-Alvarez, B.; Nakamoto, T.; Margolis, B.; McGlade, C.J.; Liddington, R.C.; Ginsberg, M.H. Integrin β cytoplasmic domain interactions with phosphotyrosine-binding domains: A structural prototype for diversity in integrin signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 2272–2277. [Google Scholar] [CrossRef] [Green Version]
- Legate, K.R.; Fässler, R. Mechanisms that regulate adaptor binding to β-integrin cytoplasmic tails. J. Cell Sci. 2009, 122, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Larjava, H.; Plow, E.F.; Wu, C. Kindlins: Essential regulators of integrin signalling and cell–matrix adhesion. EMBO Rep. 2008, 9, 1203–1208. [Google Scholar] [CrossRef]
- Wang, J.G.; Miyazu, M.; Matsushita, E.; Sokabe, M.; Naruse, K. Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem. Biophys. Res. Commun. 2001, 288, 356–361. [Google Scholar] [CrossRef]
- Lee, H.; Millward-Sadler, S.J.; Wright, M.O.; Nuki, G.; Salter, D.M. Integrin and mechanosensitive ion channel-dependent tyrosine phosphorylation of focal adhesion proteins and β-catenin in human articular chondrocytes after mechanical stimulation. J. Bone Miner. Res. 2000, 15, 1501–1509. [Google Scholar] [CrossRef]
- Fluück, M.; Carson, J.A.; Gordon, S.E.; Ziemiecki, A.; Booth, F.W. Focal adhesion proteins FAK and paxillin increase in hypertrophied skeletal muscle. Am. J. Physiol. Cell Physiol. 1999, 277, C152–C162. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK–ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-C.; Chen, Y.C.; Kuo, C.T.; Yu, H.W.; Chen, Y.Q.; Chiou, A.; Kuo, J.C. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci. Rep. 2015, 5, 18476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Guan, J.-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, Y.R.V.; Tseng, K.F.; Lai, H.Y.; Lin, C.H.; Lee, O.K. Matrix stiffness regulation of integrin-mediated mechanotransduction during osteogenic differentiation of human mesenchymal stem cells. J. Bone Miner. Res. 2011, 26, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Thodeti, C.K.; Paruchuri, S.; Meszaros, J.G. A TRP to cardiac fibroblast differentiation. Channels 2013, 7, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Zhang, W.; Shu, C.; Li, Q.; Li, M.; Li, X. Adiponectin affects vascular smooth muscle cell proliferation and apoptosis through modulation of the mitofusin-2-mediated Ras-Raf-Erk1/2 signaling pathway. Mol. Med. Rep. 2015, 12, 4703–4707. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Song, G.; Ju, Y.; Li, X.; Song, Y.; Watanabe, S. RhoA/ROCK, cytoskeletal dynamics, and focal adhesion kinase are required for mechanical stretch-induced tenogenic differentiation of human mesenchymal stem cells. J. Cell. Physiol. 2012, 227, 2722–2729. [Google Scholar] [CrossRef]
- Salasznyk, R.M.; Klees, R.F.; Williams, W.A.; Boskey, A.; Plopper, G.E. Focal adhesion kinase signaling pathways regulate the osteogenic differentiation of human mesenchymal stem cells. Exp. Cell Res. 2007, 313, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; He, J.; Li, Y.S.; Wang, X.M.; Ai, H.J.; Cui, F.Z. Activation of the ERK1/2 signaling pathway during the osteogenic differentiation of mesenchymal stem cells cultured on substrates modified with various chemical groups. BioMed Res. Int. 2013, 2013, 361906. [Google Scholar] [CrossRef]
- Ren, X.-D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar]
- Chen, B.-H.; Tzen, J.T.; Bresnick, A.R.; Chen, H.C. Roles of Rho-associated kinase and myosin light chain kinase in morphological and migratory defects of focal adhesion kinase-null cells. J. Biol. Chem. 2002, 277, 33857–33863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix stiffness–induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Swaminathan, V.; Garcia-Mata, R.; O’Brien, E.T.; Superfine, R.; Burridge, K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 2011, 13, 722–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ingber, D.E. Cell tension, matrix mechanics, and cancer development. Cancer Cell 2005, 8, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.L.; Cooke, M.E.; Alliston, T. ECM stiffness primes the TGFβ pathway to promote chondrocyte differentiation. Mol. Biol. Cell 2012, 23, 3731–3742. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H.J. Adhesion signaling–crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Naumanen, P.; Lappalainen, P.; Hotulainen, P. Mechanisms of actin stress fibre assembly. J. Microsc. 2008, 231, 446–454. [Google Scholar] [CrossRef]
- Faix, J.; Grosse, R. Staying in shape with formins. Dev. Cell 2006, 10, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Gasman, S.; Chasserot-Golaz, S.; Hubert, P.; Aunis, D.; Bader, M.F. Identification of a potential effector pathway for the trimeric Go protein associated with secretory granules Go stimulates a granule-bound phosphatidylinositol 4-kinase by activating RhoA in chromaffin cells. J. Biol. Chem. 1998, 273, 16913–16920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, B.L.; Eck, M.J. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Amann, K.J.; Higgs, H.N.; Marchand, J.B.; Kaiser, D.A.; Pollard, T.D. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature 2000, 404, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.D.; Heuser, J.A.; Pollard, T.D. The interaction of Arp2/3 complex with actin: Nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA 1998, 95, 6181–6186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilghman, R.W.; Cowan, C.R.; Mih, J.D.; Koryakina, Y.; Gioeli, D.; Slack-Davis, J.K.; Blackman, B.R.; Tschumperlin, D.J.; Parsons, J.T. Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS ONE 2010, 5, e12905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Mih, J.D.; Marinkovic, A.; Liu, F.; Sharif, A.S.; Tschumperlin, D.J. Matrix stiffness reverses the effect of actomyosin tension on cell proliferation. J. Cell Sci. 2012, 125, 5974–5983. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.A.; Yin, L.; Kothapalli, D.; Castagnino, P.; Byfield, F.J.; Xu, T.; Levental, I.; Hawthorne, E.; Janmey, P.A.; Assoian, R.K. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr. Biol. 2009, 19, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Chu, J.S.; Tsou, A.D.; Diop, R.; Tang, Z.; Wang, A.; Li, S. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-β. Biomaterials 2011, 32, 3921–3930. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Li, Y.; Li, L.; Zhang, W.; Wang, S.; Zheng, X. YAP-mediated regulation of the chondrogenic phenotype in response to matrix elasticity. J. Mol. Histol. 