Phage S144, a New Polyvalent Phage Infecting Salmonella spp. and Cronobacter sakazakii

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Phage S144 Is a Polyvalent Virus, Infecting Diverse Strains of the Enterobacteriaceae
2.2. S144 Recognizes the O-Antigen as Receptor Both in Salmonella and Cronobacter
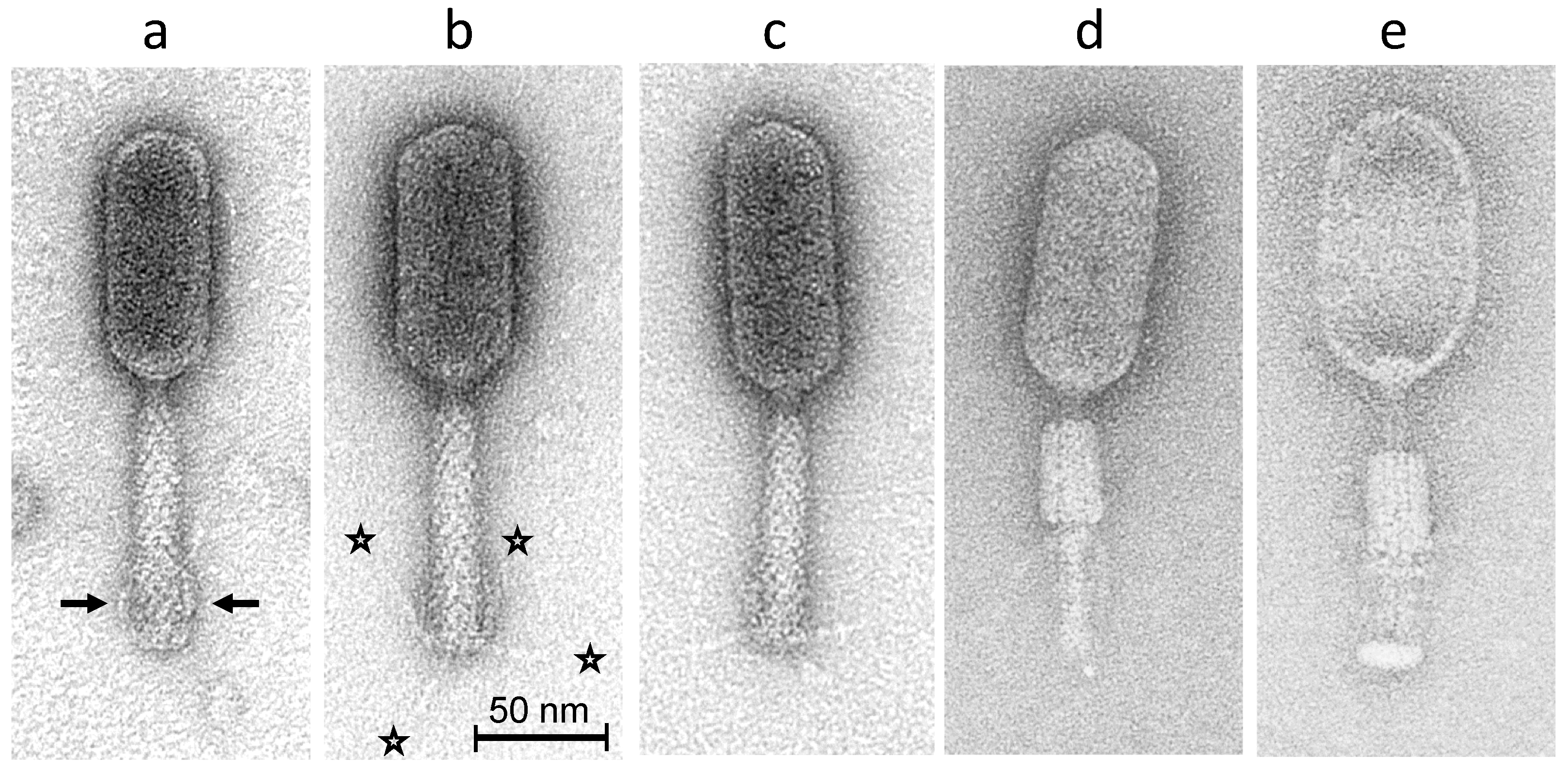
2.3. Morphology of Phage S144
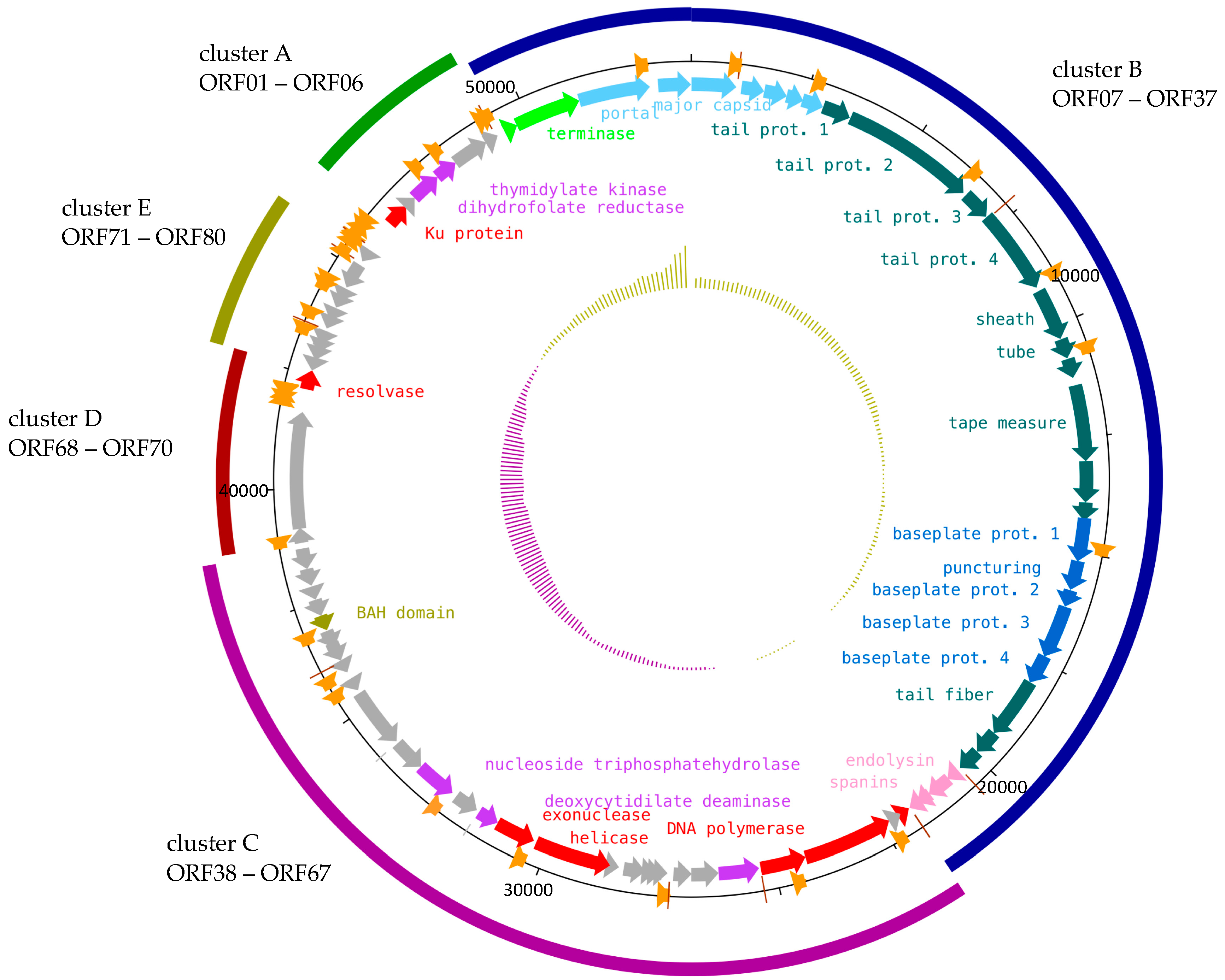
2.4. Functional Modules of Phage S144
2.5. The Genomic DNA of Phage S144 is Modified
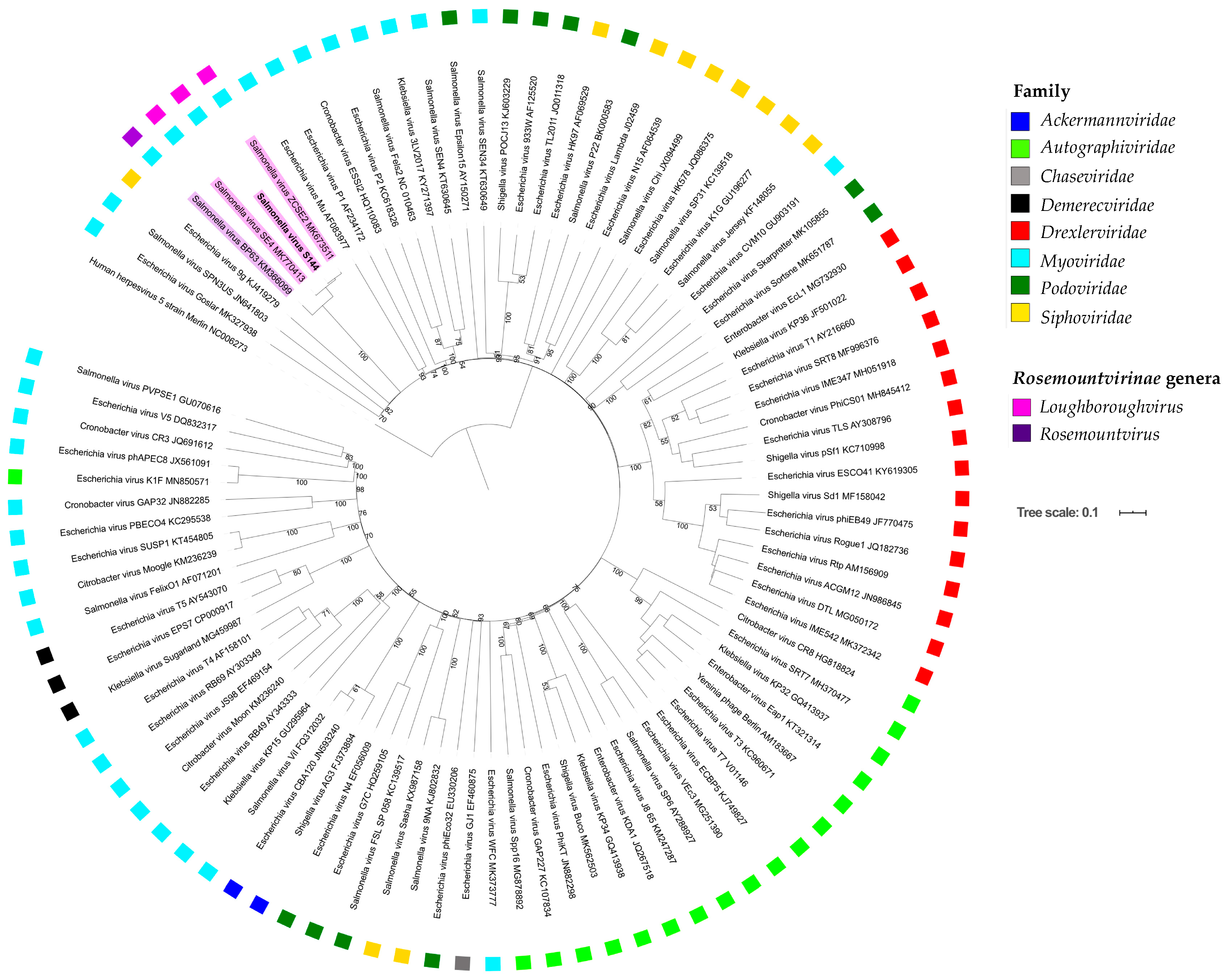
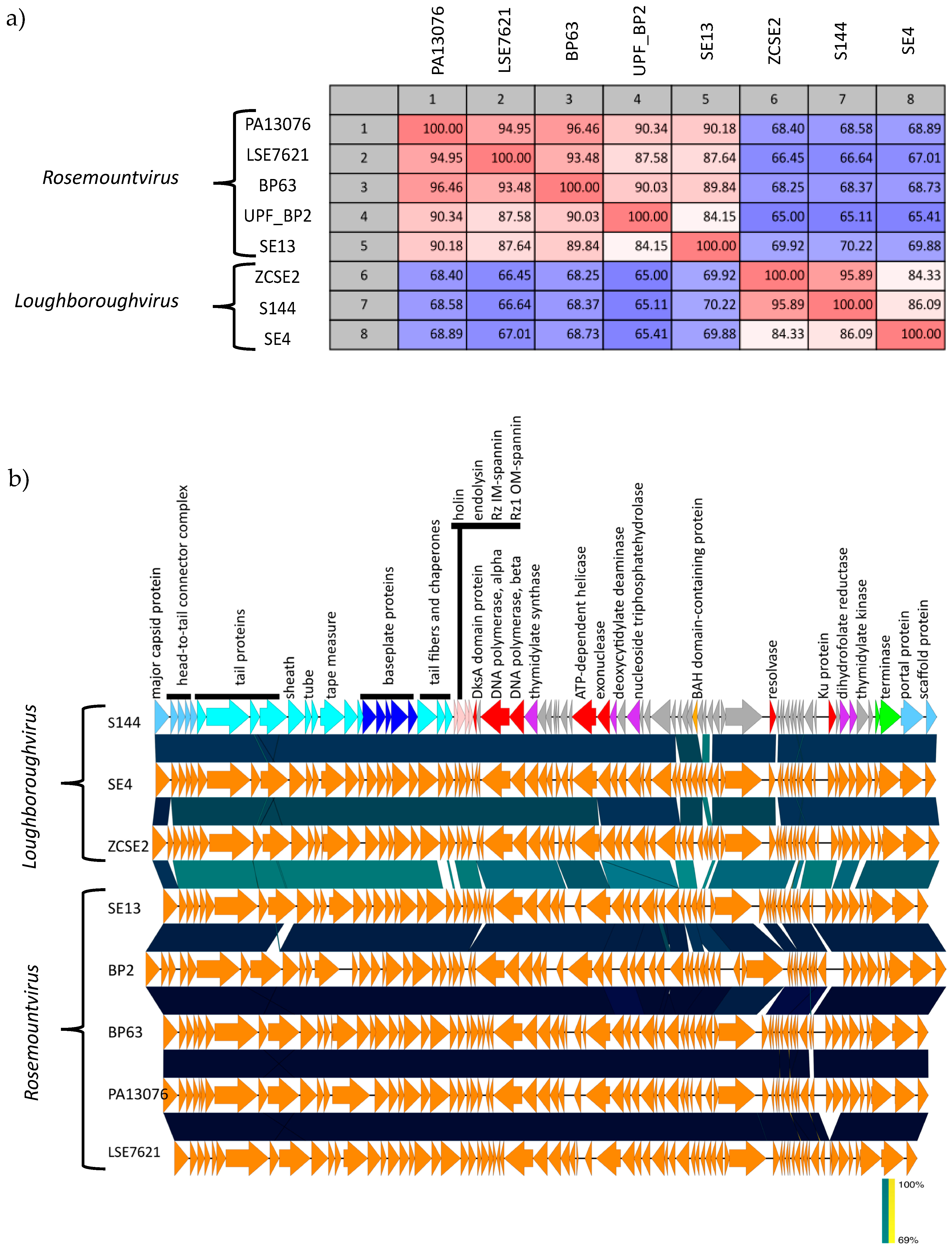
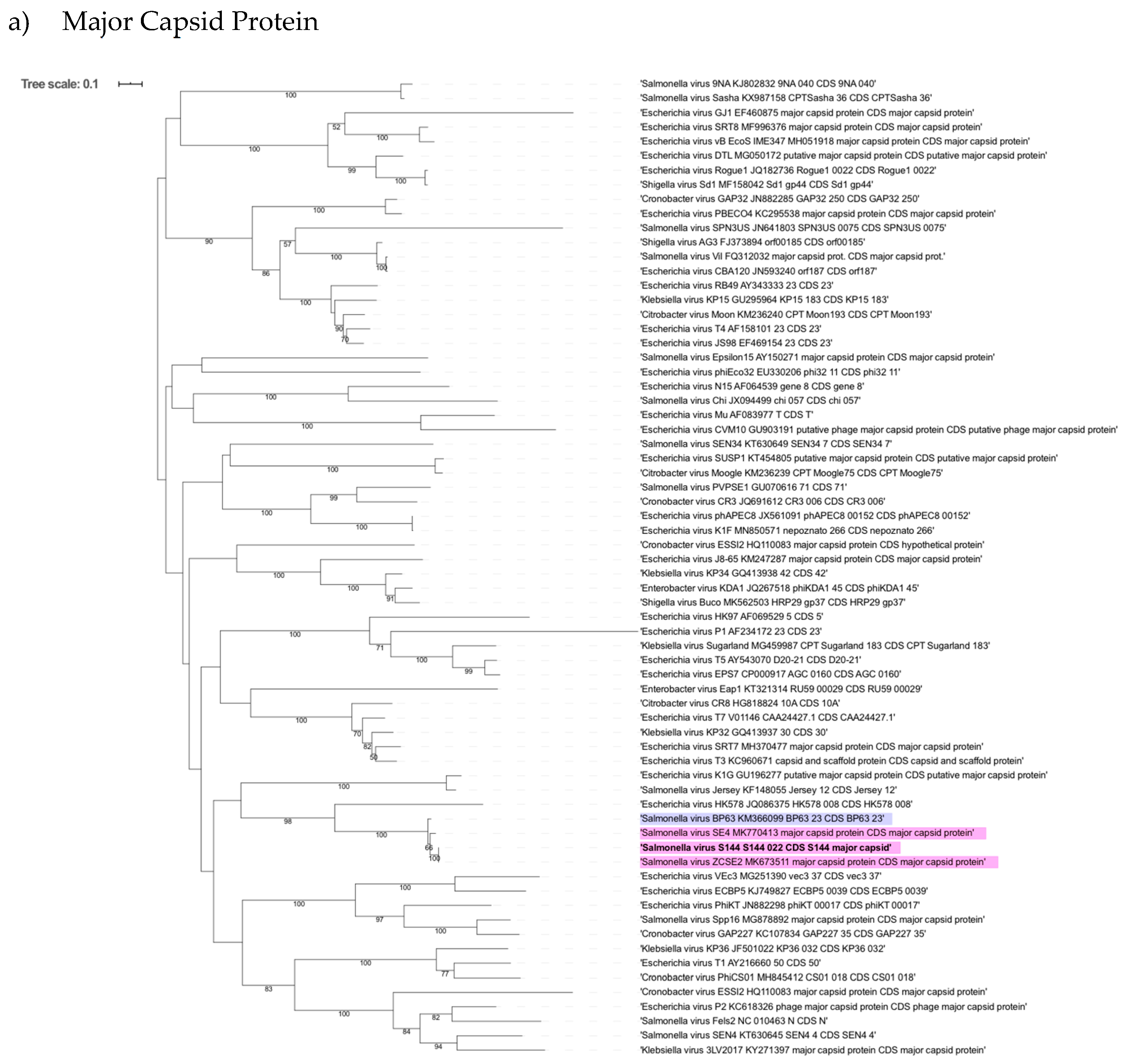
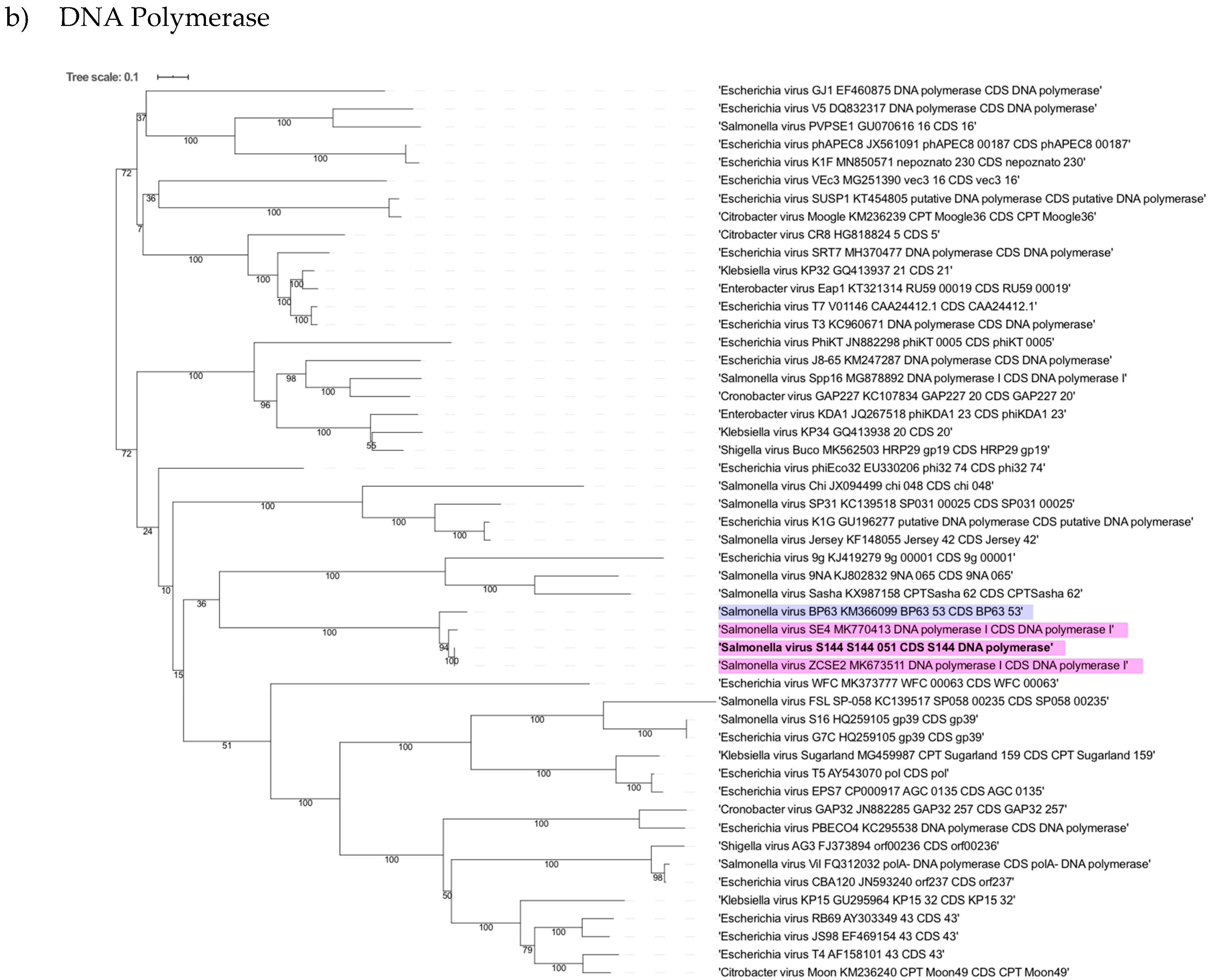
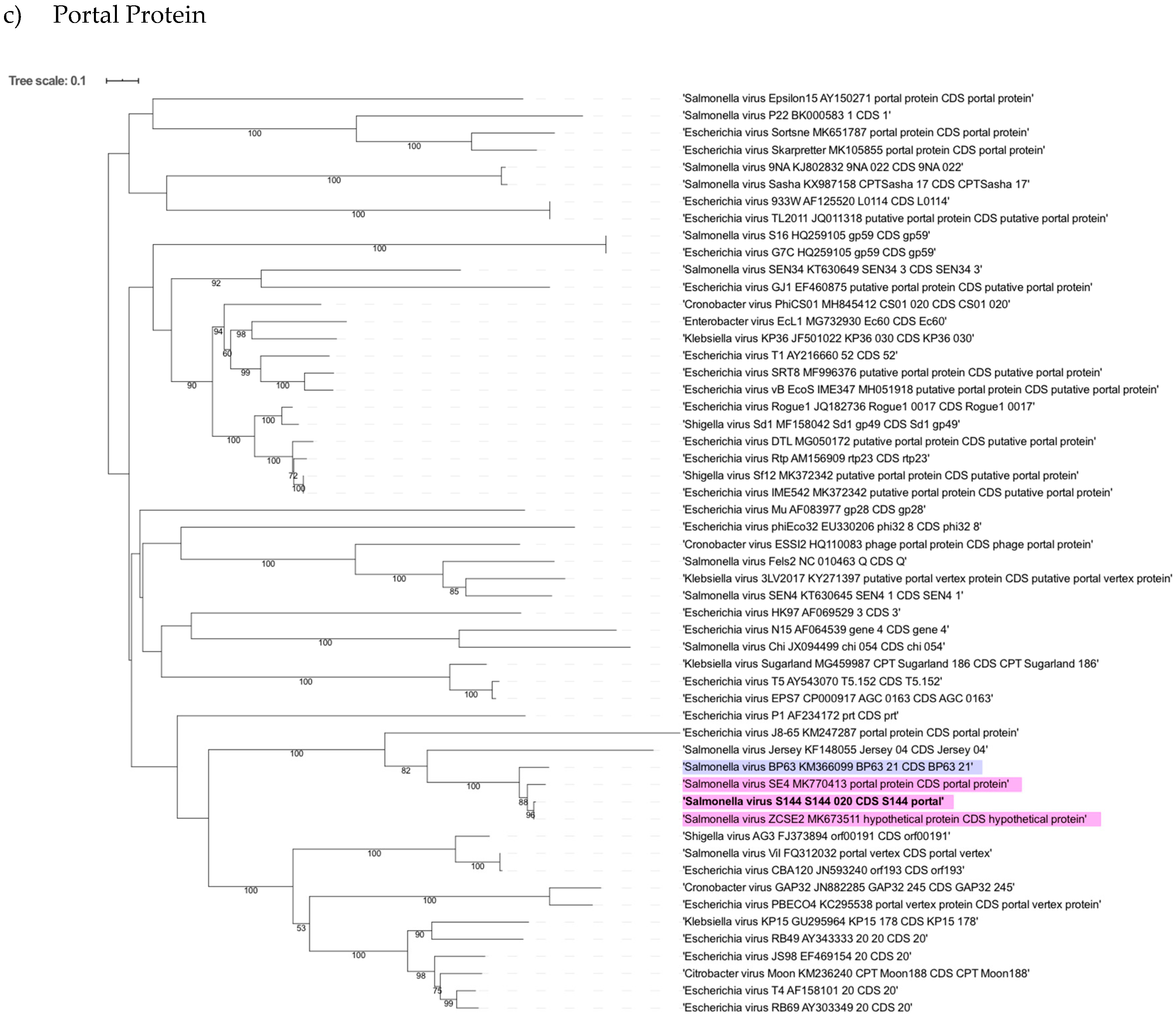
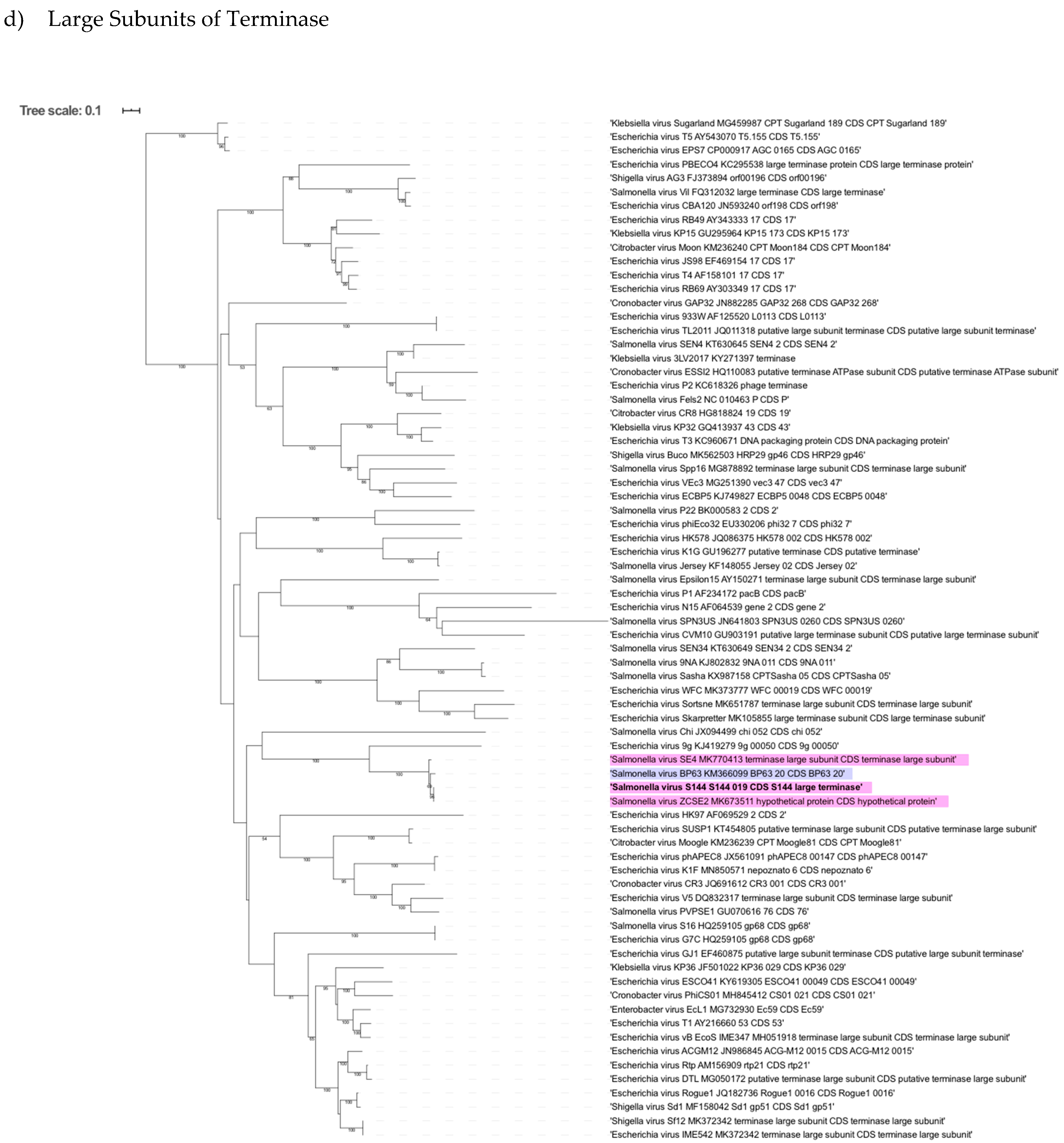
2.6. Phylogenetic Analysis Shows That S144 Is a Member of the Loughboroughvirus Genus
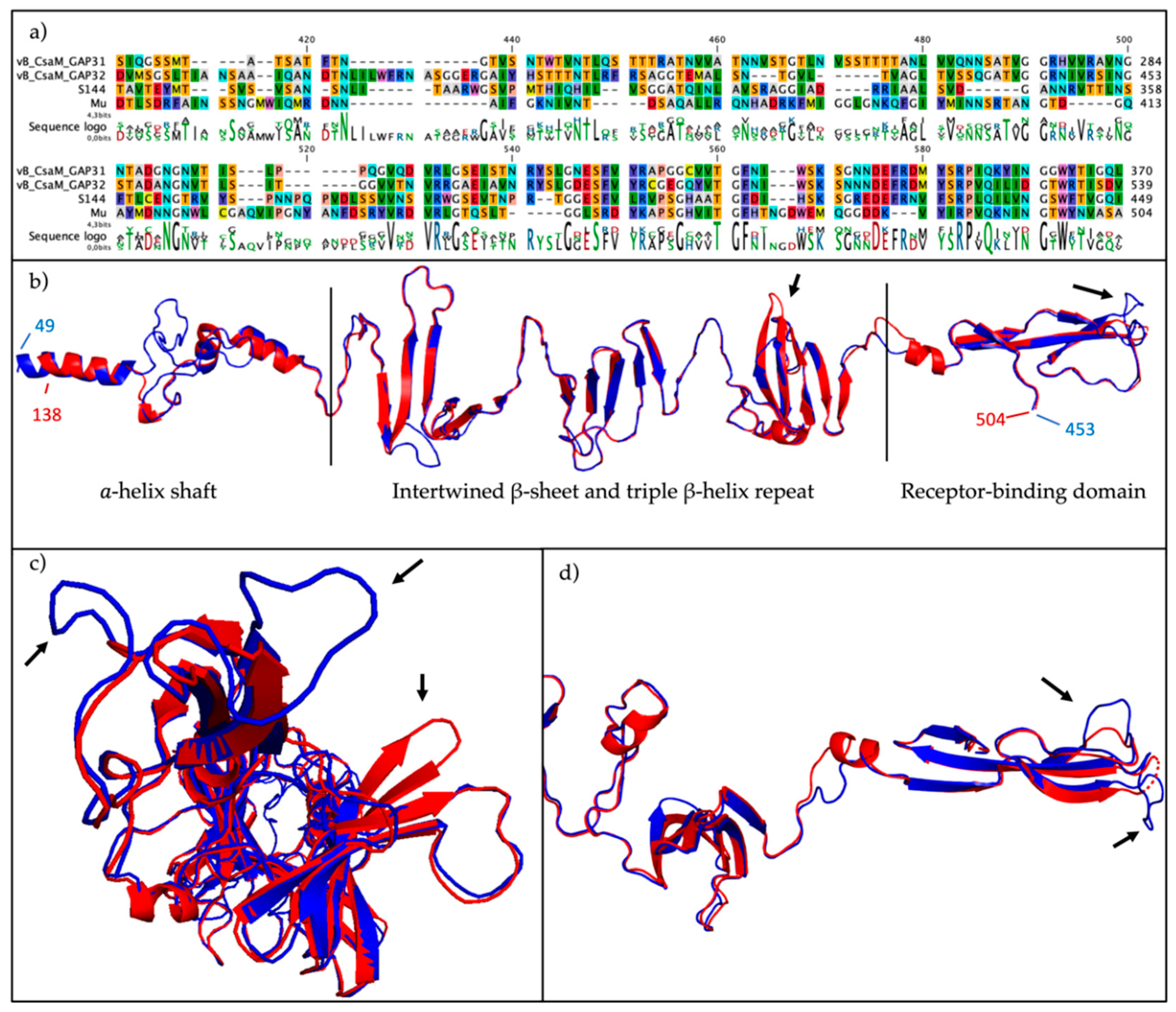
2.7. The ´Salusvirinae’ Phages Are Highly Similar Except for the Putative Receptor Binding Protein
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Phage Propagation and Plaque Assay
4.3. Transmission Electron Micrographs
4.4. Identification of the Receptor
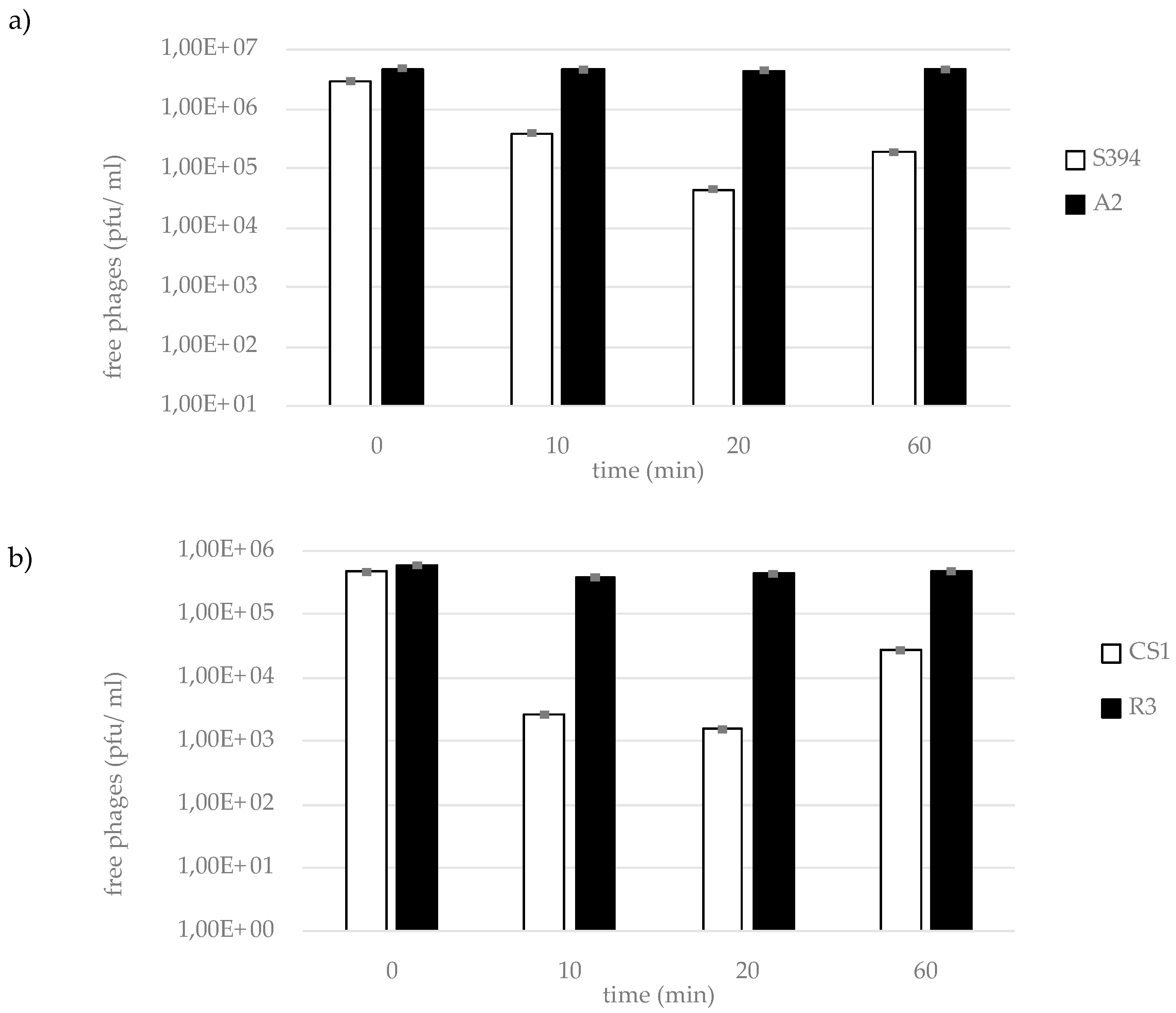
4.5. Adsorption Assay
4.6. Phage DNA Extraction and Sequencing
4.7. Genome Assembly and Bioinformatic Analysis
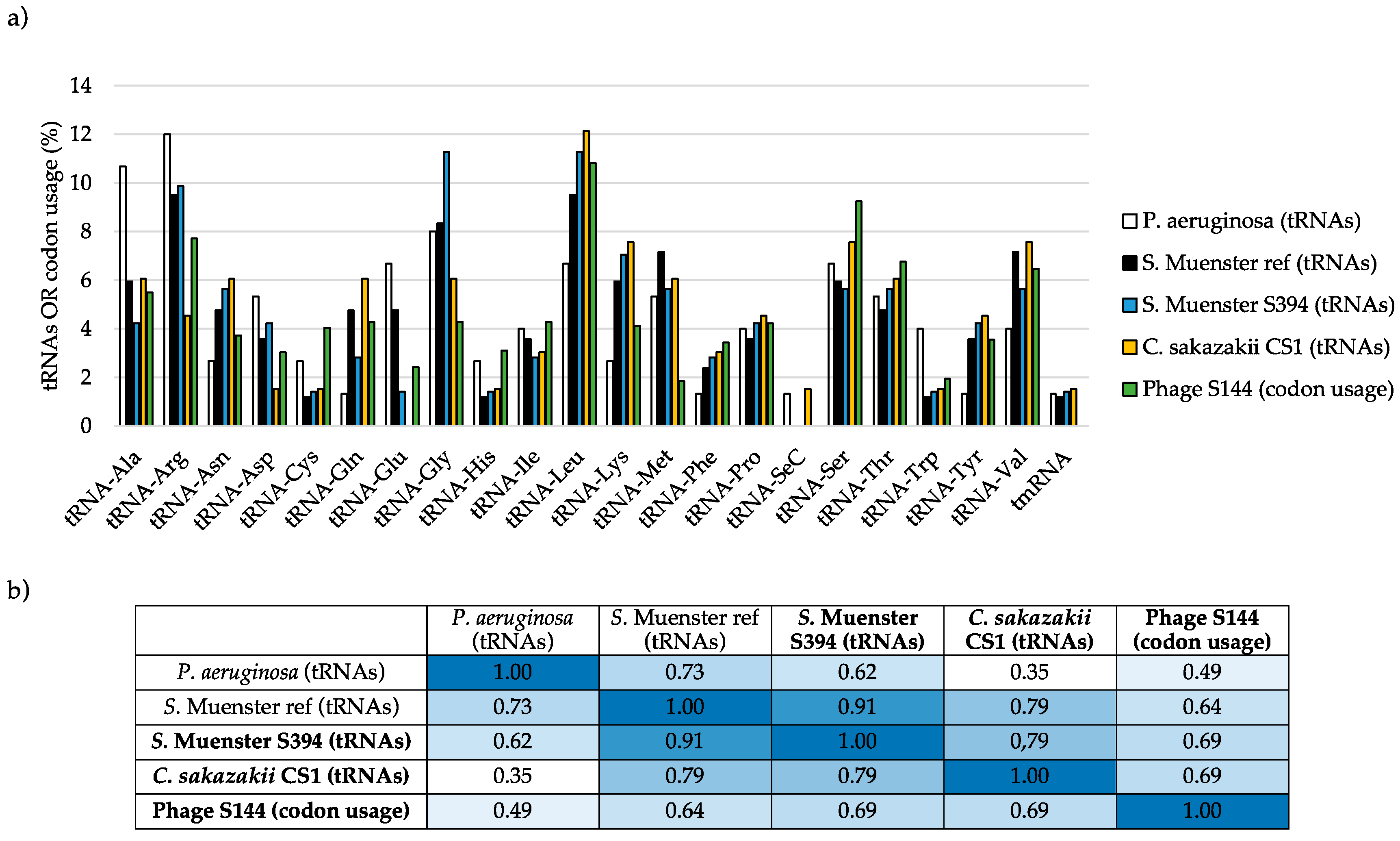
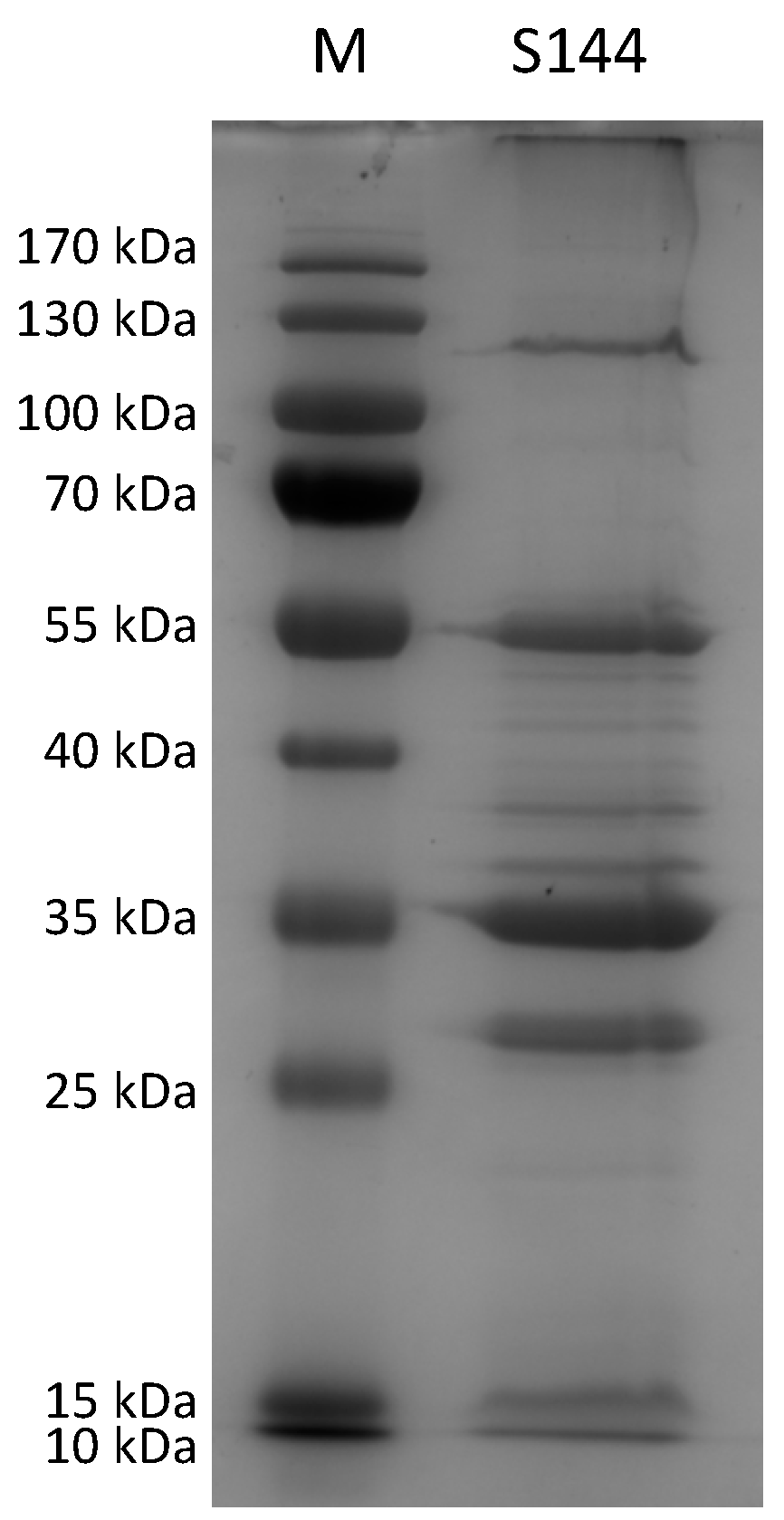
4.8. Proteome Analysis Using ESI-MS/MS
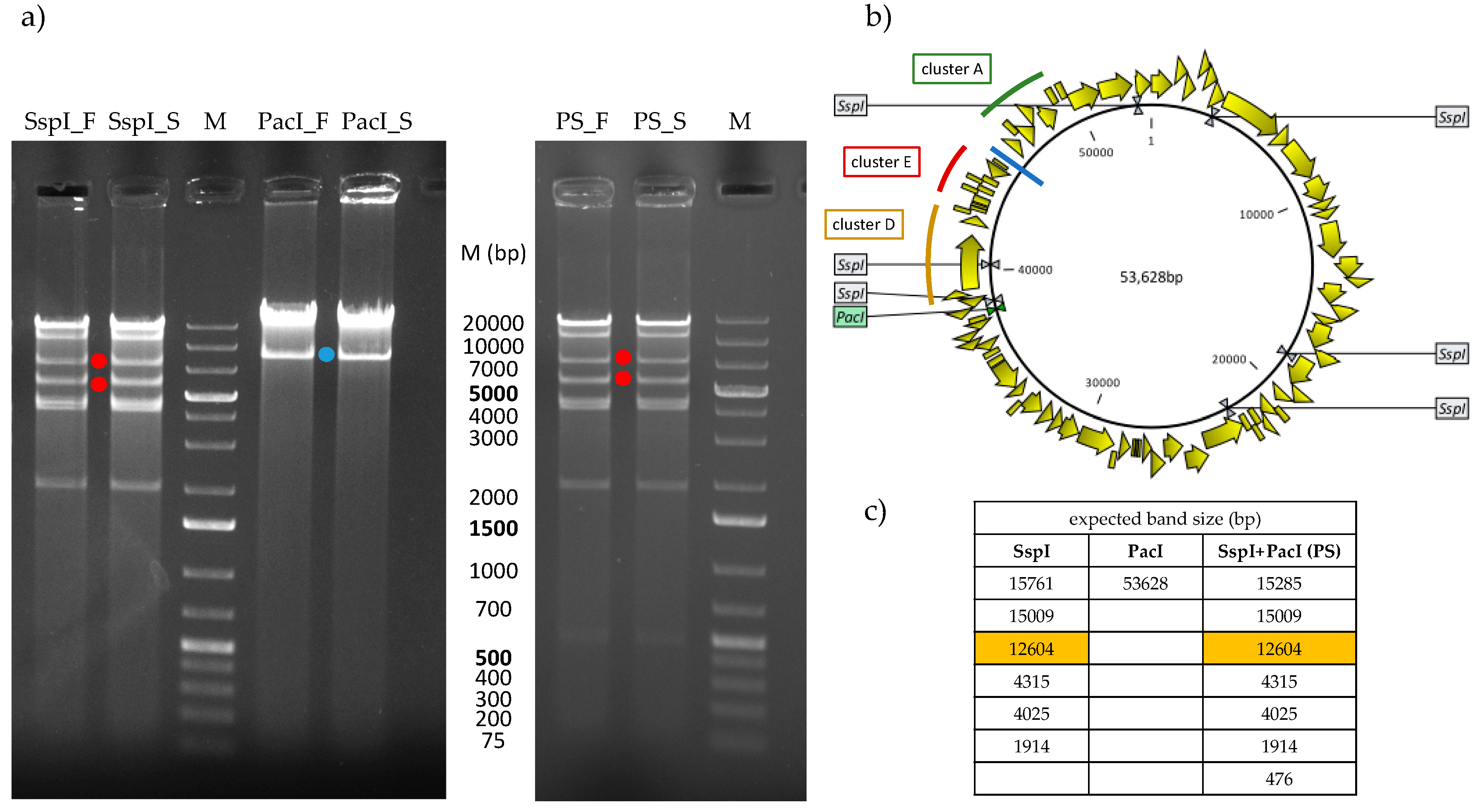
4.9. Restriction Enzyme Analysis
4.10. Taxonomy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species or serovar | KU ID | Serotype | Origin | Reference |
|---|---|---|---|---|---|
| Salmonella | Derby | EGS1 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS33 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS50 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS52 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS55 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS84 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS87 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS90 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS112 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS114 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS118 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS141 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS142 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS146 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS173 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS200 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS225 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS252 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS256 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS284 | O:4 | JEO | host range [6] |
| Salmonella | Derby | EGS310 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS28 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS38 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS42 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS43 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS69 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS73 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS96 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS121 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS126 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS136 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS152 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS191 | O:4 | JEO | host range [6] |
| Salmonella | Typhimurium | EGS304 | O:4 | JEO | host range [6] |
| Salmonella | Dublin | EGS24 | O:9 | JEO | host range [6] |
| Salmonella | Dublin | EGS40 | O:9 | JEO | host range [6] |
| Salmonella | Dublin | EGS244 | O:9 | JEO | host range [6] |
| Salmonella | Dublin | EGS306 | O:9 | JEO | host range [6] |
| Salmonella | Enteritidis | EGS45 | O:9 | JEO | host range [6] |
| Salmonella | Enteritidis | EGS47 | O:9 | JEO | host range [6] |
| Salmonella | Enteritidis | EGS48 | O:9 | JEO | host range [6] |
| Salmonella | Infantis | EGS15 | O:7 | JEO | host range, isolation and propagation [6] |
| Salmonella | Infantis | EGS108 | O:7 | JEO | host range [6] |
| Salmonella | Infantis | EGS160 | O:7 | JEO | host range [6] |
| Salmonella | Infantis | EGS208 | O:7 | JEO | host range [6] |
| Salmonella | Rough | EGS63 | n:4 | JEO | host range [6] |
| Salmonella | Rough | EGS65 | n:4 | JEO | host range [6] |
| Salmonella | Rough | EGS68 | n:4 | JEO | host range [6] |
| Salmonella | Rough | EGS105 | n:4 | JEO | host range [6] |
| Salmonella | 4.12:i:- | EGS20 | O:4 | JEO | host range [6] |
| Salmonella | 4.12:i:- | EGS61 | O:4 | JEO | host range [6] |
| Salmonella | 4.12:i:- | EGS135 | O:4 | JEO | host range [6] |
| Salmonella | 4.12:i:- | EGS162 | O:4 | JEO | host range [6] |
| Salmonella | 4.12:i:- | EGS206 | O:4 | JEO | host range [6] |
| Salmonella | 4.12:i:- | EGS235 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS82 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS165 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS129 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS156 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS167 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS169 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS193 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS287 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS307 | O:4 | JEO | host range [6] |
| Salmonella | 4.5.12:i:- | EGS248 | O:4 | JEO | host range [6] |
| Salmonella | Goettingen | EGS30 | O:9 | JEO | host range [6] |
| Salmonella | Livingstone | EGS172 | O:7 | JEO | host range [6] |
| Salmonella | London | EGS183 | O:3,10 | JEO | host range [6] |
| Salmonella | Rissen | EGS203 | O:7 | JEO | host range [6] |
| Salmonella | Brandenburg | EGS300 | O:4 | JEO | host range [6] |
| Salmonella | Bradford | EGS319 | O:4 | JEO | host range [6] |
| Salmonella | Senftenberg | EGS381 | ND | JEO | host range (this work) |
| Salmonella | Adelaide | EGS382 | ND | JEO | host range (this work) |
| Salmonella | Weslaco | EGS383 | ND | JEO | host range (this work) |
| Salmonella | Montevideo | EGS384 | O:6,7, 14 | JEO | host range (this work) |
| Salmonella | Tanger | EGS385 | O: 1,13,22 | JEO | host range (this work) |
| Salmonella | Cerro | EGS386 | ND | JEO | host range (this work) |
| Salmonella | Basel | EGS387 | ND | JEO | host range (this work) |
| Salmonella | Anatum | EGS388 | O:3,{10}{15}{15,34} | JEO | host range (this work) |
| Salmonella | Eilbek | EGS389 | ND | JEO | host range (this work) |
| Salmonella | Worthington | EGS390 | ND | JEO | host range (this work) |
| Salmonella | Onderstepoort | EGS391 | O: 1,6,14,(25) | JEO | host range (this work) |
| Salmonella | Deversoir | EGS392 | O: 45 | JEO | host range (this work) |
| Salmonella | Telaviv | EGS393 | O:28ab | JEO | host range (this work) |
| Salmonella | Muenster | EGS394 | O: 6,7 | JEO | host range and propagation (this work) |
| Salmonella | Aberdeen | EGS395 | O:11 | JEO | host range (this work) |
| Salmonella | Inverness | EGS396 | O:38 | JEO | host range (this work) |
