Steroids and Alzheimer’s Disease: Changes Associated with Pathology and Therapeutic Potential

Abstract
:1. Introduction
2. Dysregulated Brain Steroidogenesis and Steroid Concentrations Associated with AD
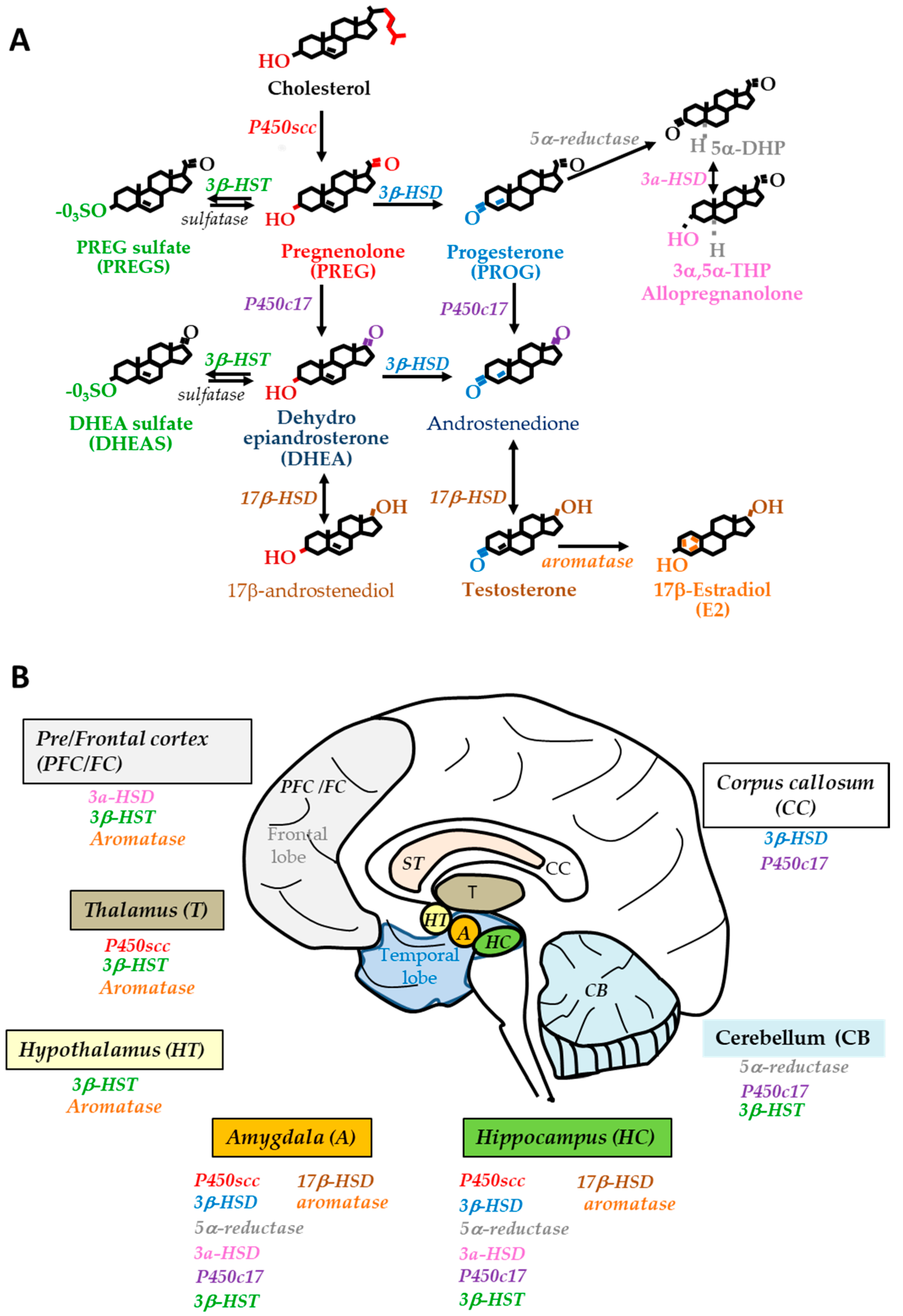
2.1. Steroidogenesis in the Human Brain
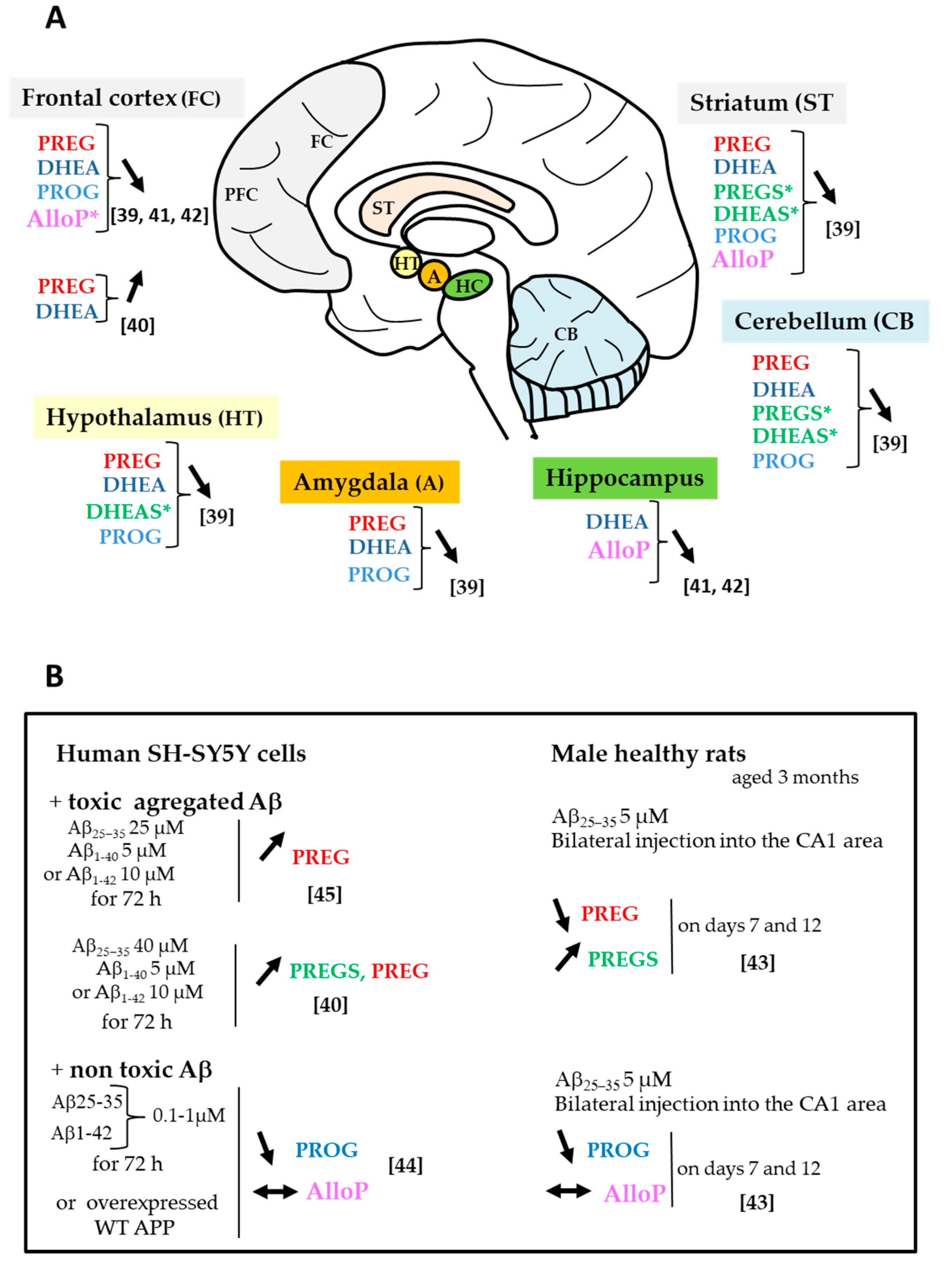
2.2. Changes in Neurosteroid and Biosynthetic Enzyme Levels

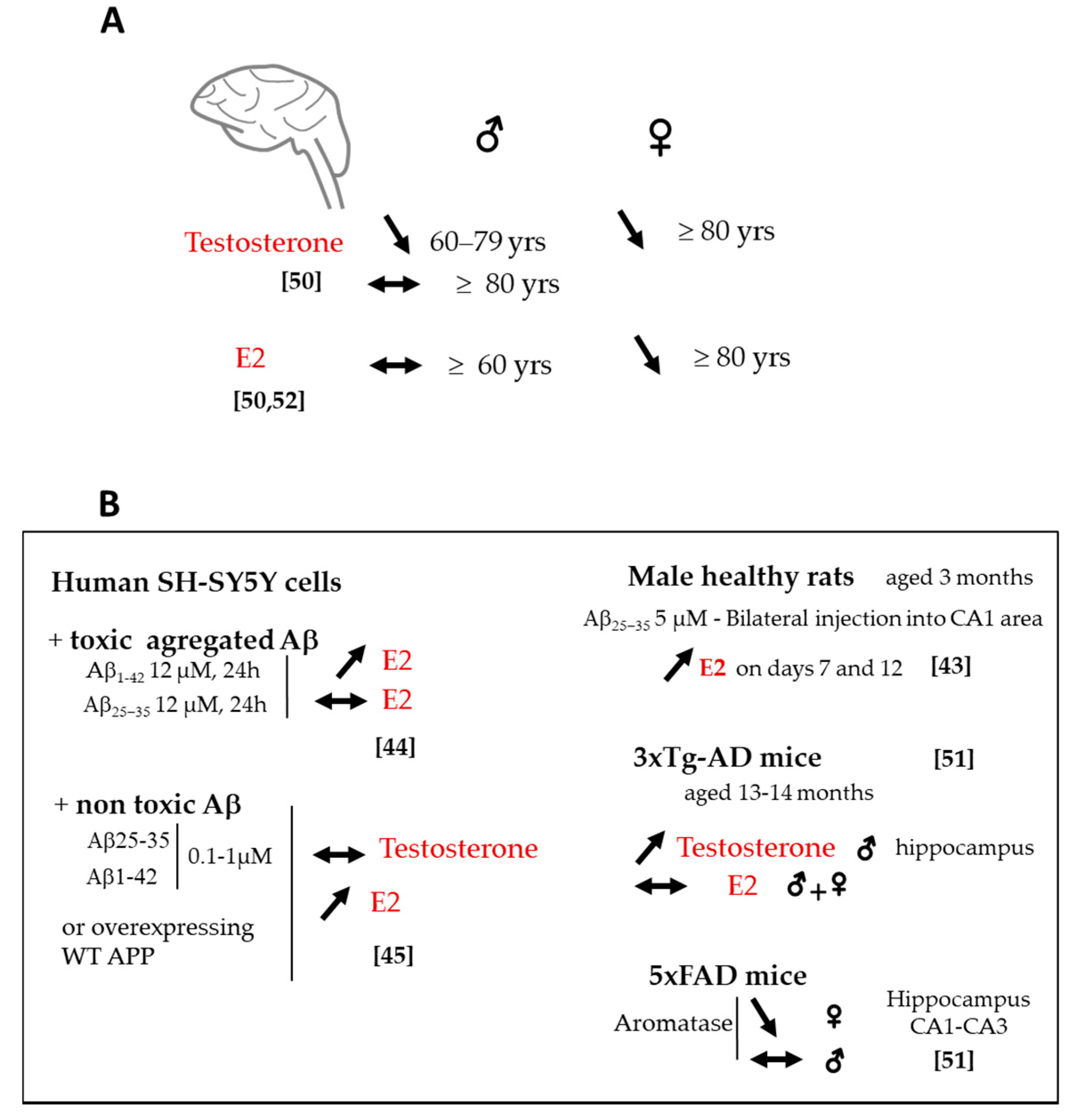
2.3. Changes in Sex Steroids and Biosynthetic Enzyme Levels
2.4. Sex Difference in Neuroactive Steroid Levels
3. Steroids and Genetics of Late-Onset AD
4. Protective Effects of Neuroactive Steroids on AD-Like Neuropathology
4.1. Amyloid-β Pathology
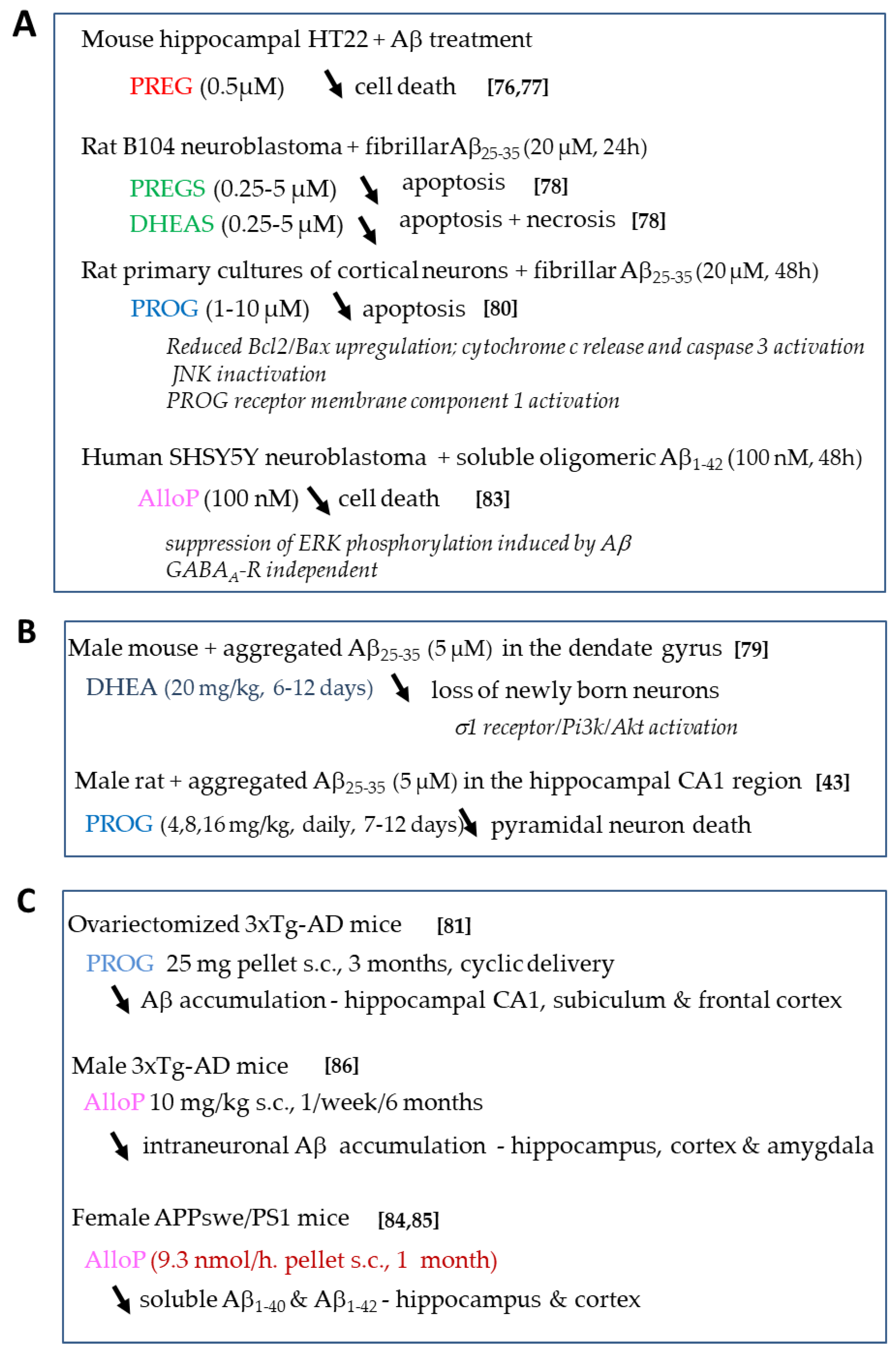
4.1.1. Effects of Neurosteroids on Aβ Toxicity
4.1.2. Effects of Sex Steroids on Aβ Toxicity
4.2. Tau Pathology
4.3. Mitochondrial Impairment
4.4. Neuroinflammation
4.5. Neurogenesis, Synaptic Failure and Memory Loss
5. Modulation of Endogenous Neuroactive Steroid Production for Protection in AD
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ApoE | apolipoprotein |
| APP | amyloid precursor protein |
| Aβ | Amyloid-β |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DHEA | dehydroepiandrosterone |
| DHEAS | dehydroepiandrosterone sulfate |
| E2 | 17β-estradiol |
| FTD | frontotemporal dementia |
| GABAA-R | γ-aminobutyric acid type A receptor |
| GC-MS | gas chromatography-mass spectrometry |
| IL-1β | interleukin-1β |
| mTOR | mechanistic target of rapamycin |
| NF-κB | nuclear factor-kappa B |
| NTFs | neurofibrillary tangles |
| P450scc | P450side chain cleavage |
| PI3K | phosphoinositide 3-kinase |
| PREG | pregnenolone |
| PREGS | pregnenolone sulfate |
| PROG | progesterone |
| PS1 | presenilin-1 |
| ROS | reactive oxygen species |
| TNFα | tumor necrosis factor alpha |
| TSPO | 18 kDa translocator protein |
| yrs. | years |
| 3 xTg-AD | triple transgenic mouse bearing the human APPSWE, TauP301L, and PS1M146V genes linked to AD and FTD |
| 3α,5α-THP | 3α,5α-tetrahydroprogesterone, allopregnanolone |
| 3α-HSD | 3α-hydrosteroid dehydrogenase |
| 3β-HSD | 3α-hydroxysteroid dehydrogenase-D5Δ5→Δ4D4 isomerase |
| 3β-HST | 3β-hydroxysteroid sulfotransferase |
| 5α-DHP | 5α-dihydroprogesterone |
| 17β-HSD | 17α-hydroxysteroid dehydrogenase |
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer brain. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bastin, C.; Delhaye, E.; Moulin, C.; Barbeau, E.J. Novelty processing and memory impairment in Alzheimer’s disease: A review. Neurosci. Biobehav. Rev. 2019, 100, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Galts, C.P.C.; Bettio, L.E.B.; Jewett, D.C.; Yang, C.C.; Brocardo, P.S.; Rodrigues, A.S.; Thacker, J.S.; Gil-Mohapel, J. Depression in neurodegenerative diseases: Common mechanisms and current treatment options. Neurosci. Biobehav. R 2019, 102, 56–84. [Google Scholar] [CrossRef]
- Rajan, K.B.; Wilson, R.S.; Weuve, J.; Barnes, L.L.; Evans, D.A. Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology 2015, 85, 898–904. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, A.L.; Chidgey, C.; Peng, P.; Masters, C.L.; Roberts, B.R. The Neurobiology and Age-Related Prevalence of the epsilon4 Allele of Apolipoprotein E in Alzheimer’s Disease Cohorts. J. Mol. Neurosci. 2016, 60, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Brinton, R.D. Neurosteroids as regenerative agents in the brain: Therapeutic implications. Nat. Rev. Endocrinol. 2013, 9, 241–250. [Google Scholar] [CrossRef]
- Melcangi, R.C.; Giatti, S.; Garcia-Segura, L.M. Levels and actions of neuroactive steroids in the nervous system under physiological and pathological conditions: Sex-specific features. Neurosci. Biobehav. Rev. 2016, 67, 25–40. [Google Scholar] [CrossRef]
- Blanco, M.J.; La, D.; Coughlin, Q.; Newman, C.A.; Griffin, A.M.; Harrison, B.L.; Salituro, F.G. Breakthroughs in neuroactive steroid drug discovery. Bioorg. Med. Chem. Lett. 2018, 28, 61–70. [Google Scholar] [CrossRef]
- Hojo, Y.; Kawato, S. Neurosteroids in Adult Hippocampus of Male and Female Rodents: Biosynthesis and Actions of Sex Steroids. Front. Endocrinol. (Lausanne) 2018, 9, 183. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Miller, W.L. Role of mitochondria in steroidogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 771–790. [Google Scholar] [CrossRef]
- Stoffel-Wagner, B. Neurosteroid metabolism in the human brain. Eur. J. Endocrinol. 2001, 145, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Romero, D.G.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P. Steroidogenic enzyme gene expression in the human brain. Mol. Cell Endocrinol. 2002, 190, 9–17. [Google Scholar] [CrossRef]
- Le Goascogne, C.; Gouezou, M.; Robel, P.; Defaye, G.; Chambaz, E.; Waterman, M.R.; Baulieu, E.E. The cholesterol side-chain cleavage complex in human brain white matter. J. Neuroendocr. 1989, 1, 153–156. [Google Scholar] [CrossRef]
- Steckelbroeck, S.; Watzka, M.; Reichelt, R.; Hans, V.H.; Stoffel-Wagner, B.; Heidrich, D.D.; Schramm, J.; Bidlingmaier, F.; Klingmuller, D. Characterization of the 5α-reductase-3α-hydroxysteroid dehydrogenase complex in the human brain. J. Clin. Endocrinol. Metab. 2001, 86, 1324–1331. [Google Scholar]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.F.; Lin, H.K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3 alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351 Pt 1, 67–77. [Google Scholar]
- Usami, N.; Yamamoto, T.; Shintani, S.; Ishikura, S.; Higaki, Y.; Katagiri, Y.; Hara, A. Substrate specificity of human 3(20) alpha-hydroxysteroid dehydrogenase for neurosteroids and its inhibition by benzodiazepines. Biol. Pharm. Bull. 2002, 25, 441–445. [Google Scholar] [CrossRef] [Green Version]
