Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches


Abstract
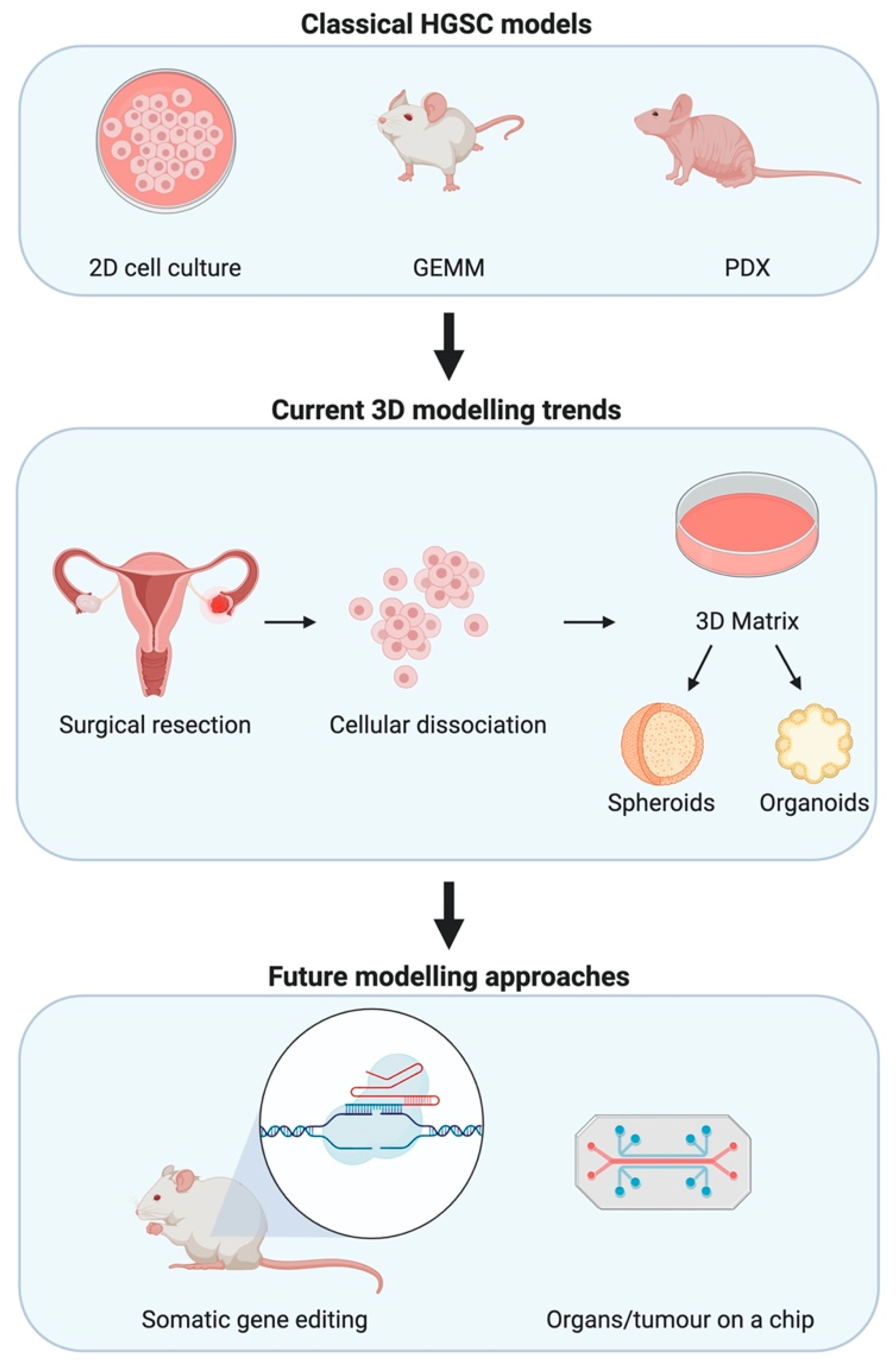
:1. Introduction
2. High-Grade Serous Ovarian Carcinoma
3. GEMMs of HGSC—Summary and Updates
4. Alternative Approaches and Future Directions
4.1. Syngeneic Cell Lines
4.2. Organoids
4.3. Somatic Gene Editing
4.4. Tumour-on-a-Chip
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HGSC | High-grade serous epithelial ovarian cancer |
| TCGA | The Cancer Genome Atlas |
| GEMM | Genetically engineered mouse model |
| OC | Ovarian cancer |
| OSE | Ovarian surface epithelia |
| Amhr2 | Anti-Mullerian hormone receptor 2 gene |
| ADCRE | Adenoviral CRE |
| Pax8 | Paired box 8 gene |
| Ovgp1 | Oviductal glycoprotein 1 gene |
| PDX | Patient-derived xenograft |
| i.p. | Intra-peritoneal |
| s.c. | Sub-cutaneous |
| CAN | Copy number alterations |
| ECM | Extracellular matrix |
| CLOVAR | Classification of Ovarian Cancer |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, B.W.; Kleihues, P. World Cancer Report; IARC Press: Lyon, France, 2003. [Google Scholar]
- Yoneda, A.; Lendorf, M.E.; Couchman, J.R.; Multhaupt, H.A. Breast and Ovarian Cancers: A Survey and Possible Roles for the Cell Surface Heparan Sulfate Proteoglycans. J. Histochem. Cytochem. 2012, 60, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Coburn, S.B.; Bray, F.; Sherman, M.E.; Trabert, B. International Patterns and Trends in Ovarian Cancer Incidence, Overall and by Histologic Subtype. Int. J. Cancer 2017, 140, 2451–2460. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, I.J.; Menon, U. Progress and Challenges in Screening for Early Detection of Ovarian Cancer. Mol. Cell Proteom. 2004, 3, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Badgwell, D.; Bast, R.C., Jr. Early Detection of Ovarian Cancer. Dis. Markers 2007, 23, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond Chemotherapy: Targeted Therapies in Ovarian Cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef]
- Shih, I.M.; Kurman, R.J. Ovarian Tumorigenesis: A Proposed Model Based on Morphological and Molecular Genetic Analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Köbel, M.; Kalloger, S.E.; Boyd, N.; McKinney, S.; Mehl, E.; Palmer, C.; Leung, S.; Bowen, N.J.; Lonescu, D.N.; Prentice, L.M.; et al. Ovarian Carcinoma Subtypes Are Different Diseases: Implications for Biomarker Studies. PLoS Med. 2008, 5, e232. [Google Scholar] [CrossRef]