| Salmonella | Bergen | EGS397 | ND | JEO | host range (this work) |
| Salmonella | Ruiru | EGS398 | ND | JEO | host range (this work) |
| Salmonella | Gaminara | EGS399 | O:16 | JEO | host range (this work) |
| Salmonella | Paratyphi B var. Java | EGS427 | ND | JEO | host range (this work) |
| Salmonella | Muenster | EGS428 | ND | JEO | host range (this work) |
| Escherichia | coli | ECOR1 | O144:H4 | [73] | host range (this work) |
| Escherichia | coli | ECOR2 | O48:H32 | [73] | host range (this work) |
| Escherichia | coli | ECOR3 | O1:H32 | [73] | host range (this work) |
| Escherichia | coli | ECOR4 | OR:H? | [73] | host range (this work) |
| Escherichia | coli | ECOR5 | O?:H6 | [73] | host range (this work) |
| Escherichia | coli | ECOR6 | O173:H? | [73] | host range (this work) |
| Escherichia | coli | ECOR7 | O8:H45 | [73] | host range (this work) |
| Escherichia | coli | ECOR8 | O86:H2 | [73] | host range (this work) |
| Escherichia | coli | ECOR9 | O167:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR10 | O6:H10 | [73] | host range (this work) |
| Escherichia | coli | ECOR11 | O10:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR12 | O?:H32 | [73] | host range (this work) |
| Escherichia | coli | ECOR13 | OR:H25 | [73] | host range (this work) |
| Escherichia | coli | ECOR14 | O71:H4 | [73] | host range (this work) |
| Escherichia | coli | ECOR15 | O25:H30 | [73] | host range (this work) |
| Escherichia | coli | ECOR16 | O9:H10 | [73] | host range (this work) |
| Escherichia | coli | ECOR17 | O29:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR18 | O?:H11 | [73] | host range (this work) |
| Escherichia | coli | ECOR19 | O89:H? | [73] | host range (this work) |
| Escherichia | coli | ECOR20 | O121:H11 | [73] | host range (this work) |
| Escherichia | coli | ECOR21 | O121:H11 | [73] | host range (this work) |
| Escherichia | coli | ECOR22 | O150:H28 | [73] | host range (this work) |
| Escherichia | coli | ECOR23 | O25.H1 | [73] | host range (this work) |
| Escherichia | coli | ECOR24 | O15:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR25 | O127:H40 | [73] | host range (this work) |
| Escherichia | coli | ECOR26 | O104:H21 | [73] | host range (this work) |
| Escherichia | coli | ECOR27 | O104:H21 | [73] | host range (this work) |
| Escherichia | coli | ECOR28 | O104:H21 | [73] | host range (this work) |
| Escherichia | coli | ECOR29 | O150:H21 | [73] | host range (this work) |
| Escherichia | coli | ECOR30 | O113:H21 | [73] | host range (this work) |
| Escherichia | coli | ECOR31 | O79:H25 | [73] | host range (this work) |
| Escherichia | coli | ECOR32 | O25:H1 | [73] | host range (this work) |
| Escherichia | coli | ECOR33 | O7:H21 | [73] | host range (this work) |
| Escherichia | coli | ECOR34 | O88:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR35 | O1:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR36 | O1:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR37 | O55:H7 | [73] | host range (this work) |
| Escherichia | coli | ECOR38 | O7:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR39 | O7:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR40 | O7:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR41 | O7:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR42 | O87:H26 | [73] | host range (this work) |
| Escherichia | coli | ECOR43 | O?:H18 | [73] | host range (this work) |
| Escherichia | coli | ECOR44 | O17:H34 | [73] | host range (this work) |
| Escherichia | coli | ECOR45 | O?:H2 | [73] | host range (this work) |
| Escherichia | coli | ECOR46 | O1:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR47 | O17:H18 | [73] | host range (this work) |
| Escherichia | coli | ECOR48 | O23:H15 | [73] | host range (this work) |
| Escherichia | coli | ECOR49 | O2:H4 | [73] | host range (this work) |
| Escherichia | coli | ECOR50 | O2:H4 | [73] | host range (this work) |
| Escherichia | coli | ECOR51 | O25:H1 | [73] | host range (this work) |
| Escherichia | coli | ECOR52 | O25:H1 | [73] | host range (this work) |
| Escherichia | coli | ECOR53 | O4:H5 | [73] | host range (this work) |
| Escherichia | coli | ECOR54 | O25:H1 | [73] | host range (this work) |
| Escherichia | coli | ECOR55 | O25:H1 | [73] | host range (this work) |
| Escherichia | coli | ECOR56 | O6:H10 | [73] | host range (this work) |
| Escherichia | coli | ECOR57 | O2:H1 | [73] | host range (this work) |
| Escherichia | coli | ECOR58 | O112:H8 | [73] | host range (this work) |
| Escherichia | coli | ECOR59 | O2:H4 | [73] | host range (this work) |
| Escherichia | coli | ECOR60 | O4:H5 | [73] | host range (this work) |
| Escherichia | coli | ECOR61 | O2:H4 | [73] | host range (this work) |
| Escherichia | coli | ECOR62 | O2:H4 | [73] | host range (this work) |
| Escherichia | coli | ECOR63 | OR:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR64 | O75:H- | [73] | host range (this work) |
| Escherichia | coli | ECOR65 | O8:H10 | [73] | host range (this work) |
| Escherichia | coli | ECOR66 | O4:H40 | [73] | host range (this work) |
| Escherichia | coli | ECOR67 | O141:H49 | [73] | host range (this work) |
| Escherichia | coli | ECOR68 | O25:H21 | [73] | host range (this work) |
| Escherichia | coli | ECOR69 | O86:H10 | [73] | host range (this work) |
| Escherichia | coli | ECOR70 | O78: NM | [73] | host range (this work) |
| Escherichia | coli | ECOR71 | O78:NM | [73] | host range (this work) |
| Escherichia | coli | ECOR72 | O144:H8 | [73] | host range (this work) |
| Escherichia | coli | NCTC12900 | O157:H7 | JEO | host range (this work) |
| Escherichia | coli | ATCC35150 | O157:H7 | JEO | host range (this work) |
| Escherichia | coli | ATCC43888 | O157:H7 | JEO | host range (this work) |
| Escherichia | coli | ATCC43895 | O157:H7 | JEO | host range (this work) |
| Escherichia | coli | JEO426 | ND | JEO | host range (this work) |
| Klebsiella | pneumonia | EGS400 | ND | JEO | host range (this work) |
| Klebsiella | pneumonia | EGS401 | ND | JEO | host range (this work) |
| Klebsiella | oxytoca | EGS402 | ND | JEO | host range (this work) |
| Enterobacter | aerogenes | EGS403 | ND | JEO | host range (this work) |
| Enterobacter | asburiae | EGS404 | ND | JEO | host range (this work) |
| Enterobacter | taylorae | EGS405 | ND | JEO | host range (this work) |
| Enterobacter | cloacae | EGS407 | ND | JEO | host range (this work) |
| Providencia | stuarti | EGS408 | ND | JEO | host range (this work) |
| Citrobacter | freundii | EGS409 | ND | JEO | host range (this work) |
| Citrobacter | koseri | EGS410 | ND | JEO | host range (this work) |
| Yersinia | enterocolitica | EGS411 | ND | JEO | host range (this work) |
| Yersinia | enterocolitica | EGS412 | ND | JEO | host range (this work) |
| Yersinia | enterocolitica | EGS413 | ND | JEO | host range (this work) |
| Yersinia | ruckeri | EGS414 | ND | JEO | host range (this work) |
| Yersinia | intermedia | EGS415 | ND | JEO | host range (this work) |
| Yersinia | frederiksenii | EGS416 | ND | JEO | host range (this work) |
| Yersinia | kristensenii | EGS417 | ND | JEO | host range (this work) |