- Higaki, Y.; Usami, N.; Shintani, S.; Ishikura, S.; El-Kabbani, O.; Hara, A. Selective and potent inhibitors of human 20 alpha-hydroxysteroid dehydrogenase (AKR1C1) that metabolizes neurosteroids derived from progesterone. Chem. Biol. Interact. 2003, 143–144, 503–513. [Google Scholar] [CrossRef]
- Falany, C.N.; Rohn-Glowacki, K.J. SULT2B1: Unique properties and characteristics of a hydroxysteroid sulfotransferase family. Drug Metab. Rev. 2013, 45, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Tamura, H. Identification and localization of two hydroxysteroid sulfotransferases in the human brain. J. Health Sci. 2002, 48, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Steckelbroeck, S.; Watzka, M.; Stoffel-Wagner, B.; Hans, V.H.; Redel, L.; Clusmann, H.; Elger, C.E.; Bidlingmaier, F.; Klingmuller, D. Expression of the 17β-hydroxysteroid dehydrogenase type 5 mRNA in the human brain. Mol. Cell Endocrinol. 2001, 171, 165–168. [Google Scholar] [CrossRef]
- Steckelbroeck, S.; Stoffel-Wagner, B.; Reichelt, R.; Schramm, J.; Bidlingmaier, F.; Siekmann, L.; Klingmuller, D. Characterization of 17β-hydroxysteroid dehydrogenase activity in brain tissue: Testosterone formation in the human temporal lobe. J. Neuroendocr. 1999, 11, 457–464. [Google Scholar] [CrossRef]
- Weill-Engerer, S.; David, J.-P.; Sazdovitch, V.; Liere, P.; Schumacher, M.; Delacourte, A.; Baulieu, E.; Akwa, Y. In vitro metabolism of dehydroepiandrosterone (DHEA) to 7α-OH-DHEA and Δ5-androstene-3β,17β-diol in specific regions of the aging brain from Alzheimer’s and nondemented patients. Brain Res. 2003, 969, 117–125. [Google Scholar] [CrossRef]
- He, X.Y.; Dobkin, C.; Yang, S.Y. 17β-Hydroxysteroid dehydrogenases and neurosteroid metabolism in the central nervous system. Mol. Cell Endocrinol. 2019, 489, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Stoffel-Wagner, B.; Watzka, M.; Steckelbroeck, S.; Schwaab, R.; Schramm, J.; Bidlingmaier, F.; Klingmuller, D. Expression of CYP19 (aromatase) mRNA in the human temporal lobe. Biochem. Biophys. Res. Commun. 1998, 244, 768–771. [Google Scholar] [CrossRef]
- Sasano, H.; Takashashi, K.; Satoh, F.; Nagura, H.; Harada, N. Aromatase in the human central nervous system. Clin. Endocrinol. (Oxf.) 1998, 48, 325–329. [Google Scholar] [CrossRef]
- Yague, J.G.; Munoz, A.; de Monasterio-Schrader, P.; Defelipe, J.; Garcia-Segura, L.M.; Azcoitia, I. Aromatase expression in the human temporal cortex. Neuroscience 2006, 138, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Hutchison, R.E.; Morris, C.M.; Hutchison, J.B. Neuroblastoma and Alzheimer’s disease brain cells contain aromatase activity. Steroids 1998, 63, 263–267. [Google Scholar] [CrossRef]
- Steckelbroeck, S.; Heidrich, D.D.; Stoffel-Wagner, B.; Hans, V.H.; Schramm, J.; Bidlingmaier, F.; Klingmuller, D. Characterization of aromatase cytochrome P450 activity in the human temporal lobe. J. Clin. Endocrinol. Metab. 1999, 84, 2795–2801. [Google Scholar] [CrossRef]
- Ishunina, T.A.; van Beurden, D.; van der Meulen, G.; Unmehopa, U.A.; Hol, E.M.; Huitinga, I.; Swaab, D.F. Diminished aromatase immunoreactivity in the hypothalamus, but not in the basal forebrain nuclei in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Steckelbroeck, S.; Watzka, M.; Reissinger, A.; Wegener-Toper, P.; Bidlingmaier, F.; Bliesener, N.; Hans, V.H.; Clusmann, H.; Ludwig, M.; Siekmann, L.; et al. Characterisation of estrogenic 17β-hydroxysteroid dehydrogenase (17β-HSD) activity in the human brain. J. Steroid Biochem. Mol. Biol. 2003, 86, 79–92. [Google Scholar] [CrossRef]
- Beyenburg, S.; Stoffel-Wagner, B.; Watzka, M.; Blumcke, I.; Bauer, J.; Schramm, J.; Bidlingmaier, F.; Elger, C.E. Expression of cytochrome P450scc mRNA in the hippocampus of patients with temporal lobe epilepsy. Neuroreport 1999, 10, 3067–3070. [Google Scholar] [CrossRef]
- Watzka, M.; Bidlingmaier, F.; Schramm, J.; Klingmuller, D.; Stoffel-Wagner, B. Sex- and age-specific differences in human brain CYP11A1 mRNA expression. J. Neuroendocr. 1999, 11, 901–905. [Google Scholar] [CrossRef]
- Luchetti, S.; Huitinga, I.; Swaab, D.F. Neurosteroid and GABA-A receptor alterations in Alzheimer’s disease, Parkinson’s disease and multiple sclerosis. Neuroscience 2011, 191, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J. Sex and the development of Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liere, P.; Schumacher, M. Mass spectrometric analysis of steroids: All that glitters is not gold. Expert Rev. Endocrinol. Metab. 2015, 10, 463–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weill-Engerer, S.; David, J.P.; Sazdovitch, V.; Liere, P.; Eychenne, B.; Pianos, A.; Schumacher, M.; Delacourte, A.; Baulieu, E.E.; Akwa, Y. Neurosteroid Quantification in Human Brain Regions: Comparison between Alzheimer’s and Nondemented Patients. J. Clin. Endocrinol. Metab. 2002, 87, 5138–5143. [Google Scholar] [CrossRef] [Green Version]
- Calan, O.G.; Akan, P.; Cataler, A.; Dogan, C.; Kocturk, S. Amyloid Beta Peptides Affect Pregnenolone and Pregnenolone Sulfate Levels in PC-12 and SH-SY5Y Cells Depending on Cholesterol. Neurochem. Res. 2016, 41, 1700–1712. [Google Scholar] [CrossRef]
- Marx, C.E.; Trost, W.T.; Shampine, L.J.; Stevens, R.D.; Hulette, C.M.; Steffens, D.C.; Ervin, J.F.; Butterfield, M.I.; Blazer, D.G.; Massing, M.W.; et al. The neurosteroid allopregnanolone is reduced in prefrontal cortex in Alzheimer’s disease. Biol. Psychiatry 2006, 60, 1287–1294. [Google Scholar] [CrossRef]