2013, 44, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Oktay, M.; Wary, K.K.; Dans, M.; Birge, R.B.; Giancotti, F.G. Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J. Cell Biol. 1999, 145, 1461–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Bergami, P.; Huang, C.; Goydos, J.S.; Yip, D.; Bar-Eli, M.; Herlyn, M.; Smalley, K.S.M.; Mahale, A.; Eroshkin, A.; Aaronson, S.; et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell 2007, 11, 447–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, A.K.; Campbell, L.E.; Castagnino, P.; Liu, W.F.; Chung, B.M.; Weaver, V.M.; Chen, C.S.; Assoian, R.K. Rac-dependent cyclin D1 gene expression regulated by cadherin-and integrin-mediated adhesion. J. Cell Sci. 2008, 121, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Dobrokhotov, O.; Samsonov, M.; Sokabe, M.; Hirata, H. Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin. Transl. Med. 2018, 7, 1–14. [Google Scholar] [CrossRef]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [Green Version]
- Sebio, A.; Lenz, H.J. Molecular Pathways: Hippo Signaling, a Critical Tumor Suppressor. Clin. Cancer Res. 2015, 21, 5002–5007. [Google Scholar] [CrossRef] [Green Version]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef]
- Pan, J.X.; Xiong, L.; Zhao, K.; Zeng, P.; Wang, B.; Tang, F.L.; Sun, D.; Guo, H.-H.; Yang, X.; Cui, S.; et al. YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating beta-catenin signaling. Bone Res. 2018, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Wu, A.; Li, P.; Li, G.; Qin, L.; Song, H.; Mak, K.K. Yap1 Regulates Multiple Steps of Chondrocyte Differentiation during Skeletal Development and Bone Repair. Cell Rep. 2016, 14, 2224–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J.; Moscona, A. Role of cell shape in growth control. Nature 1978, 273, 345–349. [Google Scholar] [CrossRef]
- Chen, C.S.; Mrksich, M.; Huang, S.; Whitesides, G.M.; Ingber, D.E. Geometric control of cell life and death. Science 1997, 276, 1425–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Chen, C.S.; Ingber, D.E. Control of cyclin D1, p27Kip1, and cell cycle progression in human capillary endothelial cells by cell shape and cytoskeletal tension. Mol. Biol. Cell 1998, 9, 3179–3193. [Google Scholar] [CrossRef] [Green Version]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.-M.; Wang, H.B.; Dembo, M.; Wang, Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, B.C.; DiMilla, P.A.; Walker, M.; Kim, S.; Wong, J.Y. Vascular smooth muscle cell durotaxis depends on substrate stiffness gradient strength. Biophys. J. 2009, 97, 1313–1322. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.; Mogha, P.; Dash, S.; Majumder, A.; Jadhav, S.; Sen, S. Matrix elasticity regulates mesenchymal stem cell chemotaxis. J. Cell Sci. 2018, 131, jcs211391. [Google Scholar] [CrossRef] [Green Version]
- Vincent, L.G.; Choi, Y.S.; Alonso-Latorre, B.; Del Álamo, J.C.; Engler, A.J. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol. J. 2013, 8, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, M.; Sugawara, M.; Yamamoto, S.; Yamaguchi, K.; Nakanishi, J. Dynamic control of cell adhesion on a stiffness-tunable substrate for analyzing the mechanobiology of collective cell migration. Biomater. Sci. 2016, 4, 933–937. [Google Scholar] [CrossRef] [Green Version]
- Chandler, E.M.; Berglund, C.M.; Lee, J.S.; Polacheck, W.J.; Gleghorn, J.P.; Kirby, B.J.; Fischbach, C. Stiffness of photocrosslinked RGD-alginate gels regulates adipose progenitor cell behavior. Biotechnol. Bioeng. 2011, 108, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.; Wells, R.; Iredale, J. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-S.; Boulaire, J.; Chan, P.P.; Chung, J.E.; Kurisawa, M. The role of stiffness of gelatin–hydroxyphenylpropionic acid hydrogels formed by enzyme-mediated crosslinking on the differentiation of human mesenchymal stem cell. Biomaterials 2010, 31, 8608–8616. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Lauer, J.C.; Selig, M.; Hanak, M.; Walters, B.; Rolauffs, B. Shaping the Cell and the Future: Recent Advancements in Biophysical Aspects Relevant to Regenerative Medicine. J. Funct. Morphol. Kinesiol. 2017, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Uynuk-Ool, T.; Rothdiener, M.; Walters, B.; Hegemann, M.; Palm, J.; Nguyen, P.; Seeger, T.; Stöckle, U.; Stegemann, J.; Aicher, W.K.; et al. The geometrical shape of mesenchymal stromal cells measured by quantitative shape descriptors is determined by the stiffness of the biomaterial and by cyclic tensile forces. J. Tissue Eng. Regen. Med. 2017, 11, 3508–3522. [Google Scholar] [CrossRef]
- Walters, B.; Uynuk-Ool, T.; Rothdiener, M.; Palm, J.; Hart, M.L.; Stegemann, J.P.; Rolauffs, B. Engineering the geometrical shape of mesenchymal stromal cells through defined cyclic stretch regimens. Sci. Rep. 2017, 7, 6640. [Google Scholar] [CrossRef] [Green Version]
- Nasser, M.; Wu, Y.; Danaoui, Y.; Ghosh, G. Engineering microenvironments towards harnessing pro-angiogenic potential of mesenchymal stem cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 102, 75–84. [Google Scholar] [CrossRef]
- Harvey, J.W. (Ed.) Chapter 8-Bone Marrow Examination. In Veterinary Hematology; W.B. Saunders: Saint Louis, MO, USA, 2012; pp. 234–259. [Google Scholar]
- Ringe, J.; Kaps, C.; Burmester, G.R.; Sittinger, M. Stem cells for regenerative medicine: Advances in the engineering of tissues and organs. Naturwissenschaften 2002, 89, 338–351. [Google Scholar] [CrossRef]
- Rowlands, A.S.; George, P.A.; Cooper-White, J.J. Directing osteogenic and myogenic differentiation of MSCs: Interplay of stiffness and adhesive ligand presentation. Am. J. Physiol. Cell Physiol. 2008, 295, C1037–C1044. [Google Scholar] [CrossRef] [Green Version]
- Colley, H.E.; Mishra, G.; Scutt, A.M.; McArthur, S.L. Plasma Polymer Coatings to Support Mesenchymal Stem Cell Adhesion, Growth and Differentiation on Variable Stiffness Silicone Elastomers. Plasma Process. Polym. 2009, 6, 831–839. [Google Scholar] [CrossRef]
- Lanniel, M.; Huq, E.; Allen, S.; Buttery, L.; Williams, P.M.; Alexander, M.R. Substrate induced differentiation of human mesenchymal stem cells on hydrogels with modified surface chemistry and controlled modulus. Soft Matter 2011, 7, 6501–6514. [Google Scholar] [CrossRef]