| Serratia | marcescens | EGS418 | ND | JEO | host range (this work) |
| Erwinia | herbicola | EGS419 | ND | JEO | host range (this work) |
| Shigella | sonnei | EGS420 | ND | JEO | host range (this work) |
| Acinetobacter | calcoaceticus | EGS421 | ND | JEO | host range (this work) |
| Acinetobacter | baumannii | EGS422 | ND | JEO | host range (this work) |
| Acinetobacter | haemolyticus | EGS423 | ND | JEO | host range (this work) |
| Acinetobacter | junii | EGS424 | ND | JEO | host range (this work) |
| Acinetobacter | johnsonii | EGS425 | ND | JEO | host range (this work) |
| Proteus | mirabilis | EGS429 | ND | JEO | host range (this work) |
| Proteus | vulgaris | EGS430 | ND | JEO | host range (this work) |
| Cronobacter | sakazakii | EGS406 | ND | JEO | host range (this work) |
| Cronobacter | sakazakii | CS1 | ND | Arla | host range and propagation (this work) |
| Cronobacter | sakazakii | CS2 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS3 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS4 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS5 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS7 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS8 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS9 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS10 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS11 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS12 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS13 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS14 | ND | Arla | host range (this work) |
| Cronobacter | sakazakii | CS15 | ND | Arla | host range (this work) |
| Genus | Species | KU ID | Serotype | Log (pfu/mL) | Use |
|---|---|---|---|---|---|
| Salmonella | Derby | EGS1 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS33 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS50 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS52 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS55 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS84 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS87 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS90 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS112 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS114 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS118 | O:4 | 6 | [6] |
| Salmonella | Derby | EGS141 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS142 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS146 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS173 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS200 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS225 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS252 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS256 | O:4 | 0 | [6] |
| Salmonella | Derby | EGS284 | O:4 | 5 | [6] |
| Salmonella | Derby | EGS310 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS28 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS38 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS42 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS43 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS69 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS73 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS96 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS121 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS126 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS136 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS152 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS191 | O:4 | 0 | [6] |
| Salmonella | Typhimurium | EGS304 | O:4 | 0 | [6] |
| Salmonella | Dublin | EGS24 | O:9 | 0 | [6] |
| Salmonella | Dublin | EGS40 | O:9 | 0 | [6] |
| Salmonella | Dublin | EGS244 | O:9 | 0 | [6] |
| Salmonella | Dublin | EGS306 | O:9 | 0 | [6] |
| Salmonella | Enteritidis | EGS45 | O:9 | 7 | [6] |
| Salmonella | Enteritidis | EGS47 | O:9 | 10 | [6] |
| Salmonella | Enteritidis | EGS48 | O:9 | 0 | [6] |
| Salmonella | Infantis | EGS15 | O:7 | 9 | [6] |
| Salmonella | Infantis | EGS108 | O:7 | 10 | [6] |
| Salmonella | Infantis | EGS160 | O:7 | 10 | [6] |
| Salmonella | Infantis | EGS208 | O:7 | 0 | [6] |
| Salmonella | Rough | EGS63 | n:4 | 0 | [6] |
| Salmonella | Rough | EGS65 | n:4 | 0 | [6] |
| Salmonella | Rough | EGS68 | n:4 | 0 | [6] |
| Salmonella | Rough | EGS105 | n:4 | 0 | [6] |
| Salmonella | 4.12:i:- | EGS20 | O:4 | 0 | [6] |
| Salmonella | 4.12:i:- | EGS61 | O:4 | 0 | [6] |
| Salmonella | 4.12:i:- | EGS135 | O:4 | 0 | [6] |
| Salmonella | 4.12:i:- | EGS162 | O:4 | 0 | [6] |
| Salmonella | 4.12:i:- | EGS206 | O:4 | 0 | [6] |
| Salmonella | 4.12:i:- | EGS235 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS82 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS165 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS129 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS156 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS167 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS169 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS193 | O:4 | 0 | [6] |
| Salmonella | 4.5.12:i:- | EGS287 | O:4 | 2 | [6] |
| Salmonella | 4.5.12:i:- | EGS307 | O:4 | 0 | [6] |
| Salmonella | 4.[5].12:i:- | EGS248 | O:4 | 0 | [6] |
| Salmonella | Goettingen | EGS30 | O:9 | 7 | [6] |
| Salmonella | Livingstone | EGS172 | O:7 | 0 | [6] |
| Salmonella | London | EGS183 | O:3,10 | 10 | [6] |
| Salmonella | Rissen | EGS203 | O:7 | 0 | [6] |
| Salmonella | Brandenburg | EGS300 | O:4 | 0 | [6] |
| Salmonella | Bradford | EGS319 | O:4 | 0 | [6] |
| Salmonella | Senftenberg | EGS381 | ND | 0 | this manuscript |
| Salmonella | Adelaide | EGS382 | ND | 0 | this manuscript |
| Salmonella | Weslaco | EGS383 | ND | 0 | this manuscript |
| Salmonella | Montevideo | EGS384 | O:6,7, 14 | 0 | this manuscript |
| Salmonella | Tanger | EGS385 | O: 1,13,22 | 8 | this manuscript |
| Salmonella | Cerro | EGS386 | ND | 0 | this manuscript |
| Salmonella | Basel | EGS387 | ND | 0 | this manuscript |
| Salmonella | Anatum | EGS388 | O: 3,{10}{15}{15,34} | 0 | this manuscript |
| Salmonella | Eilbek | EGS389 | ND | 7 | this manuscript |
| Salmonella | Worthington | EGS390 | ND | 0 | this manuscript |
| Salmonella | Onderstepoort | EGS391 | O: 1,6,14,[25] | 8 | this manuscript |
| Salmonella | Deversoir | EGS392 | O: 45 | 8 | this manuscript |
| Salmonella | Telaviv | EGS393 | O:28ab | 8 | this manuscript |
| Salmonella | Choleraesuis | EGS394 | O: 6,7 | 8 | this manuscript |
| Salmonella | Aberdeen | EGS395 | O:11 | 7 | this manuscript |
| Salmonella | Inverness | EGS396 | O:38 | 6 | this manuscript |
| Salmonella | Bergen | EGS397 | ND | 0 | this manuscript |
| Salmonella | Ruiru | EGS398 | ND | 0 | this manuscript |
| Salmonella | Gaminara | EGS399 | O:16 | 7 | this manuscript |
| Salmonella | Paratyphi B var. Java | EGS427 | ND | 0 | this manuscript |
| Salmonella | Muenster | EGS428 | ND | 7 | this manuscript |
| Escherichia | coli | ECOR1 | O144:H4 | 0 | this manuscript |
| Escherichia | coli | ECOR2 | O48:H32 | 0 | this manuscript |
| Escherichia | coli | ECOR3 | O1:H32 | 0 | this manuscript |