- Naylor, J.C.; Kilts, J.D.; Hulette, C.M.; Steffens, D.C.; Blazer, D.G.; Ervin, J.F.; Strauss, J.L.; Allen, T.B.; Massing, M.W.; Payne, V.M.; et al. Allopregnanolone levels are reduced in temporal cortex in patients with Alzheimer’s disease compared to cognitively intact control subjects. Biochim. Biophys. Acta 2010, 1801, 951–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wu, H.; Xue, G.; Ma, X.; Wu, J.; Qin, Y.; Hou, Y. Metabolic alteration of neuroactive steroids and protective effect of progesterone in Alzheimer’s disease-like rats. Neural Regen. Res. 2013, 8, 2800–2810. [Google Scholar] [PubMed]
- Schaeffer, V.; Patte-Mensah, C.; Eckert, A.; Mensah-Nyagan, A.G. Modulation of neurosteroid production in human neuroblastoma cells by Alzheimer’s disease key proteins. J. Neurobiol. 2006, 66, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Meyer, L.; Patte-Mensah, C.; Eckert, A.; Mensah-Nyagan, A.G. Dose-dependent and sequence-sensitive effects of amyloid-beta peptide on neurosteroidogenesis in human neuroblastoma cells. Neurochem. Int. 2008, 52, 948–955. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Wegiel, J.; Yang, S.Y. Intracellular oxidation of allopregnanolone by human brain type 10 17 beta-hydroxysteroid dehydrogenase. Brain Res. 2005, 1040, 29–35. [Google Scholar] [CrossRef]
- Luchetti, S.; Bossers, K.; Van de Bilt, S.; Agrapart, V.; Morales, R.R.; Frajese, G.V.; Swaab, D.F. Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1964–1976. [Google Scholar] [CrossRef]
- Aitken, L.; Quinn, S.D.; Perez-Gonzalez, C.; Samuel, I.D.; Penedo, J.C.; Gunn-Moore, F.J. Morphology-Specific Inhibition of beta-Amyloid Aggregates by 17 beta-Hydroxysteroid Dehydrogenase Type 10. Chembiochem 2016, 17, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- He, X.-Y.; Isaacs, C.; Yang, S.-Y. Roles of Mitochondrial 17β-Hydroxysteroid Dehydrogenase Type 10 in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 665–673. [Google Scholar] [CrossRef]
- Rosario, E.R.; Chang, L.; Head, E.H.; Stanczyk, F.Z.; Pike, C.J. Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Overk, C.R.; Perez, S.E.; Ma, C.; Taves, M.D.; Soma, K.K.; Mufson, E.J. Sex Steroid Levels and AD-Like Pathology in 3xTgAD Mice. J. Neuroendocr. 2013, 25, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lu, M.; Lancaster, T.; Cao, P.; Honda, S.; Staufenbiel, M.; Harada, N.; Zhong, Z.; Shen, Y.; Li, R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc. Natl. Acad. Sci. USA 2005, 102, 19198–19203. [Google Scholar] [CrossRef] [Green Version]
- Prange-Kiel, J.; Dudzinski, D.A.; Prols, F.; Glatzel, M.; Matschke, J.; Rune, G.M. Aromatase Expression in the Hippocampus of AD Patients and 5xFAD Mice. Neural Plast. 2016, 2016, 9802086. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ovejero, D.; Azcoitia, I.; Doncarlos, L.L.; Melcangi, R.C.; Garcia-Segura, L.M. Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Res. Brain Res. Rev. 2005, 48, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, M.; Iivonen, S.; Soininen, H. Aromatase enzyme and Alzheimer’s disease. Minerva Endocrinol. 2006, 31, 61–73. [Google Scholar] [PubMed]
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef]
- Nebel, R.A.; Aggarwal, N.T.; Barnes, L.L.; Gallagher, A.; Goldstein, J.M.; Kantarci, K.; Mallampalli, M.P.; Mormino, E.C.; Scott, L.; Yu, W.H.; et al. Understanding the impact of sex and gender in Alzheimer’s disease: A call to action. Alzheimers Dement. 2018, 14, 1171–1183. [Google Scholar] [CrossRef]
- Giatti, S.; Diviccaro, S.; Serafini, M.M.; Caruso, D.; Garcia-Segura, L.M.; Viviani, B.; Melcangi, R.C. Sex differences in steroid levels and steroidogenesis in the nervous system: Physiopathological role. Front. Neuroendocr. 2020, 56, 100804. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; et al. Sex differences in Alzheimer disease - the gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–469. [Google Scholar] [CrossRef]
- Corbo, R.M.; Gambina, G.; Broggio, E.; Scarabino, D.; Scacchi, R. Association study of two steroid biosynthesis genes (COMT and CYP17) with Alzheimer’s disease in the Italian population. J. Neurol. Sci. 2014, 344, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Pike, C.J. Age-related testosterone depletion and the development of Alzheimer disease. JAMA 2004, 292, 1431–1432. [Google Scholar] [CrossRef] [PubMed]
- Butler, H.T.; Warden, D.R.; Hogervorst, E.; Ragoussis, J.; Smith, A.D.; Lehmann, D.J. Association of the aromatase gene with Alzheimer’s disease in women. Neurosci. Lett. 2010, 468, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Medway, C.; Combarros, O.; Cortina-Borja, M.; Butler, H.T.; Ibrahim-Verbaas, C.A.; de Bruijn, R.F.; Koudstaal, P.J.; van Duijn, C.M.; Ikram, M.A.; Mateo, I.; et al. The sex-specific associations of the aromatase gene with Alzheimer’s disease and its interaction with IL10 in the Epistasis Project. Eur. J. Hum. Genet. 2014, 22, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Kancheva, R.; Hill, M.; Novak, Z.; Chrastina, J.; Kancheva, L.; Starka, L. Neuroactive steroids in periphery and cerebrospinal fluid. Neuroscience 2011, 191, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.; Abbiati, F.; Giatti, S.; Romano, S.; Fusco, L.; Cavaletti, G.; Melcangi, R.C. Patients treated for male pattern hair with finasteride show, after discontinuation of the drug, altered levels of neuroactive steroids in cerebrospinal fluid and plasma. J. Steroid Biochem. Mol. Biol. 2015, 146, 74–79. [Google Scholar] [CrossRef]