- Rothdiener, M.; Hegemann, M.; Uynuk-Ool, T.; Walters, B.; Papugy, P.; Nguyen, P.; Claus, V.; Seeger, T.; Stoeckle, U.; Boehme, K.A.; et al. Stretching human mesenchymal stromal cells on stiffness-customized collagen type I generates a smooth muscle marker profile without growth factor addition. Sci. Rep. 2016, 6, 35840. [Google Scholar] [CrossRef] [PubMed]
- Lorthongpanich, C.; Thumanu, K.; Tangkiettrakul, K.; Jiamvoraphong, N.; Laowtammathron, C.; Damkham, N.; U-pratya, Y.; Issaragrisil, S. YAP as a key regulator of adipo-osteogenic differentiation in human MSCs. Stem Cell Res. Ther. 2019, 10, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver-De La Cruz, J.; Nardone, G.; Vrbsky, J.; Pompeiano, A.; Perestrelo, A.R.; Capradossi, F.; Melajová, K.; Filipensky, P.; Forte, G. Substrate mechanics controls adipogenesis through YAP phosphorylation by dictating cell spreading. Biomaterials 2019, 205, 64–80. [Google Scholar] [CrossRef]
- Karystinou, A.; Roelofs, A.J.; Neve, A.; Cantatore, F.P.; Wackerhage, H.; De Bari, C. Yes-associated protein (YAP) is a negative regulator of chondrogenesis in mesenchymal stem cells. Arthritis Res. Ther. 2015, 17, 147. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.S.; Vincent, L.G.; Lee, A.R.; Dobke, M.K.; Engler, A.J. Mechanical derivation of functional myotubes from adipose-derived stem cells. Biomaterials 2012, 33, 2482–2491. [Google Scholar] [CrossRef] [Green Version]
- Saha, K.; Keung, A.J.; Irwin, E.F.; Li, Y.; Little, L.; Schaffer, D.V.; Healy, K.E. Substrate modulus directs neural stem cell behavior. Biophys. J. 2008, 95, 4426–4438. [Google Scholar] [CrossRef] [Green Version]
- Leipzig, N.D.; Shoichet, M.S. The effect of substrate stiffness on adult neural stem cell behavior. Biomaterials 2009, 30, 6867–6878. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Lee, G.; Won, Y.; Lee, M.; Kwak, J.S.; Chun, C.H.; Chun, J.S. Matrix cross-linking–mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 9424–9429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, E.; Hofmann, S.; Stok, K.S.; Notbohm, H.; Müller, R.; Rotter, N. The influence of matrix elasticity on chondrocyte behavior in 3D. J. Tissue Eng. Regen. Med. 2012, 6, e31–e42. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.P.; Gu, L.; Mooney, D.J.; Levenston, M.E.; Chaudhuri, O. Mechanical confinement regulates cartilage matrix formation by chondrocytes. Nat. Mater. 2017, 16, 1243–1251. [Google Scholar] [CrossRef]
- Schuh, E.; Kramer, J.; Rohwedel, J.; Notbohm, H.; Müller, R.; Gutsmann, T.; Rotter, N. Effect of matrix elasticity on the maintenance of the chondrogenic phenotype. Tissue Eng. Part A 2010, 16, 1281–1290. [Google Scholar] [CrossRef]
- Mahmood, T.A.; de Jong, R.; Riesle, J.; Langer, R.; van Blitterswijk, C.A. Adhesion-mediated signal transduction in human articular chondrocytes: The influence of biomaterial chemistry and tenascin-C. Exp. Cell Res. 2004, 301, 179–188. [Google Scholar] [CrossRef]
- Lee, S.; Tong, X.; Yang, F. The effects of varying poly (ethylene glycol) hydrogel crosslinking density and the crosslinking mechanism on protein accumulation in three-dimensional hydrogels. Acta Biomater. 2014, 10, 4167–4174. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, Y.; Zhao, H. The effect of matrix stiffness on biomechanical properties of chondrocytes. Acta Biochim. Biophys. Sin. 2016, 48, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuFort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: Architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Lassar, A.B. The transcriptional activity of Sox9 in chondrocytes is regulated by RhoA signaling and actin polymerization. Mol. Cell. Biol. 2009, 29, 4262–4273. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Pfleghaar, K.; Kojima, S.I.; Pack, C.G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovett, D.B.; Shekhar, N.; Nickerson, J.A.; Roux, K.J.; Lele, T.P. Modulation of Nuclear Shape by Substrate Rigidity. Cell. Mol. Bioeng. 2013, 6, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, B.M.; Johnson, E.E.P. Nuclear morphologies: Their diversity and functional relevance. Chromosoma 2017, 126, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials 2015, 48, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Wu, M.; Feng, J.; Lin, X.; Gu, Z. RhoA/Rho kinase signaling regulates transforming growth factor-beta1-induced chondrogenesis and actin organization of synovium-derived mesenchymal stem cells through interaction with the Smad pathway. Int. J. Mol. Med. 2012, 30, 1119–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Xie, J.; Deng, L.; Yang, L. Substrate stiffness together with soluble factors affects chondrocyte mechanoresponses. ACS Appl. Mater. Interfaces 2014, 6, 16106–16116. [Google Scholar] [CrossRef]
- Leipzig, N.D.; Eleswarapu, S.V.; Athanasiou, K.A. The effects of TGF-beta1 and IGF-I on the biomechanics and cytoskeleton of single chondrocytes. Osteoarthr. Cartil. 2006, 14, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Song, X.; Li, R.; Sun, L.; Gong, X.; Chen, C.; Yang, L. Zyxin-involved actin regulation is essential in the maintenance of vinculin focal adhesion and chondrocyte differentiation status. Cell Prolif. 2019, 52, e12532. [Google Scholar] [CrossRef]
- Vardouli, L.; Vasilaki, E.; Papadimitriou, E.; Kardassis, D.; Stournaras, C. A novel mechanism of TGFβ-induced actin reorganization mediated by Smad proteins and Rho GTPases. FEBS J. 2008, 275, 4074–4087. [Google Scholar] [CrossRef]
- Pascarelli, N.A.; Collodel, G.; Moretti, E.; Cheleschi, S.; Fioravanti, A. Changes in Ultrastructure and Cytoskeletal Aspects of Human Normal and Osteoarthritic Chondrocytes Exposed to Interleukin-1beta and Cyclical Hydrostatic Pressure. Int. J. Mol. Sci. 2015, 16, 26019–26034. [Google Scholar] [CrossRef] [Green Version]
- Haudenschild, D.R.; Chen, J.; Pang, N.; Steklov, N.; Grogan, S.P.; Lotz, M.K.; D’Lima, D.D. Vimentin contributes to changes in chondrocyte stiffness in osteoarthritis. J. Orthop. Res. 2011, 29, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xie, J.; Rajappa, R.; Deng, L.; Fredberg, J.; Yang, L. Interleukin-1β and tumor necrosis factor-α increase stiffness and impair contractile function of articular chondrocytes. Acta Biochim. Biophys. Sin. 2015, 47, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinall, R.L.; Lo, S.H.; Reddi, A.H. Regulation of articular chondrocyte phenotype by bone morphogenetic protein 7, interleukin 1, and cellular context is dependent on the cytoskeleton. Exp. Cell Res. 2002, 272, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rys, J.P.; DuFort, C.C.; Monteiro, D.A.; Baird, M.A.; Oses-Prieto, J.A.; Chand, S.; Burlingame, A.L.; Davidson, M.W.; Alliston, T.N. Discrete spatial organization of TGFbeta receptors couples receptor multimerization and signaling to cellular tension. Elife 2015, 4, e09300. [Google Scholar] [CrossRef]