| Escherichia | coli | ECOR4 | OR:H? | 0 | this manuscript |
| Escherichia | coli | ECOR5 | O?:H6 | 0 | this manuscript |
| Escherichia | coli | ECOR6 | O173:H? | 0 | this manuscript |
| Escherichia | coli | ECOR7 | O8:H45 | 0 | this manuscript |
| Escherichia | coli | ECOR8 | O86:H2 | 0 | this manuscript |
| Escherichia | coli | ECOR9 | O167:H- | 0 | this manuscript |
| Escherichia | coli | ECOR10 | O6:H10 | 0 | this manuscript |
| Escherichia | coli | ECOR11 | O10:H- | 0 | this manuscript |
| Escherichia | coli | ECOR12 | O?:H32 | 0 | this manuscript |
| Escherichia | coli | ECOR13 | OR:H25 | 0 | this manuscript |
| Escherichia | coli | ECOR14 | O71:H4 | 0 | this manuscript |
| Escherichia | coli | ECOR15 | O25:H30 | 0 | this manuscript |
| Escherichia | coli | ECOR16 | O9:H10 | 0 | this manuscript |
| Escherichia | coli | ECOR17 | O29:H- | 0 | this manuscript |
| Escherichia | coli | ECOR18 | O?:H11 | 0 | this manuscript |
| Escherichia | coli | ECOR19 | O89:H? | 0 | this manuscript |
| Escherichia | coli | ECOR20 | O121:H11 | 0 | this manuscript |
| Escherichia | coli | ECOR21 | O121:H11 | 0 | this manuscript |
| Escherichia | coli | ECOR22 | O150:H28 | 0 | this manuscript |
| Escherichia | coli | ECOR23 | O25.H1 | 0 | this manuscript |
| Escherichia | coli | ECOR24 | O15:H- | 0 | this manuscript |
| Escherichia | coli | ECOR25 | O127:H40 | 0 | this manuscript |
| Escherichia | coli | ECOR26 | O104:H21 | 0 | this manuscript |
| Escherichia | coli | ECOR27 | O104:H21 | 0 | this manuscript |
| Escherichia | coli | ECOR28 | O104:H21 | 0 | this manuscript |
| Escherichia | coli | ECOR29 | O150:H21 | 0 | this manuscript |
| Escherichia | coli | ECOR30 | O113:H21 | 0 | this manuscript |
| Escherichia | coli | ECOR31 | O79:H25 | 0 | this manuscript |
| Escherichia | coli | ECOR32 | O25:H1 | 0 | this manuscript |
| Escherichia | coli | ECOR33 | O7:H21 | 0 | this manuscript |
| Escherichia | coli | ECOR34 | O88:H- | 0 | this manuscript |
| Escherichia | coli | ECOR35 | O1:H- | 0 | this manuscript |
| Escherichia | coli | ECOR36 | O1:H- | 0 | this manuscript |
| Escherichia | coli | ECOR37 | O55:H7 | 0 | this manuscript |
| Escherichia | coli | ECOR38 | O7:H- | 0 | this manuscript |
| Escherichia | coli | ECOR39 | O7:H- | 0 | this manuscript |
| Escherichia | coli | ECOR40 | O7:H- | 0 | this manuscript |
| Escherichia | coli | ECOR41 | O7:H- | 0 | this manuscript |
| Escherichia | coli | ECOR42 | O87:H26 | 0 | this manuscript |
| Escherichia | coli | ECOR43 | O?:H18 | 0 | this manuscript |
| Escherichia | coli | ECOR44 | O17:H34 | 0 | this manuscript |
| Escherichia | coli | ECOR45 | O?:H2 | 0 | this manuscript |
| Escherichia | coli | ECOR46 | O1:H- | 0 | this manuscript |
| Escherichia | coli | ECOR47 | O17:H18 | 0 | this manuscript |
| Escherichia | coli | ECOR48 | O23:H15 | 0 | this manuscript |
| Escherichia | coli | ECOR49 | O2:H4 | 0 | this manuscript |
| Escherichia | coli | ECOR50 | O2:H4 | 0 | this manuscript |
| Escherichia | coli | ECOR51 | O25:H1 | 0 | this manuscript |
| Escherichia | coli | ECOR52 | O25:H1 | 0 | this manuscript |
| Escherichia | coli | ECOR53 | O4:H5 | 0 | this manuscript |
| Escherichia | coli | ECOR54 | O25:H1 | 0 | this manuscript |
| Escherichia | coli | ECOR55 | O25:H1 | 0 | this manuscript |
| Escherichia | coli | ECOR56 | O6:H10 | 0 | this manuscript |
| Escherichia | coli | ECOR57 | O2:H1 | 0 | this manuscript |
| Escherichia | coli | ECOR58 | O112:H8 | 0 | this manuscript |
| Escherichia | coli | ECOR59 | O2:H4 | 0 | this manuscript |
| Escherichia | coli | ECOR60 | O4:H5 | 0 | this manuscript |
| Escherichia | coli | ECOR61 | O2:H4 | 0 | this manuscript |
| Escherichia | coli | ECOR62 | O2:H4 | 0 | this manuscript |
| Escherichia | coli | ECOR63 | OR:H- | 0 | this manuscript |
| Escherichia | coli | ECOR64 | O75:H- | 0 | this manuscript |
| Escherichia | coli | ECOR65 | O8:H10 | 0 | this manuscript |
| Escherichia | coli | ECOR66 | O4:H40 | 0 | this manuscript |
| Escherichia | coli | ECOR67 | O141:H49 | 0 | this manuscript |
| Escherichia | coli | ECOR68 | O25:H21 | 0 | this manuscript |
| Escherichia | coli | ECOR69 | O86:H10 | 0 | this manuscript |
| Escherichia | coli | ECOR70 | O78: NM | 0 | this manuscript |
| Escherichia | coli | ECOR71 | O78:NM | 0 | this manuscript |
| Escherichia | coli | ECOR72 | O144:H8 | 0 | this manuscript |
| Escherichia | coli | NCTC12900 | O157:H7 | 0 | this manuscript |
| Escherichia | coli | ATCC35150 | O157:H7 | 0 | this manuscript |
| Escherichia | coli | ATCC43888 | O157:H7 | 0 | this manuscript |
| Escherichia | coli | ATCC43895 | O157:H7 | 0 | this manuscript |
| Escherichia | coli | EGS426 | ND | 0 | this manuscript |
| Klebsiella | pneumonia | EGS400 | ND | 0 | this manuscript |
| Klebsiella | pneumonia | EGS401 | ND | 0 | this manuscript |
| Klebsiella | oxytoca | EGS402 | ND | 0 | this manuscript |
| Enterobacter | aerogenes | EGS403 | ND | 0 | this manuscript |
| Enterobacter | asburiae | EGS404 | ND | 0 | this manuscript |
| Enterobacter | taylorae | EGS405 | ND | 0 | this manuscript |
| Enterobacter | cloacae | EGS407 | ND | 7 | this manuscript |
| Providencia | stuarti | EGS408 | ND | 0 | this manuscript |
| Citrobacter | freundii | EGS409 | ND | 0 | this manuscript |
| Citrobacter | koseri | EGS410 | ND | 0 | this manuscript |
| Yersinia | enterocolitica | EGS411 | ND | 0 | this manuscript |
| Yersinia | enterocolitica | EGS412 | ND | 0 | this manuscript |
| Yersinia | enterocolitica | EGS413 | ND | 0 | this manuscript |
| Yersinia | ruckeri | EGS414 | ND | 0 | this manuscript |
| Yersinia | intermedia | EGS415 | ND | 0 | this manuscript |
| Yersinia | frederiksenii | EGS416 | ND | 0 | this manuscript |
| Yersinia | kristensenii | EGS417 | ND | 0 | this manuscript |
| Serratia | marcescens | EGS418 | ND | 0 | this manuscript |
| Erwinia | herbicola | EGS419 | ND | 0 | this manuscript |
| Shigella | sonnei | EGS420 | ND | 0 | this manuscript |
| Acinetobacter | calcoaceticus | EGS421 | ND | 0 | this manuscript |
| Acinetobacter | baumannii | EGS422 | ND | 0 | this manuscript |
| Acinetobacter | haemolyticus | EGS423 | ND | 0 | this manuscript |
| Acinetobacter | junii | EGS424 | ND | 0 | this manuscript |
| Acinetobacter | johnsonii | EGS425 | ND | 0 | this manuscript |
| Proteus | mirabilis | EGS429 | ND | 0 | this manuscript |
| Proteus | vulgaris | EGS430 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | EGS406 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS1 | ND | 10 | this manuscript |
| Cronobacter | sakazakii | CS2 | ND | 9 | this manuscript |
| Cronobacter | sakazakii | CS3 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS4 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS5 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS7 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS8 | ND | 10 | this manuscript |
| Cronobacter | sakazakii | CS9 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS10 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS11 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS12 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS13 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS14 | ND | 0 | this manuscript |