- Martin, J.J.; Plank, E.; Jungwirth, B.; Hapfelmeier, A.; Podtschaske, A.; Kagerbauer, S.M. Weak correlations between serum and cerebrospinal fluid levels of estradiol, progesterone and testosterone in males. BMC Neurosci. 2019, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Schonknecht, P.; Pantel, J.; Klinga, K.; Jensen, M.; Hartmann, T.; Salbach, B.; Schroder, J. Reduced cerebrospinal fluid estradiol levels are associated with increased beta-amyloid levels in female patients with Alzheimer’s disease. Neurosci. Lett. 2001, 307, 122–124. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Chernick, D.; Ortiz-Valle, S.; Jeong, A.; Qu, W.; Li, L. Peripheral versus central nervous system APOE in Alzheimer’s disease: Interplay across the blood-brain barrier. Neurosci. Lett. 2019, 708, 134306. [Google Scholar] [CrossRef]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [PubMed]
- Altmann, A.; Tian, L.; Henderson, V.W.; Greicius, M.D.; Alzheimer’s Disease Neuroimaging Initiative, I. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 2014, 75, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, R.F.; Mormino, E.C.; Amariglio, R.E.; Properzi, M.J.; Rabin, J.S.; Lim, Y.Y.; Papp, K.V.; Jacobs, H.I.L.; Burnham, S.; Hanseeuw, B.J.; et al. Sex, amyloid, and APOE epsilon4 and risk of cognitive decline in preclinical Alzheimer’s disease: Findings from three well-characterized cohorts. Alzheimers Dement. 2018, 14, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Hogervorst, E.; Lehmann, D.J.; Warden, D.R.; McBroom, J.; Smith, A.D. Apolipoprotein E epsilon4 and testosterone interact in the risk of Alzheimer’s disease in men. Int. J. Geriatr. Psychiatry 2002, 17, 938–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Selkoe, D.J. Amyloid β-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef]
- Gursoy, E.; Cardounel, A.; Kalimi, M. Pregnenolone protects mouse hippocampal (HT-22) cells against glutamate and amyloid-β protein toxicity. Neurochem. Res. 2001, 26, 15–21. [Google Scholar] [CrossRef]
- Akan, P.; Kizildag, S.; Ormen, M.; Genc, S.; Oktem, M.A.; Fadiloglu, M. Pregnenolone protects the PC-12 cell line against amyloid β peptide toxicity but its sulfate ester does not. Chem. Biol. Interact. 2009, 177, 65–70. [Google Scholar] [CrossRef]
- El Bitar, F.; Meunier, J.; Villard, V.; Almeras, M.; Krishnan, K.; Covey, D.F.; Maurice, T.; Akwa, Y. Neuroprotection by the synthetic neurosteroid enantiomers ent-PREGS and ent-DHEAS against Aβ25–35 peptide-induced toxicity in vitro and in vivo in mice. Psychopharmacol. (Berl.) 2014, 231, 3293–3312. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xu, B.; Zhu, Y.; Chen, L.; Sokabe, M.; Chen, L. DHEA prevents Abeta25-35-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology 2010, 59, 323–333. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, Z.; Han, X.; Wu, H.; Yu, Y.; Wu, J.; Liu, S.; Hou, Y. Progesterone attenuates Abeta (25–35)-induced neuronal toxicity via JNK inactivation and progesterone receptor membrane component 1-dependent inhibition of mitochondrial apoptotic pathway. J. Steroid Biochem. Mol. Biol. 2015, 154, 302–311. [Google Scholar] [CrossRef]
- Carroll, J.C.; Rosario, E.R.; Villamagna, A.; Pike, C.J. Continuous and cyclic progesterone differentially interact with estradiol in the regulation of Alzheimer-like pathology in female 3xTransgenic-Alzheimer’s disease mice. Endocrinology 2010, 151, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Carroll, J.C.; Morgan, T.E.; Lin, S.; Zhao, L.; Arimoto, J.M.; Murphy, M.P.; Beckett, T.L.; Finch, C.E.; Brinton, R.D.; et al. 17β-estradiol and progesterone regulate expression of β-amyloid clearance factors in primary neuron cultures and female rat brain. Endocrinology 2012, 153, 5467–5479. [Google Scholar] [CrossRef] [Green Version]
- Mendell, A.L.; Chung, B.Y.T.; Creighton, C.E.; Kalisch, B.E.; Bailey, C.D.C.; MacLusky, N.J. Neurosteroid metabolites of testosterone and progesterone differentially inhibit ERK phosphorylation induced by amyloid beta in SH-SY5Y cells and primary cortical neurons. Brain Res. 2018, 1686, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, S.K.; Johansson, M.; Backstrom, T.; Wang, M. Chronic allopregnanolone treatment accelerates Alzheimer’s disease development in AbetaPP (Swe) PSEN1 (DeltaE9) mice. J. Alzheimers Dis. 2012, 31, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, S.K.; Johansson, M.; Backstrom, T.; Nitsch, R.M.; Wang, M. Brief but chronic increase in allopregnanolone cause accelerated AD pathology differently in two mouse models. Curr. Alzheimer Res. 2013, 10, 38–47. [Google Scholar]
- Chen, S.; Wang, J.M.; Irwin, R.W.; Yao, J.; Liu, L.; Brinton, R.D. Allopregnanolone promotes regeneration and reduces β-amyloid burden in a preclinical model of Alzheimer’s disease. PloS ONE 2011, 6, e24293. [Google Scholar] [CrossRef]
- Rosario, E.R.; Carroll, J.C.; Oddo, S.; LaFerla, F.M.; Pike, C.J. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2006, 26, 13384–13389. [Google Scholar] [CrossRef]
- Rosario, E.R.; Carroll, J.C.; Pike, C.J. Evaluation of the effects of testosterone and luteinizing hormone on regulation of β-amyloid in male 3xTg-AD mice. Brain Res. 2012, 1466, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Rosario, E.R.; Carroll, J.C.; Pike, C.J. Testosterone regulation of Alzheimer-like neuropathology in male 3xTg-AD mice involves both estrogen and androgen pathways. Brain Res. 2010, 1359, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vegeto, E.; Villa, A.; Della Torre, S.; Crippa, V.; Rusmini, P.; Cristofani, R.; Galbiati, M.; Maggi, A.; Poletti, A. The Role of Sex and Sex Hormones in Neurodegenerative Diseases. Endocr Rev. 2020, 41, 1–47. [Google Scholar] [CrossRef]