- Ayhan, A.; Kurman, R.J.; Vang, R.; Logani, S.; Seidman, J.D.; Shih, I.M. Defining the Cut Point between Low-Grade and High-Grade Ovarian Serous Carcinomas: A Clinicopathologic and Molecular Genetic Analysis. Am. J. Surg. Pathol. 2009, 33, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Howell, V.M. Genetically Engineered Mouse Models for Epithelial Ovarian Cancer: Are We There Yet? Semin. Cell Dev. Biol. 2014, 27, 106–117. [Google Scholar] [CrossRef]
- Cheasley, D.; Wakefield, M.J.; Ryland, G.L.; Allan, P.E.; Alsop, K.; Amarasinghe, K.C.; Ananda, S.; Anglesio, M.S.; Au-Yeung, G.; Bowtell, D.D.; et al. The Molecular Origin and Taxonomy of Mucinous Ovarian Carcinoma. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gemignani, M.L.; Schlaerth, A.C.; Bogomolniy, F.; Barakat, R.R.; Lin, O.; Soslow, R.; Venkatraman, E.; Boyd, J. Role of Kras and Braf Gene Mutations in Mucinous Ovarian Carcinoma. Gynecol. Oncol. 2003, 90, 378–381. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Kommoss, S.; Tolcher, M.C.; Clarke, B.; Galletta, L.; Porter, H.; Damaraju, S.; Fereday, S.; Winterhoff, B.J.; Senz, J.; et al. Molecular Characterization of Mucinous Ovarian Tumours Supports a Stratified Treatment Approach with Her2 Targeting in 19% of Carcinomas. J. Pathol. 2013, 229, 111–120. [Google Scholar] [CrossRef]
- Kelemen, L.E.; Lawrenson, K.; Tyrer, J.; Li, Q.; Lee, J.M.; Seo, J.H.; Phelan, C.M.; Beesley, J.; Chen, X.; Aben, K.K.; et al. Genome-Wide Significant Risk Associations for Mucinous Ovarian Carcinoma. Nat. Genet. 2015, 47, 888–897. [Google Scholar]
- Ren, Y.A.; Mullany, L.K.; Liu, Z.; Herron, A.J.; Wong, K.K.; Richards, J.S. Mutant P53 Promotes Epithelial Ovarian Cancer by Regulating Tumor Differentiation, Metastasis, and Responsiveness to Steroid Hormones. Cancer Res. 2016, 76, 2206–2218. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Yang, W.; et al. Arid1a Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Anglesio, M.S.; Carey, M.S.; Kobel, M.; Mackay, H.; Huntsman, D.G. Clear Cell Carcinoma of the Ovary: A Report from the First Ovarian Clear Cell Symposium, June 24th, 2010. Gynecol. Oncol. 2011, 121, 407–415. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Mandai, M.; Oura, T.; Matsumura, N.; Hamanishi, J.; Baba, T.; Matsui, S.; Murphy, S.K.; Konishi, I. Identification of an Ovarian Clear Cell Carcinoma Gene Signature That Reflects Inherent Disease Biology and the Carcinogenic Processes. Oncogene 2010, 29, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, Y.; Tsuda, H.; Inoue, T.; Berkowitz, R.S.; Mok, S.C. Pten Expression in Clear Cell Adenocarcinoma of the Ovary. Gynecol. Oncol. 2006, 101, 71–75. [Google Scholar] [CrossRef]
- McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; Yang, W.; Senz, J.; Chow, C.; Heravi-Moussavi, A.; Morin, G.B.; Mes-Masson, A.M.; Carey, M.S.; et al. Subtype-Specific Mutation of Ppp2r1a in Endometrial and Ovarian Carcinomas. J. Pathol. 2011, 223, 567–573. [Google Scholar] [CrossRef]