| Cronobacter | sakazakii | CS15 | ND | 9 | this manuscript |


| Salmonella Contig | From | To | Lenght | Product RAST | peg | Annotation According to [23] |
|---|---|---|---|---|---|---|
| contig_8 | 1973 | 2907 | 135 | putative glycosyltransferase | ||
| 21,824 | 21,843 | 20 | Cyd operon protein YbgE | |||
| 48,887 | 50,327 | 1441 | hypothetical protein; put. phosphatase | |||
| 70,525 | 70,574 | 50 | Leucine rich repeat protein | |||
| 142,368 | 167,895 | 25,528 | whole prophage | |||
| 224,361 | 224,381 | 21 | DNA translocase FtsK | |||
| 240,568 | 240,592 | 25 | putative secreted protein | |||
| 283,864 | 289,665 | 5802 | six phage proteins and three hypothetical proteins | |||
| 290,855 | 293,458 | 2604 | SopE, invertase and hypothetical protein | |||
| contig_16 | 1 | 8072 | 8072 | Alpha-fimbriae chaperone protein; Alpha-fimbriae major subunit; Alpha-fimbriae usher protein; Alpha-fimbriae tip adhesin; Transcriptional regulator; TioA protein; hypothetical protein | ||
| 114,052 | 114,144 | 93 | hypothetical protein | |||
| contig_22 | 1 | 2821 | 2821 | Lysozyme family,: hypothetical protein, phage tail fiber-like; Cytolethal distending toxin subunit B, DNase I-like; transposase | ||
| 33,364 | 33,900 | 537 | hypothetical protein | |||
| 44,450 | 53,815 | 9366 | Putative oxidoreductase YdjL; Uncharacterized MFS-type transporter YdjK; Hypothetical zinc-type alcohol dehydrogenase-like protein YdjJ; Putative aldolase YdjI; Uncharacterized sugar kinase YdjH; Uncharacterized oxidoreductase YdjG; Uncharacterized transcriptional regulator YdjF, DeoR family; Uncharacterized transcriptional regulator YdjF, DeoR family; Uncharacterized MFS-type transporter YdjE | |||
| 219,027 | 222,993 | 3967 | Cystathionine beta-lyase MalY Maltose regulon modulator; PTS system, maltose and glucose-specific IIC component / PTS system, maltose and glucose-specific IIB component; Maltose regulon regulatory protein MalI | |||
| 233,873 | 234,200 | 328 | Outer membrane porin OmpN | |||
| 311,545 | 314,813 | 3269 | hypothetical protein; transcription regulator LysR; putative oxidoreductase | |||
| 326,896 | 329,224 | 2329 | two hypotheetical proteins, hemolysin | |||
| contig_21 | 24,376 | 24,427 | 52 | Uncharacterized metal-dependent hydrolase STM4445 | ||
| 80,677 | 81,930 | 1254 | Uncharacterized protein YjfY; probable integral membrane protein Cj0014c | |||
| 94,979 | 95,102 | 124 | hypothetical protein | |||
| 145,365 | 156,714 | 11,350 | SdiA-regulated putative outer membrane protein SrgB; hypothetical proteins; K88 minor fimbrial subunit faeH precursor; Putative fimbrial chaperone protein; major pilu subunit operon regulatory protein PapB; transposase | |||
| contig_13 | 50,450 | 56,299 | 5850 | PTS system, mannitol-specific cryptic IIA component; PTS system, mannitol-specific IIC component / PTS system, mannitol-specific IIB component; Uncharacterized protein YggP; Fructose-1,6-bisphosphatase, GlpX type; Fumarate hydratase FumE; Uncharacterized sugar kinase YggC; Uncharacterized sugar kinase YggC | ||
| 103,105 | 104,327 | 1223 | hypothetical protein | |||
| contig_23 | 63,088 | 63,268 | 181 | Trp transport protein | ||
| 71,589 | 71,716 | 128 | ribosome binding factor A | |||
| 78,672 | 78,833 | 162 | hypothetical protein | |||
| 117,795 | 118,155 | 361 | putative uncharacterized protein | |||
| 137,502 | 137,555 | 54 | oxaloacetate decarboxylase | |||
| contig_9 | 4796 | 8158 | 3363 | DNA-cytosine methyltransferase; hypothetical protein | ||
| 28,620 | 29,048 | 429 | uncharacterized protein | |||
| 31,367 | 34,321 | 2955 | uncharacterized protein; Thr efflux proteins; Transcriptional regulator, AraC family | |||
| 73,912 | 74,614 | 703 | hypothetical protein | |||
| 161,073 | 161,244 | 172 | non-specific ribonucleoside hydrolase | |||
| contig_26 | 88,225 | 88,395 | 171 | uncharacterized protein YacH | ||
| 107,706 | 108,146 | 441 | hypothetical protein | |||
| 112,522 | 112,618 | 97 | Putative PTS system IIA component YadI | |||
| 116,430 | 124,419 | 7990 | Uncharacterized fimbrial-like protein YadC; Uncharacterized fimbrial-like protein YadK; Uncharacterized fimbrial-like protein YadL; Uncharacterized fimbrial-like protein YadM; Outer membrane usher protein HtrE; Fimbria adhesin EcpD; Uncharacterized fimbrial-like protein YadN | |||
| 150,755 | 154,023 | 3269 | hypothetical protein | |||
| 170,664 | 170,916 | 253 | Outer membrane protein assembly factor YaeT | |||
| contig_3 | 12,533 | 12,618 | 86 | hypothetical protein | ||
| 41,089 | 41,277 | 189 | hypothetical protein | |||
| 224,967 | 227,978 | 3012 | putative surface-exposed virulence protein | |||
| contig_4 | 59,736 | 71,228 | 11493 | Retron-type RNA-directed DNA polymerase; Zinc binding domain / DNA primase; Phage immunity repressor protein; Phage capsid and scaffold protein; hypothetical protein; integrase | ||
| contig_17 | 137,188 | 138,244 | 1057 | hypothetical protein | ||
| contig_6 | 63,478 | 63,745 | 268 | Chemotaxis protein CheV | ||
| 258,121 | 264,643 | 6523 | Uncharacterized lipoprotein YfgH; Uncharacterized protein YfgI; hypothetical proteins | |||
| 273,517 | 274,791 | 1275 | AIDA autotransporter like protein | |||
| contig_5 | 1 | 10,028 | 10,028 | CRISPR associated proteins; hypothetical proteins | ||
| 57,618 | 57,917 | 300 | Type III secretion host injection protein (YopB); Cell invasion protein Salmonella Invasion protein D | |||
| 171,596 | 173,193 | 1598 | hypothetical protein | |||
| 181,722 | 185,551 | 3830 | hypothetical proteins | |||
| contig_11 | 33,872 | 34,492 | 621 | lipase | ||
| contig_20+39 | 5049 | 5708 | 660 | O antigen biosynthesis rhamnosyltransferase RfbN | 3247 | wbaN, Rhamnosyl transferase |
| 5712 | 6797 | 1986 | Glycosyltransfearse | 3248 | wbaO, Mannosyl transferase | |
| 6790 | 7968 | 1179 | hypothetical protein | 3249 | wzy_D2,E, O-antigen polymerase | |
| 8157 | 9485 | 1329 | FIG01200878: hypothetical protein | 3250 | wzx, O-antigen flippase | |
| 9835 | 10,365 | 531 | dTDP-4-dehydrorhamnose 3,5-epimerase (EC 5.1.3.13) | 3251 | rfbC (rmlC), dTDP-4-dehydrorhamnose 3,5-epimerase | |
| 76,420 | 77,442 | 1023 | Uncharacterized fimbrial-like protein YehA | |||
| 77,458 | 79,944 | 2487 | Outer membrane usher protein YehB | |||
| 79,973 | 80,653 | 681 | Probable fimbrial chaperone YehC | |||
| 80,715 | 81,248 | 534 | Uncharacterized fimbrial-like protein YehD |
| Cronobacter Contig | From | To | Lenght | Product RAST | peg | Annotation According to [22] |
|---|---|---|---|---|---|---|
| contig_4 | 62,745 | 62,747 | 2 | Glycerate kinase (EC 2.7.1.31) | ||
| 365,022 | 365,071 | 47 | hypothetical protein | |||
| 458,333 | 458,453 | 121 | hypothetical protein | |||
| contig_1 | 6458 | 6463 | 6 | VgrG protein | ||
| 7278 | 8409 | 1132 | hypothetical protein-non coding-hypothetical protein | |||
| 8514 | 8723 | 210 | hypothetical protein | |||
| 9886 | 9899 | 14 | hypothetical protein | |||
| 10,096 | 10,191 | 96 | hypothetical protein | |||
| 10,213 | 10,223 | 11 | hypothetical protein | |||
| 105,767 | 105,932 | 166 | hypothetical protein | |||