- Carroll, J.C.; Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Oddo, S.; LaFerla, F.M.; Pike, C.J. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 2007, 27, 13357–13365. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Costa, L.; Piacentini, R.; Grassi, C.; Lanzone, A.; Gulino, A. 17β-estradiol protects cerebellar granule cells against β-amyloid-induced toxicity via the apoptotic mitochondrial pathway. Neurosci. Lett. 2014, 561, 134–139. [Google Scholar] [CrossRef]
- Perianes-Cachero, A.; Canelles, S.; Aguado-Llera, D.; Frago, L.M.; Toledo-Lobo, M.V.; Carrera, I.; Cacabelos, R.; Chowen, J.A.; Argente, J.; Arilla-Ferreiro, E.; et al. Reduction in Aβ-induced cell death in the hippocampus of 17β-estradiol-treated female rats is associated with an increase in IGF-I signaling and somatostatinergic tone. J. Neurochem. 2015, 135, 1257–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, F.P.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Tau Proteins and Tauopathies in Alzheimer’s Disease. Cell Mol. Neurobiol. 2018, 38, 965–980. [Google Scholar] [CrossRef]
- Dang, T.N.; Dobson-Stone, C.; Glaros, E.N.; Kim, W.S.; Hallupp, M.; Bartley, L.; Piguet, O.; Hodges, J.R.; Halliday, G.M.; Double, K.L.; et al. Endogenous progesterone levels and frontotemporal dementia: Modulation of TDP-43 and Tau levels in vitro and treatment of the A315T TARDBP mouse model. Dis. Model. Mech. 2013, 6, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.A.; Zhu, L.Q.; Zhang, Q.; Shi, H.R.; Wang, S.H.; Wang, Q.; Wang, J.Z. Estradiol attenuates tau hyperphosphorylation induced by upregulation of protein kinase-A. Neurochem. Res. 2008, 33, 1811–1820. [Google Scholar] [CrossRef]
- Majd, S.; Majd, Z.; Koblar, S.; Power, J. Beta estradiol and norepinephrine treatment of differentiated SH-SY5Y cells enhances tau phosphorylation at (Ser (396)) and (Ser (262)) via AMPK but not mTOR signaling pathway. Mol. Cell Neurosci. 2018, 88, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.R.; Zhu, L.Q.; Wang, S.H.; Liu, X.A.; Tian, Q.; Zhang, Q.; Wang, Q.; Wang, J.Z. 17β-estradiol attenuates glycogen synthase kinase-3β activation and tau hyperphosphorylation in Akt-independent manner. J. Neural. Transm. (Vienna) 2008, 115, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Buccarello, L.; Grignaschi, G.; Castaldo, A.M.; Di Giancamillo, A.; Domeneghini, C.; Melcangi, R.C.; Borsello, T. Sex Impact on Tau-Aggregation and Postsynaptic Protein Levels in the P301L Mouse Model of Tauopathy. J. Alzheimers Dis. 2017, 56, 1279–1292. [Google Scholar] [CrossRef]
- Levine, A.J.; Hewett, L. Estrogen replacement therapy and frontotemporal dementia. Maturitas 2003, 45, 83–88. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.C.; Han, Z.; Cascio, C.; Papadopoulos, V. Oxidative stress-mediated DHEA formation in Alzheimer’s disease pathology. Neurobiol. Aging 2003, 24, 57–65. [Google Scholar] [CrossRef]
- Sinha, M.; Saha, A.; Basu, S.; Pal, K.; Chakrabarti, S. Aging and antioxidants modulate rat brain levels of homocysteine and dehydroepiandrosterone sulphate (DHEA-S): Implications in the pathogenesis of Alzheimer’s disease. Neurosci. Lett. 2010, 483, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Saharan, S.; Mandal, P.K. The emerging role of glutathione in Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 519–529. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2012, 1822, 625–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Irwin, R.; Chen, S.; Hamilton, R.; Cadenas, E.; Brinton, R.D. Ovarian hormone loss induces bioenergetic deficits and mitochondrial β-amyloid. Neurobiol. Aging 2012, 33, 1507–1521. [Google Scholar] [CrossRef] [Green Version]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox Res. 2019, 35, 1–18. [Google Scholar] [CrossRef]
- Qian, X.; Cao, H.; Ma, Q.; Wang, Q.; He, W.; Qin, P.; Ji, B.; Yuan, K.; Yang, F.; Liu, X.; et al. Allopregnanolone attenuates Aβ25-35-induced neurotoxicity in PC12 cells by reducing oxidative stress. Int. J. Clin. Exp. Med. 2015, 8, 13610–13615. [Google Scholar]
- Costa, A.R.; Marcelino, H.; Goncalves, I.; Quintela, T.; Tomas, J.; Duarte, A.C.; Fonseca, A.M.; Santos, C.R. Sex Hormones Protect Against Amyloid-beta Induced Oxidative Stress in the Choroid Plexus Cell Line Z310. J. Neuroendocr. 2016, 28. [Google Scholar] [CrossRef]
- Irwin, R.W.; Yao, J.; Hamilton, R.T.; Cadenas, E.; Brinton, R.D.; Nilsen, J. Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology 2008, 149, 3167–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaignard, P.; Liere, P.; Therond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Role of Sex Hormones on Brain Mitochondrial Function, with Special Reference to Aging and Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 406. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Lim, Y.-A.; Mensah-Nyagan, A.G.; Götz, J.; Eckert, A. Alzheimer’s Disease, Oestrogen and Mitochondria: An Ambiguous Relationship. Mol. Neurobiol. 2012, 46, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, G.; Novakovic, M.; Savaskan, E.; Meier, F.; Baysang, G.; Brockhaus, M.; Muller-Spahn, F. The effects of beta-estradiol on SHSY5Y neuroblastoma cells during heavy metal induced oxidative stress, neurotoxicity and beta-amyloid secretion. Neuroscience 2002, 113, 849–855. [Google Scholar] [CrossRef]
- Wang, X.; Dykens, J.A.; Perez, E.; Liu, R.; Yang, S.D.; Covey, D.F.; Simpkins, J.W. Neuroprotective Effects of 17β-Estradiol and Nonfeminizing Estrogens against H2O2 Toxicity in Human Neuroblastoma SK-N-SH Cells. Mol. Pharm. 2006, 70, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Duong, P.; Tenkorang, M.A.A.; Trieu, J.; McCuiston, C.; Rybalchenko, N.; Cunningham, R.L. Neuroprotective and neurotoxic outcomes of androgens and estrogens in an oxidative stress environment. Biol. Sex. Differ. 2020, 11, 12. [Google Scholar] [CrossRef]