- Du, J.; Chen, X.; Liang, X.; Zhang, G.; Xu, J.; He, L.; Zhan, Q.; Feng, X.-Q.; Chien, S.; Yang, C. Integrin activation and internalization on soft ECM as a mechanism of induction of stem cell differentiation by ECM elasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 9466–9471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; McBeath, R.; Chen, C.S. Stem cell shape regulates a chondrogenic versus myogenic fate through Rac1 and N-cadherin. Stem Cells 2010, 28, 564–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haudenschild, D.R.; Chen, J.; Pang, N.; Lotz, M.K.; D’Lima, D.D. Rho kinase–dependent activation of SOX9 in chondrocytes. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2010, 62, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Wang, G.; Beier, F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J. Biol. Chem. 2005, 280, 11626–11634. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, V.; Dumitriu, B.; Penzo-Méndez, A.; Han, Y.; Pallavi, B. Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int. J. Biochem. Cell Biol. 2007, 39, 2195–2214. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef] [Green Version]
- Totsukawa, G.; Yamakita, Y.; Yamashiro, S.; Hartshorne, D.J.; Sasaki, Y.; Matsumura, F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 2000, 150, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Benya, P.D. Alterations in chondrocyte cytoskeletal architecture during phenotypic modulation by retinoic acid and dihydrocytochalasin B-induced reexpression. J. Cell Biol. 1988, 106, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Navarrete, R.; Lee, E.M.; Smith, K.; Hyzy, S.L.; Doroudi, M.; Williams, J.K.; Gall, K.; Boyan, B.D.; Schwartz, Z. Substrate Stiffness Controls Osteoblastic and Chondrocytic Differentiation of Mesenchymal Stem Cells without Exogenous Stimuli. PLoS ONE 2017, 12, e0170312. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ramos, P.; Mora, G.; Ripalda, P.; Vicente-Pascual, M.; Izal-Azcarate, I. Identification of signalling pathways triggered by changes in the mechanical environment in rat chondrocytes. Osteoarthr. Cartil. 2012, 20, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.M.; Matsiko, A.; Haugh, M.G.; Gleeson, J.P.; O’Brien, F.J. Mesenchymal stem cell fate is regulated by the composition and mechanical properties of collagen–glycosaminoglycan scaffolds. J. Mech. Behav. Biomed. Mater. 2012, 11, 53–62. [Google Scholar] [CrossRef]
- Bian, L.; Hou, C.; Tous, E.; Rai, R.; Mauck, R.L.; Burdick, J.A. The influence of hyaluronic acid hydrogel crosslinking density and macromolecular diffusivity on human MSC chondrogenesis and hypertrophy. Biomaterials 2013, 34, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Gegg, C.; Yang, F. The Effects of ROCK Inhibition on Mesenchymal Stem Cell Chondrogenesis Are Culture Model Dependent. Tissue Eng. Part A 2020, 26, 130–139. [Google Scholar] [CrossRef]
- Han, L.-H.; Yu, S.; Wang, T.; Behn, A.W.; Yang, F. Microribbon-Like Elastomers for Fabricating Macroporous and Highly Flexible Scaffolds that Support Cell Proliferation in 3D. Adv. Funct. Mater. 2013, 23, 346–358. [Google Scholar] [CrossRef]
- Sun, J.; Xiao, W.; Tang, Y.; Li, K.; Fan, H. Biomimetic interpenetrating polymer network hydrogels based on methacrylated alginate and collagen for 3D pre-osteoblast spreading and osteogenic differentiation. Soft Matter 2012, 8, 2398–2404. [Google Scholar] [CrossRef]
- Caplan, A.I.; Correa, D. The MSC: An injury drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Karbaat, L.; Wu, L.; Leijten, J.; Both, S.K.; Karperien, M. Trophic Effects of Mesenchymal Stem Cells in Tissue Regeneration. Tissue Eng. Part B Rev. 2017, 23, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Hofer, H.R.; Tuan, R.S. Secreted trophic factors of mesenchymal stem cells support neurovascular and musculoskeletal therapies. Stem Cell Res. Ther 2016, 7, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdeen, A.A.; Weiss, J.B.; Lee, J.; Kilian, K.A. Matrix composition and mechanics direct proangiogenic signaling from mesenchymal stem cells. Tissue Eng. Part A 2014, 20, 2737–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mohri, H.; Wu, Y.; Mohanty, S.; Ghosh, G. Impact of matrix stiffness on fibroblast function. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 74, 146–151. [Google Scholar] [CrossRef]
- Kusuma, G.D.; Carthew, J.; Lim, R.; Frith, J.E. Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem Cells Dev. 2017, 26, 617–631. [Google Scholar] [CrossRef]
- Li, J.; Wu, Y.; Schimmel, N.; Al-Ameen, M.A.; Ghosh, G. Breast cancer cells mechanosensing in engineered matrices: Correlation with aggressive phenotype. J. Mech. Behav. Biomed. Mater. 2016, 61, 208–220. [Google Scholar] [CrossRef]
- Yang, H.; Cheam, N.M.J.; Cao, H.; Lee, M.K.H.; Sze, S.K.; Tan, N.S.; Tay, C.Y. Materials Stiffness-Dependent Redox Metabolic Reprogramming of Mesenchymal Stem Cells for Secretome-Based Therapeutic Angiogenesis. Adv. Healthc. Mater. 2019, 8, 1900929. [Google Scholar] [CrossRef]
- Seib, F.P.; Prewitz, M.; Werner, C.; Bornhäuser, M. Matrix elasticity regulates the secretory profile of human bone marrow-derived multipotent mesenchymal stromal cells (MSCs). Biochem. Biophys. Res. Commun. 2009, 389, 663–667. [Google Scholar] [CrossRef]
- Schulze-Tanzil, G. Activation and dedifferentiation of chondrocytes: Implications in cartilage injury and repair. Ann. Anat. Anat. Anz. 2009, 191, 325–338. [Google Scholar] [CrossRef]
- Caron, M.M.; Emans, P.J.; Coolsen, M.M.; Voss, L.; Surtel, D.A.; Cremers, A.; van Rhijn, L.W.; Welting, T.J.M. Redifferentiation of dedifferentiated human articular chondrocytes: Comparison of 2D and 3D cultures. Osteoarthr. Cartil. 2012, 20, 1170–1178. [Google Scholar] [CrossRef] [Green Version]