| 105,999 | 106,098 | 100 | hypothetical protein | |||
| 106,147 | 106,170 | 24 | hypothetical protein | |||
| 291,675 | 291,697 | 23 | hypothetical protein | |||
| 299,568 | 299,652 | 85 | EF hand domain protein | |||
| 299,670 | 299,754 | 85 | EF hand domain protein | |||
| 299,790 | 299,826 | 37 | hypothetical protein | |||
| 668,809 | 668,870 | 62 | hypothetical protein | |||
| 680,943 | 681,141 | 199 | hypothetical protein | |||
| 681,144 | 681,614 | 471 | hypothetical protein | |||
| 747,237 | 747,243 | 7 | hypothetical protein | |||
| 747,498 | 747,987 | 490 | hypothetical protein | 744 | wehL, glycosyltransferase | |
| 748,471 | 751,728 | 3257 | hypothetical protein | 745 | wehK, glycosyltransferase | |
| hypothetical protein | 746 | wehJ, glycosyl transferase | ||||
| hypothetical protein | 747 | wzy, O-antigen polymerase | ||||
| predicted glycosyl transferase | 748 | wdaN, glycosyl transferase | ||||
| Glycosyl transferase, group 2 family protein | 749 | |||||
| 751,992 | 753,948 | 1956 | Glycosyl transferase, group 2 family protein | 749 | ||
| Membrane protein involved in the export of O- antigen, teichoic acid lipoteichoic acids | 750 | wzx, O antigen flippase | ||||
| dTDP-4-dehydrorhamnose 3,5-epimerase (EC 5.1.3.13) | 751 | rmlC, dTDP-6-deoxy-D-glucose-3,5 epimerase | ||||
| contig_8 | 188 | 299 | 112 | hypothetical protein | ||
| 373 | 391 | 19 | hypothetical protein | |||
| 1696 | 1850 | 155 | Retron-type RNA-directed DNA polymerase | |||
| 1908 | 2235 | 328 | Retron-type RNA-directed DNA polymerase | |||
| 12,403 | 12,456 | 54 | hypothetical protein | |||
| 65,487 | 65,654 | 168 | hypothetical protein | |||
| 193,244 | 193,398 | 155 | hypothetical protein | |||
| 434,360 | 434,493 | 134 | hypothetical protein | |||
| 519,306 | 519,588 | 283 | hypothetical protein | |||
| 520,419 | 520,724 | 306 | hypothetical protein | |||
| 520,975 | 521,362 | 388 | hypothetical protein | |||
| contig_2 | 48,377 | 49,964 | 1588 | hypothetical protein | ||
| 50,769 | 51,114 | 346 | hypothetical protein | |||
| 140,889 | 140,939 | 51 | Exopolysaccharide phosphotransferase SCO6023 (EC 2.7.-.-) | 1735 | ||
| 141,013 | 141,080 | 68 | Exopolysaccharide phosphotransferase SCO6023 (EC 2.7.-.-) | 1735 | ||
| 141,860 | 142,540 | 681 | Beta-1,3-glucosyltransferase | 1736 | ||
| 143,653 | 143,805 | 153 | hypothetical protein | 1737 | ||
| 144,395 | 145,226 | 832 | Capsular polysaccharide export system protein KpsS | 1738 | ||
| contig_9 | 108,824 | 109,127 | 304 | hypothetical protein | ||
| contig_11 | 1490 | 1496 | 7 | hypothetical protein | ||
| 2890 | 2897 | 8 | hypothetical protein | |||
| 2909 | 2985 | 77 | hypothetical protein | |||
| 9254 | 9439 | 186 | acyltransferase family protein | |||
| contig_3 | 41,695 | 41,885 | 191 | hypothetical protein | ||
| 191,953 | 191,971 | 19 | hypothetical protein | |||
| contig_7 | 41,705 | 41,715 | 11 | Transcriptional regulator, LysR family | ||
| 41,759 | 41,804 | 46 | Transcriptional regulator, LysR family | |||
| 48,859 | 48,914 | 56 | hypothetical protein | |||
| 50,602 | 50,766 | 165 | hypothetical protein | |||
| 50,808 | 50,928 | 121 | hypothetical protein | |||
| 99,427 | 99,516 | 90 | hypothetical protein | |||
| 125,451 | 126,002 | 552 | hypothetical protein |



| RE | RS | met. Lambda | unmet. Lambda | Expected RS for unmet. Lambda | S144 on C. sakazakii | S144 on S. Muenster | S144 on S. Infantis | Expected RS for S144 |
|---|---|---|---|---|---|---|---|---|
| XhoI | C↓TCGAG | 1 | 1 | 1 | 0 | 0 | 0 | 2 |
| GAGCT↑C | ||||||||
| EheI | GGC↓GCC | 1 | 1 | 1 | 0 | 0 | 0 | 5 |
| CCG↑CGG | ||||||||
| SspI | AAT↓ATT | 1 | 1 | 20 | 1 | 1 | 1 | 6 |
| TTA↑TAA | ||||||||
| Bsp120I | G↓GGCCC | 1 | 1 | 1 | 0 | 0 | 0 | 3 |
| CCCGG↑G | ||||||||
| Cfr42I | CCGC↓GG | 1 | 1 | 4 | 0 | 0 | 0 | 3 |
| GG↑CGCC | ||||||||
| SspDI | G↓GCGCC | 1 | 1 | 1 | 0 | 0 | 0 | 5 |
| CCGCG↑G | ||||||||
| MauBI | CG↓CGCGCG | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| GCGCGC↑GC | ||||||||
| PacI | TTAAT↓TAA | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| AAT↑TAATT | ||||||||
| NotI | GC↓GGCCGC | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| CGCCGG↑CG |




References
- Switt, A.I.M.; Sulakvelidze, A.; Wiedmann, M.; Kropinski, A.M.; Wishart, D.S.; Poppe, C.; Liang, Y. Salmonella phages and prophages: Genomics, taxonomy, and applied aspects. In Salmonella: Methods and Protocols, Methods in Molecular Biology; Schatten, H., Eisenstark, A., Eds.; Springer Science+Business Media: New York, NY, USA, 2015; Volume 1225, pp. 237–287. [Google Scholar] [CrossRef]
- Leiman, P.G.; Arisaka, F.; Van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, N.M.I.; Prokhorov, N.S.; Guerrero-Ferreira, R.; Shneider, M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kunisawa, T.; Kanaya, S.; Kutter, E. Comparison of synonymous codon distribution patterns of bacteriophage and host genomes. DNA Res. 1998, 5, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, D.M. Transcriptional control in the prereplicative phase of T4 development. Virol. J. 2010, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Gencay, Y.E.; Gambino, M.; From Prüssing, T.; Brøndsted, L. The genera of bacteriophages and their receptors are the major determinants of host range. Environ. Microbiol. 2019, 21, 2095–2111. [Google Scholar] [CrossRef]
- Ross, A.; Ward, S.; Hyman, P. More is better: Selecting for broad host range bacteriophages. Front. Microbiol. 2016, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hyman, P. Phages for phage therapy: Isolation, characterization, and host range breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Wongsuntornpoj, S.; Switt, A.I.M.; Bergholz, P.; Wiedmann, M.; Chaturongakul, S. Salmonella phages isolated from dairy farms in Thailand show wider host range than a comparable set of phages isolated from U.S. dairy farms. Vet. Microbiol. 2014, 172, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Paolozzi, L.; Ghelardini, P. The bacteriophage Mu. In The Bacteriophages; Calendar, R., Abedon, S.T., Eds.; Oxford University Press: Oxford, UK, 2006; pp. 469–496. [Google Scholar]
- Schwarzer, D.; Buettner, F.F.R.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.; Bowman, V.D.; Stummeyer, K.; Mühlenhoff, M.; et al. A Multivalent Adsorption Apparatus Explains the Broad Host Range of Phage phi92: A Comprehensive Genomic and Structural Analysis. J. Virol. 2012, 86, 10384–10398. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, I.; Osada, K.; Azam, A.H.; Asakawa, H.; Miyanaga, K.; Tanji, Y. The presence of two receptor-binding proteins contributes to the wide host range of staphylococcal twort-like phages. Appl. Environ. Microbiol. 2016, 82, 5763–5774. [Google Scholar] [CrossRef] [Green Version]
- Plattner, M.; Shneider, M.M.; Arbatsky, N.P.; Shashkov, A.S.; Chizhov, A.O.; Nazarov, S.; Prokhorov, N.S.; Taylor, N.M.; Buth, S.A.; Gambino, M.; et al. Structure and Function of the Branched Receptor-Binding Complex of Bacteriophage CBA120. J. Mol. Boil. 2019, 431, 3718–3739. [Google Scholar] [CrossRef] [PubMed]