- Schuessel, K.; Leutner, S.; Cairns, N.J.; Muller, W.E.; Eckert, A. Impact of gender on upregulation of antioxidant defence mechanisms in Alzheimer’s disease brain. J. Neural. Transm. (Vienna) 2004, 111, 1167–1182. [Google Scholar]
- Vina, J.; Lloret, A. Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-beta peptide. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S527–S533. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Singh, A.K.; Mishra, G.; Maurya, A.; Awasthi, R.; Kumari, K.; Thakur, A.; Rai, A.; Rai, G.K.; Sharma, B.; Kulkarni, G.T.; et al. Role of TREM2 in Alzheimer’s Disease and its Consequences on beta- Amyloid, Tau and Neurofibrillary Tangles. Curr. Alzheimer Res. 2019, 16, 1216–1229. [Google Scholar] [CrossRef] [PubMed]
- Hashemiaghdam, A.; Mroczek, M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J. Neuroimmunol. 2020, 341, 577185. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Wang, X.; Sun, S.; Xue, G.; Li, J.; Hou, Y. Progesterone exerts neuroprotective effects against Abeta-induced neuroinflammation by attenuating ER stress in astrocytes. Int. Immunopharmacol. 2016, 33, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Uchoa, M.F.; Moser, V.A.; Pike, C.J. Interactions between inflammation, sex steroids, and Alzheimer’s disease risk factors. Front. Neuroendocr. 2016, 43, 60–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vegeto, E.; Belcredito, S.; Ghisletti, S.; Meda, C.; Etteri, S.; Maggi, A. The endogenous estrogen status regulates microglia reactivity in animal models of neuroinflammation. Endocrinology 2006, 147, 2263–2272. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Shen, Y.; Yang, L.B.; Lue, L.F.; Finch, C.; Rogers, J. Estrogen enhances uptake of amyloid beta-protein by microglia derived from the human cortex. J. Neurochem. 2000, 75, 1447–1454. [Google Scholar] [CrossRef]
- Yun, J.; Yeo, I.J.; Hwang, C.J.; Choi, D.Y.; Im, H.S.; Kim, J.Y.; Choi, W.R.; Jung, M.H.; Han, S.B.; Hong, J.T. Estrogen deficiency exacerbates Aβ-induced memory impairment through enhancement of neuroinflammation, amyloidogenesis and NF-kB activation in ovariectomized mice. Brain Behav. Immun. 2018, 73, 282–293. [Google Scholar] [CrossRef]
- Yao, P.L.; Zhuo, S.; Mei, H.; Chen, X.F.; Li, N.; Zhu, T.F.; Chen, S.T.; Wang, J.M.; Hou, R.X.; Le, Y.Y. Androgen alleviates neurotoxicity of beta-amyloid peptide (Aβ) by promoting microglial clearance of Aβ and inhibiting microglial inflammatory response to Aβ. CNS Neurosci. Ther. 2017, 23, 855–865. [Google Scholar] [CrossRef]
- Laurent, C.; Buee, L.; Blum, D. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21–33. [Google Scholar] [CrossRef]
- Terada, T.; Yokokura, M.; Obi, T.; Bunai, T.; Yoshikawa, E.; Ando, I.; Shimada, H.; Suhara, T.; Higuchi, M.; Ouchi, Y. In vivo direct relation of tau pathology with neuroinflammation in early Alzheimer’s disease. J. Neurol. 2019, 266, 2186–2196. [Google Scholar] [CrossRef]
- Bellucci, A.; Westwood, A.J.; Ingram, E.; Casamenti, F.; Goedert, M.; Spillantini, M.G. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am. J. Pathol. 2004, 165, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, A.; Bugiani, O.; Ghetti, B.; Spillantini, M.G. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis. 2011, 8, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, J.R.; Avila, J.; Bolos, M. Dephosphorylated rather than hyperphosphorylated Tau triggers a pro-inflammatory profile in microglia through the p38 MAPK pathway. Exp. Neurol. 2018, 310, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; Bryant, A.; Hu, M.; Robbins, A.B.; Hopp, S.C.; Hyman, B.T. Partial reduction of microglia does not affect tau pathology in aged mice. J. Neuroinflammation 2018, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Romero-Molina, C.; Navarro, V.; Sanchez-Varo, R.; Jimenez, S.; Fernandez-Valenzuela, J.J.; Sanchez-Mico, M.V.; Munoz-Castro, C.; Gutierrez, A.; Vitorica, J.; Vizuete, M. Distinct Microglial Responses in Two Transgenic Murine Models of TAU Pathology. Front. Cell Neurosci. 2018, 12, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatt, A.; Lee, H.; Williams, G.; Thuret, S.; Ballard, C. Expression of neurogenic markers in Alzheimer’s disease: A systematic review and metatranscriptional analysis. Neurobiol. Aging 2019, 76, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Yang, R.; Chang, F.; Chen, L.; Xie, G.; Sokabe, M. Neurosteroid PREGS protects neurite growth and survival of newborn neurons in the hippocampal dentate gyrus of APPswe/PS1dE9 mice. Curr. Alzheimer Res. 2012, 9, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Irwin, R.W.; Wang, J.M.; Chen, S.; Brinton, R.D. Neuroregenerative mechanisms of allopregnanolone in Alzheimer’s disease. Front. Endocrinol. (Lausanne) 2011, 2, 117. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, R.; Wainwright, S.R.; Galea, L.A. Sex hormones and adult hippocampal neurogenesis: Regulation, implications, and potential mechanisms. Front. Neuroendocr. 2016, 41, 129–152. [Google Scholar] [CrossRef] [Green Version]