- Dehne, T.; Karlsson, C.; Ringe, J.; Sittinger, M.; Lindahl, A. Chondrogenic differentiation potential of osteoarthritic chondrocytes and their possible use in matrix-associated autologous chondrocyte transplantation. Arthritis Res. Ther. 2009, 11, R133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurich, M.; Hofmann, G.O.; Best, N.; Rolauffs, B. Induced Redifferentiation of Human Chondrocytes from Articular Cartilage Lesion in Alginate Bead Culture After Monolayer Dedifferentiation: An Alternative Cell Source for Cell-Based Therapies? Tissue Eng. Part A 2018, 24, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Schuh, E.; Hofmann, S.; Stok, K.; Notbohm, H.; Müller, R.; Rotter, N. Chondrocyte redifferentiation in 3D: The effect of adhesion site density and substrate elasticity. J. Biomed. Mater. Res. A 2012, 100, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Levett, P.A.; Melchels, F.P.; Schrobback, K.; Hutmacher, D.W.; Malda, J.; Klein, T.J. Chondrocyte redifferentiation and construct mechanical property development in single-component photocrosslinkable hydrogels. J. Biomed. Mater. Res. A 2014, 102, 2544–2553. [Google Scholar] [CrossRef]
- Jimenez, G.; López-Ruiz, E.; Kwiatkowski, W.; Montañez, E.; Arrebola, F.; Carrillo, E.; Gray, P.C.; Belmonte, J.C.I.; Choe, S.; Perán, M.; et al. Activin A/BMP2 chimera AB235 drives efficient redifferentiation of long term cultured autologous chondrocytes. Sci. Rep. 2015, 5, 16400. [Google Scholar] [CrossRef] [Green Version]
- Hsieh-Bonassera, N.D.; Wu, I.; Lin, J.K.; Schumacher, B.L.; Chen, A.C.; Masuda, K.; Bugbee, W.D.; Sah, R.L. Expansion and redifferentiation of chondrocytes from osteoarthritic cartilage: Cells for human cartilage tissue engineering. Tissue Eng. Part A 2009, 15, 3513–3523. [Google Scholar] [CrossRef] [Green Version]
- Babur, B.K.; Ghanavi, P.; Levett, P.; Lott, W.B.; Klein, T.; Cooper-White, J.J.; Crawford, R.; Doran, M.R. The interplay between chondrocyte redifferentiation pellet size and oxygen concentration. PLoS ONE 2013, 8, e58865. [Google Scholar] [CrossRef] [Green Version]
- Meretoja, V.V.; Dahlin, R.L.; Wright, S.; Kasper, F.K.; Mikos, A.G. Articular chondrocyte redifferentiation in 3D co-cultures with mesenchymal stem cells. Tissue Eng. Part C Methods 2014, 20, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Kisiday, J.D. Expansion of Chondrocytes for Cartilage Tissue Engineering: A Review of Chondrocyte Dedifferentiation and Redifferentiation as a Function of Growth in Expansion Culture. Regen. Med. Front. 2020, 2, e200002. [Google Scholar]
- Sanz-Ramos, P.; Mora, G.; Vicente-Pascual, M.; Ochoa, I.; Alcaine, C.; Moreno, R.; Doblaré, M.; Izal-Azcárate, I. Response of sheep chondrocytes to changes in substrate stiffness from 2 to 20 Pa: Effect of cell passaging. Connect. Tissue Res. 2013, 54, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Levinson, C.; Hertl, M.; Broguiere, N.; Brück, O.; Mustjoki, S.; Gerstenberg, A.; Weber, D.; Salzmann, G.; Steinwachs, M.; et al. Characterization of polydactyly chondrocytes and their use in cartilage engineering. Sci. Rep. 2019, 9, 4275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zu, Y.; Li, J.; Du, S.; Xu, Y.; Zhang, L.; Jiang, L.; Wang, Z.; Chien, S.; Yang, C. Extracellular matrix stiffness dictates Wnt expression through integrin pathway. Sci. Rep. 2016, 6, 20395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, J.; Ryu, J.H.; Kim, J.H.; Chun, C.H.; Chun, J.S. alpha-Catenin inhibits beta-catenin-T-cell factor/lymphoid enhancing factor transcriptional activity and collagen type II expression in articular chondrocytes through formation of Gli3R.alpha-catenin.beta-catenin ternary complex. J. Biol. Chem. 2012, 287, 11751–11760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.G.; Yu, S.S.; Ryu, J.H.; Jeon, H.B.; Yoo, Y.J.; Eom, S.H.; Chun, J.S. Regulation of beta-catenin signaling and maintenance of chondrocyte differentiation by ubiquitin-independent proteasomal degradation of alpha-catenin. J. Biol. Chem. 2005, 280, 12758–12765. [Google Scholar] [CrossRef] [Green Version]
- Nuernberger, S.; Cyran, N.; Albrecht, C.; Redl, H.; Vécsei, V.; Marlovits, S. The influence of scaffold architecture on chondrocyte distribution and behavior in matrix-associated chondrocyte transplantation grafts. Biomaterials 2011, 32, 1032–1040. [Google Scholar] [CrossRef]
- Panni, A.S.; Del Regno, C.; Mazzitelli, G.; D’Apolito, R.; Corona, K.; Vasso, M. Good clinical results with autologous matrix-induced chondrogenesis (Amic) technique in large knee chondral defects. Knee Surg. Sports Traumatol. Arthrosc. 2018, 26, 1130–1136. [Google Scholar]
- Zak, L.; Albrecht, C.; Wondrasch, B.; Widhalm, H.; Vekszler, G.; Trattnig, S.; Marlovits, S.; Aldrian, S. Results 2 years after matrix-associated autologous chondrocyte transplantation using the Novocart 3D scaffold: An analysis of clinical and radiological data. Am. J. Sports Med. 2014, 42, 1618–1627. [Google Scholar] [CrossRef]
- Müller, P.E.; Gallik, D.; Hammerschmid, F.; Baur-Melnyk, A.; Pietschmann, M.F.; Zhang, A.; Niethammer, T.R. Third-generation autologous chondrocyte implantation after failed bone marrow stimulation leads to inferior clinical results. Knee Surg. Sports Traumatol. Arthrosc. 2020, 28, 470–477. [Google Scholar] [CrossRef]
- Niethammer, T.R.; Limbrunner, K.; Betz, O.B.; Gülecyüz, M.F.; Pietschmann, M.F.; Feist, M.; Müller, P.E. Analysis of the autologous chondrocyte quality of matrix-based autologous chondrocyte implantation in the knee joint. Int. Orthop. 2016, 40, 205–212. [Google Scholar] [CrossRef]
- Crawford, D.C.; DeBerardino, T.M.; Williams, R.J., III. NeoCart, an autologous cartilage tissue implant, compared with microfracture for treatment of distal femoral cartilage lesions: An FDA phase-II prospective, randomized clinical trial after two years. JBJS 2012, 94, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.E.; Williams, R.J., III; DeBerardino, T.M.; Taylor, D.C.; Ma, C.B.; Kane, M.S.; Crawford, D.C. Magnetic resonance imaging characterization and clinical outcomes after NeoCart surgical therapy as a primary reparative treatment for knee cartilage injuries. Am. J. Sports Med. 2017, 45, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Broese, M.; Simon, A.; Liodakis, E.; Ettinger, M.; Guenther, D.; Zeichen, J.; Krettek, C.; Jagodzinski, M.; Haasper, C. CaReS®(MACT) versus microfracture in treating symptomatic patellofemoral cartilage defects: A retrospective matched-pair analysis. J. Orthop. Sci. 2013, 18, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Saris, D.; Price, A.; Widuchowski, W.; Bertrand-Marchand, M.; Caron, J.; Drogset, J.O.; Emans, P.; Podskubka, A.; Tsuchida, A.; Kili, S.; et al. Matrix-applied characterized autologous cultured chondrocytes versus microfracture: Two-year follow-up of a prospective randomized trial. Am. J. Sports Med. 2014, 42, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Brittberg, M.; Recker, D.; Ilgenfritz, J.; Saris, D.B.; SUMMIT Extension Study Group. Matrix-applied characterized autologous cultured chondrocytes versus microfracture: Five-year follow-up of a prospective randomized trial. Am. J. Sports Med. 2018, 46, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Delcogliano, M.; de Caro, F.; Scaravella, E.; Ziveri, G.; De Biase, C.F.; Marotta, D.; Marenghi, P.; Delcogliano, A. Use of innovative biomimetic scaffold in the treatment for large osteochondral lesions of the knee. Knee Surg. Sports Traumatol. Arthrosc. 2014, 22, 1260–1269. [Google Scholar] [CrossRef]