- Bamford, D.; Caldentey, J.; Bamford, J.K. Bacteriophage Prd1: A Broad Host Range Dsdna Tectivirus With an Internal Membrane. Adv. Virus Res. 1995, 45, 281–319. [Google Scholar] [CrossRef] [PubMed]
- Ziedaite, G.; Daugelavicius, R.; Bamford, J.K.H.; Bamford, D.H. The holin protein of bacteriophage PRD1 forms a pore for small-molecule and endolysin translocation. J. Bacteriol. 2005, 187, 5397–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzer, D.; Browning, C.; Stummeyer, K.; Oberbeck, A.; Mühlenhoff, M.; Gerardy-Schahn, R.; Leiman, P.G. Structure and biochemical characterization of bacteriophage phi92 endosialidase. Virology 2015, 477, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroel, J.; Kleinheinz, K.A.; Jurtz, V.I.; Zschach, H.; Lund, O.; Nielsen, M.; Larsen, M.V. HostPhinder: A phage host prediction tool. Viruses 2016, 8, 116. [Google Scholar] [CrossRef]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef] [Green Version]
- Guibourdenche, M.; Roggentin, P.; Mikoleit, M.; Fields, P.I.; Bockemühl, J.; Grimont, P.A.D.; Weill, F.X. Supplement 2003-2007 (No. 47) to the White-Kauffmann-Le Minor scheme. Res. Microbiol. 2010, 161, 26–29. [Google Scholar] [CrossRef]
- Iversen, C.; Mullane, N.; McCardell, B.; Tall, B.D.; Lehner, A.; Fanning, S.; Stephan, R.; Joosten, H. Cronobacter gen. nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1, and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. lausannensis subsp. nov. and Cronobacter dublinensis subsp. lactaridi subsp. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 1442–1447. [Google Scholar] [CrossRef]
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [CrossRef]
- Jarvis, K.G.; Grim, C.J.; Franco, A.A.; Gopinath, G.; Sathyamoorthy, V.; Hu, L.; Sadowski, J.A.; Lee, C.S.; Tall, B.D. Molecular Characterization of Cronobacter Lipopolysaccharide O-Antigen Gene Clusters and Development of Serotype-Specific PCR Assays. Appl. Environ. Microbiol. 2011, 77, 4017–4026. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Knirel, Y.A.; Feng, L.; Perepelov, A.V.; Senchenkova, S.N.; Reeves, P.R.; Wang, L. Structural diversity in Salmonella O antigens and its genetic basis. FEMS. Microbiol. Rev. 2014, 38, 56–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, B.; Brown, D.T. Function of gene 49 of bacteriophage T4. II. Analysis of intracellular development and the structure of very fast sedimenting DNA. J. Virol. 1976, 18, 1000–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosig, G. Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu. Rev. Genet. 1998, 32, 379–413. [Google Scholar] [CrossRef] [PubMed]
- Sharples, G.J. The X philes: Structure-specific endonucleases that resolve Holliday junctions. Mol. Microbiol. 2001, 39, 823–834. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Weller, G.R.; Doherty, A.J.; Jackson, S.P. The Gam protein of bacteriophage Mu is an orthologue of eukaryotic Ku. EMBO. Rep. 2003, 4, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.J.; Barker, M.M.; Ross, W.; Schneider, D.A.; Webb, C.; Foster, J.W.; Gourse, R.L. DksA: A critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 2004, 118, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Łyzén, R.; Maitra, A.; Milewska, K.; Kochanowska-ŁYzén, M.; James Hernandez, V.; Szalewska-Pa-Lasz, A. The dual role of DksA protein in the regulation of Escherichia coli pArgX promoter. Nucleic Acids Res. 2016, 44, 10316–10325. [Google Scholar] [CrossRef] [Green Version]
- Neuhard, J.; Kelin, R. Chapter II, Biosynthesis and conversions of pyrimidines. In Escherichia Coli and Salmonella: Cellular and Molecular Biology, 2nd ed.; Neidhardt, F.C., Curtiss, R., Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, B., Reznikoff, W.S., Riley, M., Eds.; ASM Press: Washington, DC, USA, 1996; pp. 580–599. [Google Scholar]
- Daws, T.D.; Fuchs, J.A. Isolation and characterization of an Escherichia coli mutant deficient in dTMP kinase activity. J. Bacteriol. 1984, 157, 440–444. [Google Scholar] [CrossRef] [Green Version]
- Reynes, J.P.; Tiraby, M.; Baron, M.; Drocourt, D.; Tiraby, G. Escherichia coli thymidylate kinase: Molecular cloning, nucleotide sequence, and genetic organization of the corresponding tmk locus. J. Bacteriol. 1996, 178, 2804–2812. [Google Scholar] [CrossRef] [Green Version]
- Leipe, D.D.; Koonin, E.V.; Aravind, L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: Multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J. Mol. Biol. 2004, 343, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Belfort, M.; Maley, G.F.; Maley, F. Characterization of the Escherichia coli thyA gene and its amplified thymidylate synthetase product. Proc. Natl. Acad. Sci. USA 1983, 80, 1858–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, K.; Tremblay, D.M.; Delaquis, P.; Goodridge, L.; Levesque, R.C.; Moineau, S.; Suttle, C.A.; Wang, S. Diversity and host specificity revealed by biological characterization and whole genome sequencing of bacteriophages infecting Salmonella enterica. Viruses 2019, 11, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, L.E.; Veljkovic, M. and Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- North, O.I.; Sakai, K.; Yamashita, E.; Nakagawa, A.; Iwazaki, T.; Büttner, C.R.; Takeda, S.; Davidson, A.R. Phage tail fibre assembly proteins employ a modular structure to drive the correct folding of diverse fibres. Nat. Microbiol. 2019, 4, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage lysis: Do we have the hole story yet? Curr. Opin. Microbiol. 2013, 16, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, R. Phage spanins: Diversity, topological dynamics and gene convergence. BMC Bioinform. 2018, 19, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; van Sinderen, D. Bacteriophage orphan DNA methyltransferases: Insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef] [Green Version]
- Callebaut, I.; Courvalin, J.C.; Mornon, J.P. The BAH (bromo-adjacent homology) domain: A link between DNA methylation, replication and transcriptional regulation. FEBS. Lett. 1999, 446, 189–193. [Google Scholar] [CrossRef]
- Oliver, A.W.; Jones, S.A.; Roe, S.M.; Matthews, S.; Goodwin, G.H.; Pearl, L.H. Crystal structure of the proximal BAH domain of the polybromo protein. Biochem. J. 2005, 389, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.; Taha, O.; El-Sherif, H.; Connerton, I.; Hooton, S.P.; Bassim, N.; Connerton, I.F.; El-Shibiny, A. Bacteriophage ZCSE2 is a Potent Antimicrobial against Salmonella enterica Serovars: Ultrastructure, Genomics and Efficacy. Viruses 2020, 12, 424. [Google Scholar] [CrossRef] [Green Version]
- Mandeville, R. Phagelux GRAS Application for SalmoPro®. 2015. Available online: https://www.fda.gov/media/95017/download (accessed on 1 April 2020).
- Pottker, E.S.; Webber, B.; Rizzo, N.N.; Pontin, K.; Peixoto, C.D.S.; Levadowski, R.; Mistura, E.; Santos, L.R.; Rodrigues, L.B.; Nascimento, V.P. New Lithic Bacteriophage to Biocontrol of Salmonella Enterica. In Anais do III Congreso Paranaense de Microbiologia, Londrina. 2018, Volume 2. Available online: https://proceedings.science/cpm-2018/papers/new-lithic-bacteriophage-to-biocontrol-of-salmonella-enterica (accessed on 1 April 2020).