- Catasús, L.; Bussaglia, E.; Rodríguez, I.; Gallardo, A.; Pons, C.; Irving, J.A.; Prat, J. Molecular Genetic Alterations in Endometrioid Carcinomas of the Ovary: Similar Frequency of Beta-Catenin Abnormalities but Lower Rate of Microsatellite Instability and Pten Alterations Than in Uterine Endometrioid Carcinomas. Hum. Pathol. 2004, 35, 1360–1368. [Google Scholar] [CrossRef]
- Dinulescu, D.M.; Ince, T.A.; Quade, B.J.; Shafer, S.A.; Crowley, D.; Jacks, T. Role of K-Ras and Pten in the Development of Mouse Models of Endometriosis and Endometrioid Ovarian Cancer. Nat. Med. 2005, 11, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Hendrix-Lucas, N.; Kuick, R.; Zhai, Y.; Schwartz, D.R.; Akyol, A.; Hanash, S.; Misek, D.E.; Katabuchi, H.; Williams, B.O.; et al. Mouse Model of Human Ovarian Endometrioid Adenocarcinoma Based on Somatic Defects in the Wnt/Β-Catenin and Pi3k/Pten Signaling Pathways. Cancer Cell 2007, 11, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Zhai, Y.; Kuick, R.; Karnezis, A.N.; Garcia, P.; Naseem, A.; Hu, T.C.; Fearon, E.R.; Cho, K.R. Impact of Oviductal Versus Ovarian Epithelial Cell of Origin on Ovarian Endometrioid Carcinoma Phenotype in the Mouse. J. Pathol. 2016, 240, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nakayama, K.; Ishibashi, T.; Ishikawa, N.; Ishikawa, M.; Katagiri, H.; Minamoto, T.; Sato, E.; Sanuki, K.; Yamashita, H.; et al. Kras/Braf Analysis in Ovarian Low-Grade Serous Carcinoma Having Synchronous All Pathological Precursor Regions. Int. J. Mol. Sci. 2016, 17, 625. [Google Scholar] [CrossRef] [Green Version]
- Singer, G.; Oldt, R., 3rd; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, I.M. Mutations in Braf and Kras Characterize the Development of Low-Grade Ovarian Serous Carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [Green Version]
- Mullany, L.K.; Fan, H.Y.; Liu, Z.; White, L.D.; Marshall, A.; Gunaratne, P.; Anderson, M.L.; Creighton, C.J.; Xin, L.; Deavers, M.; et al. Molecular and Functional Characteristics of Ovarian Surface Epithelial Cells Transformed by Krasg12d and Loss of Pten in a Mouse Model in Vivo. Oncogene 2011, 30, 3522–3536. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; DeFazio, A.; et al. Driver Mutations in Tp53 Are Ubiquitous in High Grade Serous Carcinoma of the Ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of Brca1 and Brca2 Mutations with Survival, Chemotherapy Sensitivity, and Gene Mutator Phenotype in Patients with Ovarian Cancer. Jama 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Milea, A.; George, S.H.; Matevski, D.; Jiang, H.; Madunic, M.; Berman, H.K.; Gauthier, M.L.; Gallie, B.; Shaw, P.A. Retinoblastoma Pathway Deregulatory Mechanisms Determine Clinical Outcome in High-Grade Serous Ovarian Carcinoma. Mod. Pathol. 