- Rutgers, M.; Saris, D.B.; Vonk, L.A.; van Rijen, M.H.; Akrum, V.; Langeveld, D.; van Boxtel, A.; Dhert, W.J.; Creemers, L.B. Effect of collagen type I or type II on chondrogenesis by cultured human articular chondrocytes. Tissue Eng. Part A 2013, 19, 59–65. [Google Scholar] [CrossRef]
- Chowdhury, T.T.; Schulz, R.M.; Rai, S.S.; Thuemmler, C.B.; Wuestneck, N.; Bader, A.; Homandberg, G.A. Biomechanical modulation of collagen fragment-induced anabolic and catabolic activities in chondrocyte/agarose constructs. Arthritis Res. Ther. 2010, 12, R82. [Google Scholar] [CrossRef] [Green Version]
- Jennings, L.; Wu, L.; King, K.B.; Hämmerle, H.; Cs-Szabo, G.; Mollenhauer, J. The effects of collagen fragments on the extracellular matrix metabolism of bovine and human chondrocytes. Connect. Tissue Res. 2001, 42, 71–86. [Google Scholar] [CrossRef]
- Billinghurst, R.C.; Dahlberg, L.; Ionescu, M.; Reiner, A.; Bourne, R.; Rorabeck, C.; Mitchell, P.; Hambor, J.; Diekmann, O.; Schesche, H.; et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J. Clin. Investig. 1997, 99, 1534–1545. [Google Scholar] [CrossRef] [Green Version]
- Appleton, C.T.; Usmani, S.E.; Bernier, S.M.; Aigner, T.; Beier, F. Transforming growth factor alpha suppression of articular chondrocyte phenotype and Sox9 expression in a rat model of osteoarthritis. Arthritis Rheum. 2007, 56, 3693–3705. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Liu, S.; Yang, B.; Hajal, C.; Changede, R.; Hu, J.; Wolfenson, H.; Hone, J.; Sheetz, M. EGFR and HER2 activate rigidity sensing only on rigid matrices. Nat. Mater. 2017, 16, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Tsai, Y.H.; Deng, W.P.; Shih, S.N.; Fang, C.L.; Burch, J.G.; Chen, W.H.; Lai, W.F. Type I and II collagen regulation of chondrogenic differentiation by mesenchymal progenitor cells. J. Orthop. Res. 2005, 23, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Tamaddon, M.; Burrows, M.; Ferreira, S.A.; Dazzi, F.; Apperley, J.F.; Bradshaw, A.; Brand, D.D.; Czernuszka, J.; Gentleman, E. Monomeric, porous type II collagen scaffolds promote chondrogenic differentiation of human bone marrow mesenchymal stem cells in vitro. Sci. Rep. 2017, 7, 43519. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Gong, T.; Xie, J.; Lin, S.; Liu, Y.; Zhou, T.; Lin, Y. Softening substrates promote chondrocytes phenotype via RhoA/ROCK pathway. ACS Appl. Mater. Interfaces 2016, 8, 22884–22891. [Google Scholar] [CrossRef]
- Annamalai, R.T.; Mertz, D.R.; Daley, E.L.; Stegemann, J.P. Collagen Type II enhances chondrogenic differentiation in agarose-based modular microtissues. Cytotherapy 2016, 18, 263–277. [Google Scholar] [CrossRef] [Green Version]
- Porcellini, G.; Merolla, G.; Campi, F.; Pellegrini, A.; Bodanki, C.S.; Paladini, P. Arthroscopic treatment of early glenohumeral arthritis. J. Orthop. Traumatol. 2013, 14, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Brix, M.O.; Stelzeneder, D.; Chiari, C.; Koller, U.; Nehrer, S.; Dorotka, R.; Windhager, R.; Domayer, S.E. Treatment of full-thickness chondral defects with hyalograft C in the knee: Long-term results. Am. J. Sports Med. 2014, 42, 1426–1432. [Google Scholar] [CrossRef]
- Thier, S.; Weiss, C.; Fickert, S. Arthroscopic autologous chondrocyte implantation in the hip for the treatment of full-thickness cartilage defects: A case series of 29 patients and review of the literature. SICOT J. 2017, 3, 72. [Google Scholar] [CrossRef] [Green Version]
- McCormick, F.; Cole, B.J.; Nwachukwu, B.; Harris, J.D.; Adkisson, H.D., IV; Farr, J. Treatment of focal cartilage defects with a juvenile allogeneic 3-dimensional articular cartilage graft. Oper. Tech. Sports Med. 2013, 21, 95–99. [Google Scholar] [CrossRef] [Green Version]
- De Windt, T.S.; Saris, D.B.; Slaper-Cortenbach, I.C.; van Rijen, M.H.; Gawlitta, D.; Creemers, L.B.; de Weger, R.A.; Dhert, W.J.A.; Vonk, L.A. Direct Cell-Cell Contact with Chondrocytes Is a Key Mechanism in Multipotent Mesenchymal Stromal Cell-Mediated Chondrogenesis. Tissue Eng. Part A 2015, 21, 2536–2547. [Google Scholar] [CrossRef] [PubMed]
- Von der Mark, K.; Gauss, V.; Von Der Mark, H.; Müller, P. Relationship between cell shape and type of collagen synthesised as chondrocytes lose their cartilage phenotype in culture. Nature 1977, 267, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Dessau, W.; Vertel, B.M.; von der Mark, H.; von der Mark, K. Extracellular matrix formation by chondrocytes in monolayer culture. J. Cell Biol. 1981, 90, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, C.; Tichy, B.; Nürnberger, S.; Hosiner, S.; Zak, L.; Aldrian, S.; Marlovits, S. Gene expression and cell differentiation in matrix-associated chondrocyte transplantation grafts: A comparative study. Osteoarthr. Cartil. 2011, 19, 1219–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benya, P.D.; Shaffer, J.D. Dedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell 1982, 30, 215–224. [Google Scholar] [CrossRef]
- Schneider, U.; Rackwitz, L.; Andereya, S.; Siebenlist, S.; Fensky, F.; Reichert, J.; Löer, I.; Barthel, T.; Rudert, M.; Nöth, U. A prospective multicenter study on the outcome of type I collagen hydrogel-based autologous chondrocyte implantation (CaReS) for the repair of articular cartilage defects in the knee. Am. J. Sports Med. 2011, 39, 2558–2565. [Google Scholar] [CrossRef]
- Lee, J.; Abdeen, A.A.; Kilian, K.A. Rewiring mesenchymal stem cell lineage specification by switching the biophysical microenvironment. Sci. Rep. 2014, 4, 5188. [Google Scholar] [CrossRef] [Green Version]
- Metavarayuth, K.; Sitasuwan, P.; Zhao, X.; Lin, Y.; Wang, Q. Influence of Surface Topographical Cues on the Differentiation of Mesenchymal Stem Cells in Vitro. ACS Biomater. Sci. Eng. 2016, 2, 142–151. [Google Scholar] [CrossRef]