- Ponti, G.; Farinetti, A.; Marraudino, M.; Panzica, G.; Gotti, S. Sex Steroids and Adult Neurogenesis in the Ventricular-Subventricular Zone. Front. Endocrinol. (Lausanne) 2018, 9, 156. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Liang, K.S.; Wang, X.J.; Zhou, X.Y.; Sun, J.; Zhou, S.N. Chronic Estradiol Administration During the Early Stage of Alzheimer’s Disease Pathology Rescues Adult Hippocampal Neurogenesis and Ameliorates Cognitive Deficits in Aβ1–42 Mice. Mol. Neurobiol. 2017, 54, 7656–7669. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacology 2019, 195, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhou, R.; Chen, L.; Cai, W.; Tomimoto, H.; Sokabe, M.; Chen, L. Pregnenolone sulfate enhances survival of adult-generated hippocampal granule cells via sustained presynaptic potentiation. Neuropharmacology 2011, 60, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gibbs, T.; Farb, D. Pregnenolone sulfate as a modulator of synaptic plasticity. Psychopharmacology 2014, 231, 3537–3556. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Ishizuka, Y.; Yamazaki, H.; Shirao, T. Allopregnanolone increases mature excitatory synapses along dendrites via protein kinase A signaling. Neuroscience 2015, 305, 139–145. [Google Scholar] [CrossRef]
- Clements, L.; Harvey, J. Activation of oestrogen receptor α induces a novel form of LTP at hippocampal temporoammonic-CA1 synapses. Br. J. Pharm. 2020, 177, 642–655. [Google Scholar] [CrossRef]
- Hara, Y.; Crimins, J.L.; Puri, R.; Wang, A.C.J.; Motley, S.E.; Yuk, F.; Ramos, T.M.; Janssen, W.G.M.; Rapp, P.R.; Morrison, J.H. Estrogen Alters the Synaptic Distribution of Phospho-GluN2B in the Dorsolateral Prefrontal Cortex While Promoting Working Memory in Aged Rhesus Monkeys. Neuroscience 2018, 394, 303–315. [Google Scholar] [CrossRef]
- Hatanaka, Y.; Hojo, Y.; Mukai, H.; Murakami, G.; Komatsuzaki, Y.; Kim, J.; Ikeda, M.; Hiragushi, A.; Kimoto, T.; Kawato, S. Rapid increase of spines by dihydrotestosterone and testosterone in hippocampal neurons: Dependence on synaptic androgen receptor and kinase networks. Brain Res. 2015, 1621, 121–132. [Google Scholar] [CrossRef]
- Fattoretti, P.; Malatesta, M.; Mariotti, R.; Zancanaro, C. Testosterone administration increases synaptic density in the gyrus dentatus of old mice independently of physical exercise. Exp. Gerontol. 2019, 125, 110664. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Capani, F.; Sortino, M.A. Early β-Amyloid-induced Synaptic Dysfunction Is Counteracted by Estrogen in Organotypic Hippocampal Cultures. Curr. Alzheimer Res. 2016, 13, 631–640. [Google Scholar] [CrossRef]
- Lau, C.F.; Ho, Y.S.; Hung, C.H.; Wuwongse, S.; Poon, C.H.; Chiu, K.; Yang, X.; Chu, L.W.; Chang, R.C. Protective effects of testosterone on presynaptic terminals against oligomeric beta-amyloid peptide in primary culture of hippocampal neurons. Biomed. Res. Int. 2014, 2014, 103906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirova, A.M.; Bays, R.B.; Lagalwar, S. Working memory and executive function decline across normal aging, mild cognitive impairment, and Alzheimer’s disease. Biomed. Res. Int. 2015, 2015, 748212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, T.; Su, T.P.; Privat, A. Sigma1 (σ1) receptor agonists and neurosteroids attenuate β25-35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience 1998, 83, 413–428. [Google Scholar] [CrossRef]
- Yang, R.; Chen, L.; Wang, H.; Xu, B.; Tomimoto, H. Anti-amnesic effect of neurosteroid PREGS in Aβ25-35-injected mice through σ1 receptor- and α7 nAChR-mediated neuroprotection. Neuropharmacology 2012, 63, 1042–1050. [Google Scholar] [CrossRef]
- Frye, C.A.; Walf, A.A. Effects of progesterone administration and APPswe+PSEN1Deltae9 mutation for cognitive performance of mid-aged mice. Neurobiol. Learn. Mem. 2008, 89, 17–26. [Google Scholar] [CrossRef]
- Singh, C.; Liu, L.; Wang, J.M.; Irwin, R.W.; Yao, J.; Chen, S.; Henry, S.; Thompson, R.F.; Brinton, R.D. Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol. Aging 2012, 33, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Dye, R.V.; Miller, K.J.; Singer, E.J.; Levine, A.J. Hormone replacement therapy and risk for neurodegenerative diseases. Int. J. Alzheimers Dis. 2012, 2012, 258454. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Rubio, G.; Herrera-Perez, J.J.; Hernandez-Hernandez, O.T.; Martinez-Mota, L. Relationship between androgen deficiency and memory impairment in aging and Alzheimer’s disease. Actas Esp. Psiquiatr. 2017, 45, 227–247. [Google Scholar]
- Osmanovic-Barilar, J.; Salkovic-Petrisi, M. Evaluating the Role of Hormone Therapy in Postmenopausal Women with Alzheimer’s Disease. Drugs Aging 2016, 33, 787–808. [Google Scholar] [CrossRef] [Green Version]
- Arbo, B.D.; Ribeiro, M.F.; Garcia-Segura, L.M. Development of new treatments for Alzheimer’s disease based on the modulation of translocator protein (TSPO). Ageing Res. Rev. 2019, 54, 100943. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocr. 2018, 30. [Google Scholar] [CrossRef]
- Liere, P.; Pianos, A.; Oudinet, J.-P.; Schumacher, M.; Akwa, Y. Differential effects of the 18-kDa translocator protein (TSPO) ligand etifoxine on steroidogenesis in rat brain, plasma and steroidogenic glands: Pharmacodynamic studies. Psychoneuroendocrinology 2017, 83, 122–134. [Google Scholar] [CrossRef]
- Janssen, B.; Vugts, D.J.; Funke, U.; Molenaar, G.T.; Kruijer, P.S.; van Berckel, B.N.; Lammertsma, A.A.; Windhorst, A.D. Imaging of neuroinflammation in Alzheimer’s disease, multiple sclerosis and stroke: Recent developments in positron emission tomography. Biochim. Biophys. Acta 2016, 1862, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol. Ther. 2019, 194, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Da Pozzo, E.; Giacomelli, C.; Barresi, E.; Costa, B.; Taliani, S.; Passetti Fda, S.; Martini, C. Targeting the 18-kDa translocator protein: Recent perspectives for neuroprotection. Biochem. Soc. Trans. 2015, 43, 559–565. [Google Scholar] [CrossRef]