2014, 27, 991–1001. [Google Scholar] [CrossRef] [Green Version]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-Grade Serous Carcinomas Arise in the Mouse Oviduct Via Defects Linked to the Human Disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both Fallopian Tube and Ovarian Surface Epithelium Are Cells-of-Origin for High-Grade Serous Ovarian Carcinoma. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilks, C.B.; Ionescu, D.N.; Kalloger, S.E.; Köbel, M.; Irving, J.; Clarke, B.; Santos, J.; Le, N.; Moravan, V.; Swenerton, K. Tumor Cell Type Can Be Reproducibly Diagnosed and Is of Independent Prognostic Significance in Patients with Maximally Debulked Ovarian Carcinoma. Hum. Pathol. 2008, 39, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D. The Genesis and Evolution of High-Grade Serous Ovarian Cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically Relevant Gene Signatures of High-Grade Serous Ovarian Carcinoma. J. Clin. Invest. 2013, 123, 517–525. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and Therapeutic Relevance of Molecular Subtypes in High-Grade Serous Ovarian Cancer. J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Jing, Y.; Zhang, M.; Zhang, Z.; Ma, P.; Peng, H.; Shi, K.; Gao, W.Q.; Zhuang, G. Stroma-Associated Master Regulators of Molecular Subtypes Predict Patient Prognosis in Ovarian Cancer. Sci. Rep. 2015, 5, 16066. [Google Scholar] [CrossRef] [Green Version]
- Schwede, M.; Waldron, L.; Mok, S.C.; Wei, W.; Basunia, A.; Merritt, M.A.; Mitsiades, C.S.; Parmigiani, G.; Harrington, D.P.; Quackenbush, J.; et al. The Impact of Stroma Admixture on Molecular Subtypes and Prognostic Gene Signatures in Serous Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.H.; Bast, R.C. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [Green Version]
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian Surface Epithelium: Biology, Endocrinology, and Pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Jarboe, E.; Folkins, A.; Nucci, M.R.; Kindelberger, D.; Drapkin, R.; Miron, A.; Lee, Y.; Crum, C.P. Serous Carcinogenesis in the Fallopian Tube: A Descriptive Classification. Int. J. Gynecol. Pathol. 