- Anderson, H.J.; Sahoo, J.K.; Ulijn, R.V.; Dalby, M.J. Mesenchymal Stem Cell Fate: Applying Biomaterials for Control of Stem Cell Behavior. Front. Bioeng. Biotechnol. 2016, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Noriega, S.E.; Hasanova, G.I.; Schneider, M.J.; Larsen, G.F.; Subramanian, A. Effect of fiber diameter on the spreading, proliferation and differentiation of chondrocytes on electrospun chitosan matrices. Cells Tissues Organs 2012, 195, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, P.; Andereya, S.; Angele, P.; Ateschrang, A.; Aurich, M.; Baumann, M.; Behrens, P.; Bosch, U.; Erggelet, C.; Fickert, S.; et al. Autologous chondrocyte implantation (ACI) for cartilage defects of the knee: A guideline by the working group “Tissue Regeneration” of the German Society of Orthopaedic Surgery and Traumatology (DGOU). Z. Orthop. Unfall. 2013, 151, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Angele, P.; Fritz, J.; Albrecht, D.; Koh, J.; Zellner, J. Defect type, localization and marker gene expression determines early adverse events of matrix-associated autologous chondrocyte implantation. Injury 2015, 46 (Suppl. S4), S2–S9. [Google Scholar] [CrossRef]
- Niemeyer, P.; Feucht, M.J.; Fritz, J.; Albrecht, D.; Spahn, G.; Angele, P. Cartilage repair surgery for full-thickness defects of the knee in Germany: Indications and epidemiological data from the German Cartilage Registry (KnorpelRegister DGOU). Arch. Orthop. Trauma Surg. 2016, 136, 891–897. [Google Scholar] [CrossRef]
- Aurich, M.; Hofmann, G.O.; Rolauffs, B. Tissue engineering-relevant characteristics of ex vivo and monolayer-expanded chondrocytes from the notch versus trochlea of human knee joints. Int. Orthop. 2017, 41, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Aurich, M.; Anders, J.; Trommer, T.; Liesaus, E.; Seifert, M.; Schömburg, J.; Rolauffs, B.; Wagner, A.; Mollenhauer, J. Histological and cell biological characterization of dissected cartilage fragments in human osteochondritis dissecans of the femoral condyle. Arch. Orthop. Trauma Surg. 2006, 126, 606–614. [Google Scholar] [CrossRef]
- Aurich, M.; Hofmann, G.O.; Gras, F.; Rolauffs, B. Human osteochondritis dissecans fragment-derived chondrocyte characteristics ex vivo, after monolayer expansion-induced de-differentiation, and after re-differentiation in alginate bead culture. BMC Musculoskelet. Disord. 2018, 19, 168. [Google Scholar] [CrossRef]
- Rothdiener, M.; Uynuk-Ool, T.; Südkamp, N.; Aurich, M.; Grodzinsky, A.J.; Kurz, B.; Rolauffs, B. Human osteoarthritic chondrons outnumber patient- and joint-matched chondrocytes in hydrogel culture-Future application in autologous cell-based OA cartilage repair? J. Tissue Eng. Regen. Med. 2018, 12, e1206–e1220. [Google Scholar] [CrossRef]
- Cole, A.A.; Kuettner, K.E. Molecular basis for differences between human joints. Cell. Mol. Life Sci. 2002, 59, 19–26. [Google Scholar] [CrossRef]
- Cole, A.A.; Margulis, A.; Kuettner, K.E. Distinguishing ankle and knee articular cartilage. Foot Ankle Clin. 2003, 8, 305–316. [Google Scholar] [CrossRef]
- Eger, W.; Aurich, M.; Schumacher, B.L.; Mollenhauer, J.; Kuettner, K.E.; Cole, A.A. Differences in metabolism of chondrocytes of the knee and ankle joint. Z. Orthop. Ihre Grenzgeb. 2003, 141, 18–20. [Google Scholar]
- Eger, W.; Schumacher, B.L.; Mollenhauer, J.; Kuettner, K.E.; Cole, A.A. Human knee and ankle cartilage explants: Catabolic differences. J. Orthop. Res. 2002, 20, 526–534. [Google Scholar] [CrossRef]
- Murata, M.; Yudoh, K.; Shimizu, H.; Beppu, M.; Nakamura, H.; Kato, T.; Masuko, K. Layilin, a talin-binding hyaluronan receptor, is expressed in human articular chondrocytes and synoviocytes and is down-regulated by interleukin-1beta. Mod. Rheumatol. 2013, 23, 478–488. [Google Scholar] [CrossRef]
- Lv, H.; Li, L.; Sun, M.; Zhang, Y.; Chen, L.; Rong, Y.; Li, Y. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res. Ther. 2015, 6, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, M.; Raute, K.; Hummel, B.; Madl, J.; Creusen, G.; Thomas, O.S.; Christen, E.H.; Hotz, N.; Gübeli, R.J.; Engesser, R.; et al. Phytochrome-Based Extracellular Matrix with Reversibly Tunable Mechanical Properties. Adv. Mater. 2019, 31, e1806727. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.A.; Rolauffs, B. Onset and Progression of Human Osteoarthritis-Can Growth Factors, Inflammatory Cytokines, or Differential miRNA Expression Concomitantly Induce Proliferation, ECM Degradation, and Inflammation in Articular Cartilage? Int. J. Mol. Sci. 2018, 19, 2282. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef] [Green Version]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Sabra, H.; Brunner, M.; Mandati, V.; Wehrle-Haller, B.; Lallemand, D.; Ribba, A.S.; Chevalier, G.; Guardiola, P.; Block, M.R.; Bouvard, D. beta1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J. Biol. Chem. 2017, 292, 19179–19197. [Google Scholar] [CrossRef] [Green Version]
- Sero, J.E.; Bakal, C. Multiparametric Analysis of Cell Shape Demonstrates that beta-PIX Directly Couples YAP Activation to Extracellular Matrix Adhesion. Cell Syst. 2017, 4, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, X.; Jing, X.; Li, M.; Ren, Y.; Chen, J.; Yang, C.; Wu, H.; Guo, F. Hypoxia promotes maintenance of the chondrogenic phenotype in rat growth plate chondrocytes through the HIF-1alpha/YAP signaling pathway. Int. J. Mol. Med. 2018, 42, 3181–3192. [Google Scholar]
- Wang, L.; Zhang, Z.; Yu, X.; Huang, X.; Liu, Z.; Chai, Y.; Yang, L.; Wang, Q.; Li, M.; Zhao, J.; et al. Unbalanced YAP-SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene 2019, 38, 2042–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaas, A.; Velasco, J.; Gorski, D.J.; Li, J.; Cole, A.; Christopherson, K.; Sandy, J.D. The relationship between fibrogenic TGFbeta1 signaling in the joint and cartilage degradation in post-injury osteoarthritis. Osteoarthr. Cartil. 2011, 19, 1081–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thielen, N.G.M.; van der Kraan, P.M.; van Caam, A.P.M. TGFβ/BMP Signaling Pathway in Cartilage Homeostasis. Cells 2019, 8, 969. [Google Scholar] [CrossRef] [Green Version]
- Van der Kraan, P.M.; Goumans, M.J.; Davidson, E.B.; Ten Dijke, P. Age-dependent alteration of TGF-beta signalling in osteoarthritis. Cell Tissue Res. 2012, 347, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Vite, A.; Zhang, C.; Yi, R.; Emms, S.; Radice, G.L. alpha-Catenin-dependent cytoskeletal tension controls Yap activity in the heart. Development 2018, 145, dev149823. [Google Scholar] [CrossRef] [Green Version]