- Arbo, B.D.; Marques, C.V.; Ruiz-Palmero, I.; Ortiz-Rodriguez, A.; Ghorbanpoor, S.; Arevalo, M.A.; Garcia-Segura, L.M.; Ribeiro, M.F. 4′-Chlorodiazepam is neuroprotective against amyloid-β through the modulation of survivin and bax protein expression in vitro. Brain Res. 2016, 1632, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Lejri, I.; Grimm, A.; Halle, F.; Abarghaz, M.; Klein, C.; Maitre, M.; Schmitt, M.; Bourguignon, J.J.; Mensah-Nyagan, A.G.; Bihel, F.; et al. TSPO Ligands Boost Mitochondrial Function and Pregnenolone Synthesis. J. Alzheimers Dis. 2019, 72, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Grimm, A.; Lejri, I.; Halle, F.; Schmitt, M.; Gotz, J.; Bihel, F.; Eckert, A. Mitochondria modulatory effects of new TSPO ligands in a cellular model of tauopathies. J. Neuroendocr. 2020, 32, e12796. [Google Scholar] [CrossRef] [Green Version]
- Barron, A.M.; Garcia-Segura, L.M.; Caruso, D.; Jayaraman, A.; Lee, J.W.; Melcangi, R.C.; Pike, C.J. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 8891–8897. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.; Pike, C.J. TSPO ligand PK11195 improves Alzheimer-related outcomes in aged female 3xTg-AD mice. Neurosci. Lett. 2018, 683, 7–12. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DHEA | Allopregnanolone | E2 |
|---|---|---|
| In vitro Forebrain mitochondria from male Swiss mouse + Aβ1–42 (4 µM) or Aβ25–35 (50 µM) for 20 min DHEA (3, 10 or 30 µM, 20 min) ↓ mitochondrial respiration dysfunction through σ1 receptor mechanism ↓ increased ROS production [108] | In vitro PC12 cells + aggregated Aβ25–35 (20 µM, 24 h) Allopregnanolone pretreatment (10 µM, 2 h) ↓ ROS generation ↓ SOD activity [109] | In vitro Choroid plexus explants or cell line Z310 + Aβ1–42 (0.66 µM, 24 h) E2 pretreatment (1 µM, 8–12 h)↓ ROS production, ↓ Aβ uptake [110] In vivo Ovariectomized 3xTg-AD mice E2 treatment (0.25 mg continuous 90-day pellet) prevented in isolated forebrain mitochondria: ↓ respiration, ↓ energy deficits ↓ Aβ load, ↓ lipid peroxidation [107] |
| Steroid Treatment | Models of Aβ-induced Inflammation | Inflammatory Response against Aβ Neurotoxicity |
|---|---|---|
| PROG Pretreatment 4, 8 or 16 mg/kg, i.p. daily, for 7 and 12 days after Aβ injection | In vivo Male rats + aggregated Aβ25–35 5 µM in the hippocampal CA1 region | ↓ the upregulation of TNFα and IL-1β induced by Aβ [43] ↓ endoplasmic reticulum stress markers PERK/elF2α |
| E2 pretreatment pellets 0.01 mg s.c. daily, from 5 to 10–14 months of age E2 pretreatment 100 nM, 48 h E2 pretreatment 10 µM, 60 min | In vivo Ovariectomized APP23 mice, 10–14 months of age (early stage of disease) In vitro Human cortical microglia + fluorescein-Aβ1–42 100 nM Microglial BV-2 cell + aggregated Aβ1–42 1 µM | ↓ Mac-1 positive inflammatory plaques ↑ microglia clearance of Aβ [125] ↑ microglia uptake of Aβ, through non-classical estrogen receptor [126] ↓ Aβ-induced NF-κB [125,127] |
| Pretreatment Testosterone 100 nM Dihydrotestosterone 10 nM Pretreatment Testosterone 200 µg Dihydrotestosterone 100 µg s.c. every day for 2 weeks Pretreatment Dihydrotestosterone0.5 and 1 nM for 6 h | Murine microglia N9 cell line + aggregated Aβ1–42 (2 µM) for 30 min Male C57BL/6 mice + aggregated Aβ1–42 (1 µM) into the CA1 region Murine microglia N9 cell line + aggregated Aβ1–42 1 µM for 1 h or 24 h | ↓ Aβ-induced proinflammatory cytokine IL-1β via suppression of NF-κB and p38 activation by Aβ [128] ↓ Aβ-induced proinflammatory cytokine IL-1β [128] ↑ microglia Aβ uptake through upregulating FPR2 ↑ microglia clearance of Aβ through upregulating ECE-1c [128] |
| Impaired Neurogenesis | Aβ-Induced Memory Loss |
|---|---|
| • Male APPswe/PS1dE9 mice PREGS ↑ survival & maturation of newborn cells [137] Allopregnanolone 10 mg/kg 1/week/6 months at 3 months of age ↑ survival of newly generated cells in hippocampal subgranular zone [86] • Rat B104 neuroblastoma cells PREGS or DHEAS 5 µM, 24 h ↓ Aβ25–35 (20 µM)-induced decrease in neurite outgrowth [78] • 3 xTg-AD mice aged 6 and 9 months Allopregnanolone 10 mg/kg s.c. ↑ neurogenesis (BrDU+ neural progenitor survival) 1 h later [157] | • Male healthy mice + aggregated Aβ25–35 (3 nmol i.c.v) PREGS, DHEAS, DHEA (5, 10, 20 mg/kg s.c. respectively) ↓memory Aβ-induced deficits in short-term memory (spontaneous alternation in the Y-maze test) and in long-term memory (step-down passive avoidance test), 7 and 14 days post-Aβ infusion, respectively, through σ1 receptor activation [154] • Male healthy mice + aggregated Aβ25–35 (9 nmol i.c.v) Pretreatment by PREGS or DHEAS 0.5 nmol i.c.v 6 h before Aβ infusion ↓ memory deficits in short-term memory (spontaneous alternation test) and in long-term memory (step-through passive avoidance test) [78] • Ovariectomized APPswe/PS1dE9 mice PROG pellet (25 mg, 90-day release) ↑ short-term memory performance (object recognition T-maze test) [156] Allopreganolone (4.7 nmol or 9.3 nmol) for 12 weeks ↑ learning deficits in the Morris water maze [84] •3 xTg-AD mice aged 6 & 9 months Allopregnanolone 10 mg/kg s.c. 7 days prior to the start of the learning trials restored maximal learning capacity in the trace eyeblink conditioning, except at 12 months of age [157] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akwa, Y. Steroids and Alzheimer’s Disease: Changes Associated with Pathology and Therapeutic Potential. Int. J. Mol. Sci. 2020, 21, 4812. https://doi.org/10.3390/ijms21134812
Akwa Y. Steroids and Alzheimer’s Disease: Changes Associated with Pathology and Therapeutic Potential. International Journal of Molecular Sciences. 2020; 21(13):4812. https://doi.org/10.3390/ijms21134812
Chicago/Turabian StyleAkwa, Yvette. 2020. "Steroids and Alzheimer’s Disease: Changes Associated with Pathology and Therapeutic Potential" International Journal of Molecular Sciences 21, no. 13: 4812. https://doi.org/10.3390/ijms21134812