- Bao, H.; Zhang, P.; Zhang, H.; Zhou, Y.; Zhang, L.; Wang, R. Bio-control of Salmonella Enteritidis in foods using bacteriophages. Viruses 2015, 7, 4836–4853. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Liu, Y.; Peng, L.; Cai, X.; Shen, L.; Duan, M.; Ning, Y.; Liu, S.; Li, C.; Liu, Y.; et al. Characterization of the narrow-spectrum bacteriophage LSE7621 towards Salmonella Enteritidis and its biocontrol potential on lettuce and tofu. LWT 2020, 118, 108791. [Google Scholar] [CrossRef]
- Kulikov, E.E.; Golomidova, A.K.; Letarova, M.A.; Kostryukova, E.S.; Zelenin, A.S.; Prokhorov, N.S.; Letarov, A.V. Genomic sequencing and biological characteristics of a novel Escherichia coli bacteriophage 9g, a putative representative of a new siphoviridae genus. Viruses 2014, 6, 5077–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol. 2009, 502, 1–18. [Google Scholar] [CrossRef]
- Ackermann, H.W.; DuBow, M.S. Phage multiplication. In Viruses of Prokaryotes: General Properties of Bacteriophages; Ackermann, H.W., DuBow, M.S., Eds.; CRC Press: Boca Raton, FL, USA, 1987; Volume 1, pp. 49–85. [Google Scholar]
- El Haddad, L.; Ben Abdallah, N.; Plante, P.-L.; Dumaresq, J.; Katsarava, R.; Labrie, S.; Corbeil, J.; St-Gelais, D.; Moineau, S. Improving the Safety of Staphylococcus aureus Polyvalent Phages by Their Production on a Staphylococcus xylosus Strain. PLoS ONE 2014, 9, e102600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigel, C.; Seitz, H. Bacteriophage replication modules. FEMS Microbiol. Rev. 2006, 30, 321–381. [Google Scholar] [CrossRef] [PubMed]
- Tomita, F.; Takahashi, I. A novel enzyme, dCTP deaminase, found in Bacillus subtilis infected with phage PBS 1. Biochim. Biophys. Acta 1969, 179, 18–27. [Google Scholar] [CrossRef]
- Yamada, T.; Satoh, S.; Ishikawa, H.; Fujiwara, A.; Kawasaki, T.; Fujie, M.; Ogata, H. A jumbo phage infecting the phytopathogen Ralstonia solanacearum defines a new lineage of the Myoviridae family. Virology 2010, 398, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 Genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [Green Version]
- Kutter, E.; Bryan, D.; Ray, G.; Brewster, E.; Blasdel, B.; Guttman, B. From host to phage metabolism: Hot tales of phage T4′s takeover of E. coli. Viruses 2018, 10, 387. [Google Scholar] [CrossRef] [Green Version]
- Ochman, H.; Lawrence, J.G. Phylogenetics and the amelioration of bacterial genomes. In Escherichia Coli and Salmonella: Cellular and Molecular Biology, 2nd ed.; Neidhardt, F.C., Curtis, R., Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, B., Reznikoff, W.S., Riley, M., Eds.; ASM Press: Washington, DC, USA, 1996; Volume 2, pp. 2627–2637. [Google Scholar]
- Kim, S.; Kim, Y.T.; Yoon, H.; Lee, J.H.; Ryu, S. The complete genome sequence of Cronobacter sakazakii ATCC 29544T, a food-borne pathogen, isolated from a child’s throat. Gut Pathog. 2017, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ma, Y.; Wang, Y.; Yang, H.; Shen, W.; Chen, X. Transcription regulation mechanisms of bacteriophages. Bioengineered 2014, 5, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, A. Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol. 2008, 66, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.-H. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acid Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Broeker, N.K.; Kiele, F.; Casjens, S.R.; Gilcrease, E.B.; Thalhammer, A.; Koetz, J.; Barbirz, S. In vitro studies of lipopolysaccharide-mediated DNA release of podovirus HK620. Viruses 2018, 10, 289. [Google Scholar] [CrossRef] [Green Version]
- Andres, D.; Hanke, C.; Baxa, U.; Seul, A.T.; Barbirz, S.; Seckler, R. Tailspike interactions with lipopolysaccharide effect DNA ejection from phage P22 particles in vitro. J. Biol. Chem. 2010, 285, 36768–36775. [Google Scholar] [CrossRef] [Green Version]
- Hyman, P.; van Raaij, M. Bacteriophage T4 long tail fiber domains. Biophys. Rev. 2018, 10, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Hashemolhosseini, S.; Stierhof, Y.D.; Hindennach, I.; Henning, U. Characterization of the helper proteins for the assembly of tail fibers of coliphages T4 and lambda. J. Bacteriol. 1996, 178, 6258–6265. [Google Scholar] [CrossRef] [Green Version]
- Bruessow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [Green Version]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tetart, F.; Desplats, C.; Krisch, H.M. Genome Plasticity in the Distal Tail Fiber Locus of the T-even Bacteriophage: Recombination between Conserved Motifs Swaps Adhesin Specificity. J. Mol. Biol. 1998, 282, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Cambillau, C. A Common Evolutionary Origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 2011, 75, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grose, J.H.; Casjens, S.R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 2015, 468, 421–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasifar, R.; Kropinski, A.M.; Sabour, P.M.; Ackermann, H.W.; Alanis Villa, A.; Abbasifar, A.; Griffiths, M.W. Genome sequence of Cronobacter sakazakii myovirus vB_CsaM_GAP31. J. Virol. 2012, 86, 13830–13831. [Google Scholar] [CrossRef] [Green Version]
- Abbasifar, R.; Griffiths, M.W.; Sabour, P.M.; Ackermann, H.W.; Vandersteegen, K.; Lavigne, R.; Nobene, J.-P.; Villaa, A.A.; Abbasifara, A.; Nash, J.H.E.; et al. Supersize me: Cronobacter sakazakii phage GAP32. Virology 2014, 460, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochman, H.; Selander, R.K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 1984, 157, 690–693. [Google Scholar] [CrossRef] [Green Version]
- Carlson, K. Working with bacteriophages: Common techniques and methodological approaches. In Bacteriophage Biology and Applications, 1st ed.; Kutter, E., Sulakvelidze, A., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 437–494. [Google Scholar]
- Kim, M.; Ryu, S. Spontaneous and transient defence against bacteriophage by phase-variable glucosylation of O-antigen in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2012, 86, 411–425. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 1st ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Available online: https://www.cshlpress.com/pdf/sample/2013/MC4/MC4FM.pdf (accessed on 1 April 2020).
- Lee, E.; Helt, G.; Reese, J.T.; Munoz-Torres, M.; Childers, C.; Buels, R.; Stein, L.; Holmes, I.H.; Elsik, C.G.; Lewis, S. Web Apollo: A web-based genomic annotation editing platform. Genome Boil. 2013, 14, R93. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses. Nucleic Acids Res. 2018, 46, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, M.; Rocha, M.; Oliveira, H.; Dias, O. Predicting promoters in phage genomes using machine learning models. Adv. Intell. Syst. Comput. 2020, 1005, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Mijalis, E.; Rasche, E. Find Spanin Workflow. Available online: https://cpt.tamu.edu/galaxy-pub/u/elenimijalis/w/imported-findspanin-v20-apollo (accessed on 1 January 2018).
- Carver, T.; Thomson, N.; Bleasby, A.; Berriman, M.; Parkhill, J. DNA Plotter: Circular and linear interactive genome visualization. Bioinformatics 2009, 25, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Stothard, P. The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Prot. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, J.; Li, R.; Shi, Q.; Xue, Z.; Zhang, Y. ThreaDomEx: A unified platform for predicting continuous and discontinuous protein domains by multiple-threading and segment assembly. Nucleic Acids Res. 2017, 45, W400–W407. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Goker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; Van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Lobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]





| Genus | Species or Serovar | Infected/Tested Strains | Reference |
|---|---|---|---|
| Salmonella | Derby | 2/21 | [6] |
| Salmonella | Typhimurium | 0/13 | [6] |
| Salmonella | Dublin | 0/4 | [6] |
| Salmonella | Enteritidis | 2/3 | [6] |
| Salmonella | Infantis | 3/4 | [6] |
| Salmonella | Rough | 0/4 | [6] |
| Salmonella | 4.12:i:- | 0/6 | [6] |
| Salmonella | 4.5.12:i:- | 1/9 | [6] |
| Salmonella | 4.5.12:i:- | 1/1 | [6] |
| Salmonella | Goettingen | 1/1 | [6] |
| Salmonella | Livingstone | 0/1 | [6] |
| Salmonella | London | 1/1 | [6] |
| Salmonella | Rissen | 0/1 | [6] |
| Salmonella | Brandenburg | 0/1 | [6] |
| Salmonella | Bradford | 0/1 | [6] |
| Salmonella | Senftenberg | 0/1 | this work |
| Salmonella | Adelaide | 0/1 | this work |
| Salmonella | Weslaco | 0/1 | this work |
| Salmonella | Montevideo | 0/1 | this work |
| Salmonella | Tanger | 1/1 | this work |
| Salmonella | Cerro | 0/1 | this work |
| Salmonella | Basel | 0/1 | this work |
| Salmonella | Anatum | 0/1 | this work |
| Salmonella | Eilbek | 1/1 | this work |
| Salmonella | Worthington | 0/1 | this work |
| Salmonella | Onderstepoort | 1/1 | this work |
| Salmonella | Deversoir | 1/1 | this work |
| Salmonella | Telaviv | 1/1 | this work |
| Salmonella | Choleraesuis | 1/1 | this work |
| Salmonella | Aberdeen | 1/1 | this work |
| Salmonella | Inverness | 1/1 | this work |
| Salmonella | Bergen | 0/1 | this work |
| Salmonella | Ruiru | 0/1 | this work |
| Salmonella | Gaminara | 1/1 | this work |
| Salmonella | Paratyphi B var. Java | 0/1 | this work |
| Salmonella | Muenster | 1/1 | this work |
| Escherichia | coli | 0/77 | this work |
| Klebsiella | spp. | 0/3 | this work |
| Enterobacter | spp. | 1/4 | this work |
| Providencia | stuarti | 0/1 | this work |
| Citrobacter | spp. | 0/2 | this work |
| Yersinia | spp. | 0/1 | this work |
| Serratia | marcescens | 0/1 | this work |
| Erwinia | herbicola | 0/1 | this work |
| Shigella | sonnei | 0/1 | this work |
| Acinetobacter | spp. | 0/1 | this work |
| Proteus | mirabilis | 0/2 | this work |
| Cronobacter | sakazakii | 4/15 | this work |
| ORF | Product | Functional Category | Cluster | Proteins | |||
|---|---|---|---|---|---|---|---|
| gp | MW (kDa) | Unique Peptides | Coverage (%) | ||||
| ORF03 | put. dihydrofolate reductase | nt biosynthesis | A | gp03 | 25 | 3 | 25 |
| ORF08 | terminase, large subunit | packaging | B | gp08 | 54 | 2 | 4 |
| ORF09 | portal prot. | capsid morphogenesis | B | gp09 | 58 | 12 | 27 |
| ORF10 | scaffold prot. | capsid morphogenesis | B | gp10 | 27 | 2 | 5 |
| ORF11 | major capsid prot. | capsid morphogenesis | B | gp11 | 35 | 14 | 49 |
| ORF12 | put. head-to-tail connector complex 1 | capsid morphogenesis | B | gp12 | 18 | 10 | 49 |
| ORF14 | put. head-to-tail connector complex 3 | capsid morphogenesis | B | gp14 | 14 | 5 | 52 |
| ORF15 | put. head-to-tail connector complex 4 | capsid morphogenesis | B | gp15 | 17 | 3 | 22 |
| ORF16 | put. tail prot. 1 | tail morphogenesis | B | gp16 | 21 | 3 | 24 |
| ORF17 | put. tail prot. 2 | tail morphogenesis | B | gp17 | 111 | 25 | 33 |
| ORF18 | put. tail prot. 3 | tail morphogenesis | B | gp18 | 22 | 17 | 80 |
| ORF20 | put. sheath prot. | tail morphogenesis | B | gp20 | 42 | 9 | 32 |
| ORF21 | put. tube prot. | tail morphogenesis | B | gp21 | 16 | 6 | 26 |
| ORF23 | put. tape measure prot. | tail morphogenesis | B | gp23 | 60 | 10 | 26 |
| ORF24 | conserved hyp. prot. | tail morphogenesis | B | gp24 | 32 | 11 | 31 |
| ORF25 | conserved hyp. prot. | tail morphogenesis | B | gp25 | 14 | 4 | 27 |
| ORF26 | put. baseplate prot. 1 | baseplate morphogenesis | B | gp26 | 35 | 9 | 32 |
| ORF27 | put. puncturing prot. | baseplate morphogenesis | B | gp27 | 23 | 6 | 50 |
| ORF28 | put. baseplate prot. 2 | baseplate morphogenesis | B | gp28 | 14 | 5 | 45 |
| ORF29 | put. baseplate prot. 3 | baseplate morphogenesis | B | gp29 | 42 | 10 | 30 |
| ORF30 | put. baseplate prot. 4 | baseplate morphogenesis | B | gp30 | 24 | 7 | 33 |
| ORF31 | put. tail fiber prot. | tail morphogenesis | B | gp31 | 48 | 9 | 24 |
| ORF35 | put. endolysin | lysis | B | gp35 | 20 | 5 | 28 |
| ORF36 | put. IM-spanin | lysis | B | gp36 | 12 | 5 | 38 |
| ORF37 | put. OM-spanin | lysis | B | gp37 | 10 | 1 | 10 |
| ORF40 | hyp. prot. | hyp. | C | gp40 | |||
| ORF43 | thymidylate synthase | nt biosynthesis | C | gp43 | 33 | 5 | 21 |
| ORF54 | conserved hyp. prot. | hyp. | C | gp54 | 19 | 2 | 21 |
| ORF55 | put. P-loop with nucleoside triphosphatehydrolase | nt biosynthesis | C | gp55 | 33 | 2 | 9 |
| ORF58 | conserved hyp. prot. | hyp. | C | gp58 | 49 | 5 | 16 |
| ORF69 | conserved hyp. prot. | hyp. | D | gp69 | 96 | 2 | 4 |
| ORF79 | conserved hyp. prot. | hyp. | E | gp79 | 21 | 2 | 14 |
| Genus | Phage | Morphotype | Capsid Shape | Coding (%) | GC (%) | Genome Size (bp) | ORFs | tRNAs | GenBank Acc. No. | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Loughboroughvirus | Salmonella phage S144 | myovirus | prolate, 101 × 44 nm | 93.4 | 46 | 53,628 | 80 | 0 | MT663719 | this work |
| Salmonella phage SE4 | myovirus | ND | 92.2 | 45 | 53,494 | 76 | 0 | MK770413.1 | [35] | |
| Salmonella phage ZCSE2 | myovirus | prolate 84 × 35 nm | 92.0 | 46 | 53,965 | 78 | 0 | MK673511.1 | [43] | |
| Rosemountvirus | Salmonella phage BP63 | myovirus | ND | 93.2 | 46 | 52,437 | 76 | 0 | KM366099.1 | [44] |
| Salmonella phage SE13 | myovirus | ND | 91.0 | 46 | 52,438 | 73 | 0 | MK770411.1 | [35] | |
| Salmonella phage UPF_BP2 | myovirus | ND | 85.2 | 46 | 54,894 | 70 | 0 | KX826077.1 | [45] | |
| Salmonella phage vB_SenM_PA13076 | myovirus | oval Ø 66 nm | 89.2 | 46 | 52,474 | 68 | 0 | MF740800.1 | [46] | |
| Salmonella phage LSE7621 | myovirus | symmetrical Ø 56 nm | 92.1 | 46 | 50,936 | 72 | 0 | MK568062.1 | [47] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambino, M.; Nørgaard Sørensen, A.; Ahern, S.; Smyrlis, G.; Gencay, Y.E.; Hendrix, H.; Neve, H.; Noben, J.-P.; Lavigne, R.; Brøndsted, L. Phage S144, a New Polyvalent Phage Infecting Salmonella spp. and Cronobacter sakazakii. Int. J. Mol. Sci. 2020, 21, 5196. https://doi.org/10.3390/ijms21155196
Gambino M, Nørgaard Sørensen A, Ahern S, Smyrlis G, Gencay YE, Hendrix H, Neve H, Noben J-P, Lavigne R, Brøndsted L. Phage S144, a New Polyvalent Phage Infecting Salmonella spp. and Cronobacter sakazakii. International Journal of Molecular Sciences. 2020; 21(15):5196. https://doi.org/10.3390/ijms21155196
Chicago/Turabian StyleGambino, Michela, Anders Nørgaard Sørensen, Stephen Ahern, Georgios Smyrlis, Yilmaz Emre Gencay, Hanne Hendrix, Horst Neve, Jean-Paul Noben, Rob Lavigne, and Lone Brøndsted. 2020. "Phage S144, a New Polyvalent Phage Infecting Salmonella spp. and Cronobacter sakazakii" International Journal of Molecular Sciences 21, no. 15: 5196. https://doi.org/10.3390/ijms21155196