- Seddiki, R.; Narayana GH, N.S.; Strale, P.O.; Balcioglu, H.E.; Peyret, G.; Yao, M.; Le, A.P.; Lim, C.T.; Yan, J.; Ladoux, B.; et al. Force-dependent binding of vinculin to alpha-catenin regulates cell-cell contact stability and collective cell behavior. Mol. Biol. Cell 2018, 29, 380–388. [Google Scholar] [CrossRef]
- Yonemura, S.; Wada, Y.; Watanabe, T.; Nagafuchi, A.; Shibata, M. alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Wen, K.K.; Rubenstein, P.A.; DeMali, K.A. Vinculin nucleates actin polymerization and modifies actin filament structure. J. Biol. Chem. 2009, 284, 30463–30473. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, Y.; Zhang, M.; Heallen, T.; Leach, J.; Tao, G.; Xiao, Y.; Bai, Y.; Li, W.; Willerson, J.T.; Martin, J.F. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Sci. Signal. 2015, 8, ra41. [Google Scholar] [CrossRef] [Green Version]
- Albert, P.J.; Schwarz, U.S. Modeling cell shape and dynamics on micropatterns. Cell Adh. Migr. 2016, 10, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Iskratsch, T.; Wolfenson, H.; Sheetz, M.P. Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 2014, 15, 825–833. [Google Scholar] [CrossRef]
- Paluch, E.K.; Nelson, C.M.; Biais, N.; Fabry, B.; Moeller, J.; Pruitt, B.L.; Wollnik, C.; Kudryasheva, G.; Rehfeldt, F.; Federle, W. Mechanotransduction: Use the force(s). BMC Biol. 2015, 13, 47. [Google Scholar] [CrossRef] [Green Version]
- Strzyz, P. Mechanotransduction: May the force be with you. Nat. Rev. Mol. Cell Biol. 2016, 17, 533. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
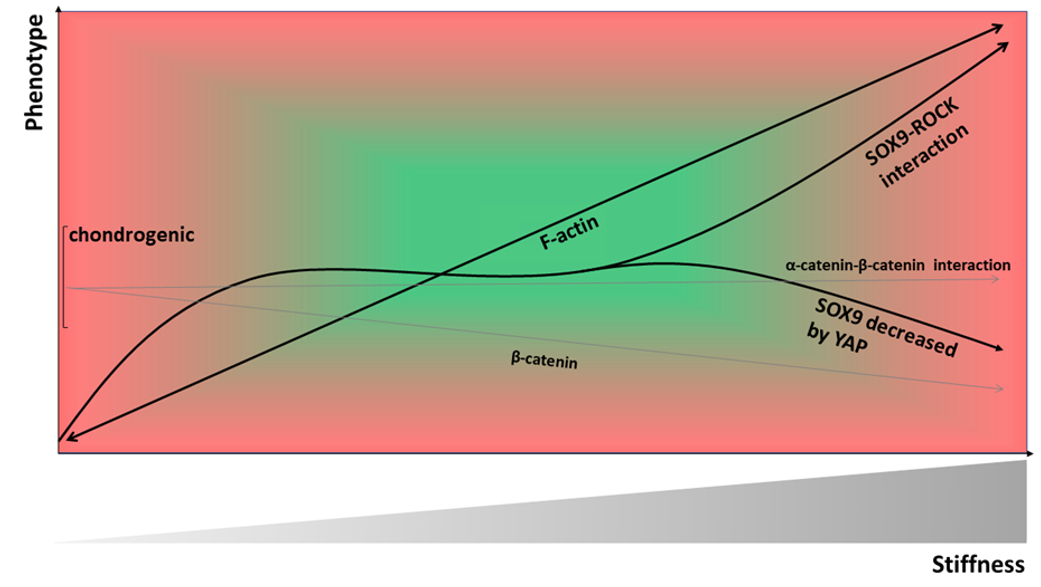{kind=link}
| Product Name | Type of Material | Stiffness Data | Morphology | Gene Expression | Porosity | Reference |
|---|---|---|---|---|---|---|
| Chondro-Gide® | 3D Hyaluronan web | NA | Some spherical; mainly elongated; polygonal | De-differentiated phenotype | High (up to 200 µm) | [228,229] |
| Hyalograft C | Autologous chondrocytes grown on a 3D hyaluronan-based scaffold | NA | Spherical, elongated, polygonal | Lower ACAN and collagen type II expression | High (up to 200 µm) | [249,250] |
| MACI® | Membrane of type I/III collagen | NA | Elongated-fibroblast like cell shape | High collagen type I | [236,237] | |
| Novocart®3D | Type I collagen sponges with bilayer structure | NA | Mainly spherical | High expression of collagen type II, little collagen type I, X | High (10-100 µm) | [230,231,232] |
| NeoCart® | Collagen type I loaded into sponges of same material | NA | NA | NA | NA | [233,234] |
| Novocart Inject | Autologous CHs, hydrogel is a combination of human albumin and hyaluronic acid | NA | NA | NA | NA | [251] |
| RevaFlex™ (formerly DeNovo ET®) | Hyaline neocartilage discs composed of allogenic juvenile CHs | NA | NA | NA | NA | [252] |
| Product Name | Type of Material | Stiffness Data | Morphology | Gene Expression | Porosity | Reference |
|---|---|---|---|---|---|---|
| CaReS® | Collagen type I | NA | Spheroid, many elongated, polygonal | High collagen type II and ACAN | Low | [235] |
| MaioRegen® | Collagen type Iand hydroxyapatite | NA | NA | NA | NA | [238] |
| Key Molecule | Cell Type | Phenotype |
|---|---|---|
| ROCK-SOX9 | CH | stress fiber-inducing effect of ROCK leads to de-differentiation of CH phenotype at supra-chondrogenic stiffnesses [190] |
| RhoA/ROCK/myosin II | CH | high material stiffness increases expression of stress fibers, which leads to a downregulation of collagen type II, but upregulation of SOX9 low material stiffness/disruption of actin network restores the chondrogenic phenotype [163] |
| MSC | high material stiffness causes high cross-linking density of fibers → stiffness-specific upregulation of distinct lineage genes [121] | |
| ATDC5 | high material stiffness leads to upregulation of SOX9 [190] | |
| YAP/TAZ | CH | high stiffness leads to nuclear accumulation of YAP/TAZ and a degenerative CH phenotype [162] YAP inactivation restores collagen type II levels [122] |
| MSC | soft substrate leads to YAP/TAZ accumulation in the cytoplasm → no proliferation [127]/chondrogenic differentiation [122] stiff substrate leads to active YAP/TAZ in the nucleus → induces proliferation [127] and osteogenic differentiation [121] | |
| TGF-β | CH | low stiffness + TGF-β lead to elevated levels of chondrogenic gene expression [190] higher stiffness + TGF-β increase cell stiffness and lead to higher SOX9 expression [190] |
| MSC | differential effects of TGF-β modulated by stiffness soft material stiffness + TGF-β → chondrogenic differentiation [121] medium material stiffness + TGF-β → myogenic differentiation [121] | |
| Lamin A | MSC | soft material stiffness induces low lamin-A expression → adipogenic differentiation [173] high material stiffness induces high lamin-A expression → osteogenic differentiation [173] |
| Wnt/β-catenin | CH/MSC | high material stiffness leads to accumulation of β-catenin and de-differentiation of CHs [225] |
| α-catenin | CH | counteracts the β-catenin mediated inhibition of collagen type II expression [227] |
| IL-1β Rac1/cyclin D1 | CH | elevated levels of IL-1β increase cellular stiffness [184] |
| CH/ MSC | high material stiffness leads to upregulation of cyclin D1 mediated by Rac1, inducing S-phase entry and proliferation [125] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selig, M.; Lauer, J.C.; Hart, M.L.; Rolauffs, B. Mechanotransduction and Stiffness-Sensing: Mechanisms and Opportunities to Control Multiple Molecular Aspects of Cell Phenotype as a Design Cornerstone of Cell-Instructive Biomaterials for Articular Cartilage Repair. Int. J. Mol. Sci. 2020, 21, 5399. https://doi.org/10.3390/ijms21155399
Selig M, Lauer JC, Hart ML, Rolauffs B. Mechanotransduction and Stiffness-Sensing: Mechanisms and Opportunities to Control Multiple Molecular Aspects of Cell Phenotype as a Design Cornerstone of Cell-Instructive Biomaterials for Articular Cartilage Repair. International Journal of Molecular Sciences. 2020; 21(15):5399. https://doi.org/10.3390/ijms21155399
Chicago/Turabian StyleSelig, Mischa, Jasmin C. Lauer, Melanie L. Hart, and Bernd Rolauffs. 2020. "Mechanotransduction and Stiffness-Sensing: Mechanisms and Opportunities to Control Multiple Molecular Aspects of Cell Phenotype as a Design Cornerstone of Cell-Instructive Biomaterials for Articular Cartilage Repair" International Journal of Molecular Sciences 21, no. 15: 5399. https://doi.org/10.3390/ijms21155399