2008, 27, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih, I.M. Tp53 Mutations in Serous Tubal Intraepithelial Carcinoma and Concurrent Pelvic High-Grade Serous Carcinoma—Evidence Supporting the Clonal Relationship of the Two Lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.Y.; Liu, Z.; Paquet, M.; Wang, J.; Lydon, J.P.; DeMayo, F.J.; Richards, J.S. Cell Type–Specific Targeted Mutations of Kras and Pten Document Proliferation Arrest in Granulosa Cells Versus Oncogenic Insult to Ovarian Surface Epithelial Cells. Cancer Res. 2009, 69, 6463–6472. [Google Scholar] [CrossRef] [Green Version]
- Jamin, S.P.; Arango, N.A.; Mishina, Y.; Hanks, M.C.; Behringer, R.R. Requirement of Bmpr1a for Müllerian Duct Regression During Male Sexual Development. Nat. Genet. 2002, 32, 408–410. [Google Scholar] [CrossRef]
- Xing, D.; Scangas, G.; Nitta, M.; He, L.; Xu, X.; Ioffe, Y.J.; Aspuria, P.J.; Hedvat, C.Y.; Anderson, M.L.; Oliva, E.; et al. A Role for Brca1 in Uterine Leiomyosarcoma. Cancer Res. 2009, 69, 8231–8235. [Google Scholar] [CrossRef] [Green Version]
- Flesken-Nikitin, A.; Choi, K.C.; Eng, J.P.; Shmidt, E.N.; Nikitin, A.Y. Induction of Carcinogenesis by Concurrent Inactivation of P53 and Rb1 in the Mouse Ovarian Surface Epithelium. Cancer Res. 2003, 63, 3459–3463. [Google Scholar]
- Šale, S.; Orsulic, S. Models of Ovarian Cancer Metastasis: Murine Models. Drug Discov. Today Dis. Models 2006, 3, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Mittag, J.; Winterhager, E.; Bauer, K.; Grummer, R. Congenital Hypothyroid Female Pax8-Deficient Mice Are Infertile Despite Thyroid Hormone Replacement Therapy. Endocrinology 2007, 148, 719–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, N.J.; Logani, S.; Dickerson, E.B.; Kapa, L.B.; Akhtar, M.; Benigno, B.B.; McDonald, J.F. Emerging Roles for Pax8 in Ovarian Cancer and Endosalpingeal Development. Gynecol. Oncol. 2007, 104, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Tacha, D.; Zhou, D.; Cheng, L. Expression of Pax8 in Normal and Neoplastic Tissues: A Comprehensive Immunohistochemical Study. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Laury, A.R.; Hornick, J.L.; Perets, R.; Krane, J.F.; Corson, J.; Drapkin, R.; Hirsch, M.S. Pax8 Reliably Distinguishes Ovarian Serous Tumors from Malignant Mesothelioma. Am. J. Surg. Pathol. 2010, 34, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, E.B.; Verhage, H.G.; Jaffe, R.C. Complementary Deoxyribonucleic Acid Cloning and Molecular Characterization of an Estrogen-Dependent Human Oviductal Glycoprotein1. Biol. Reprod. 1994, 51, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCool, K.W.; Freeman, Z.T.; Zhai, Y.; Wu, R.; Hu, K.; Liu, C.J.; Tomlins, S.A.; Fearon, E.R.; Magnuson, B.; Kuick, R.; et al. Murine Oviductal High-Grade Serous Carcinomas Mirror the Genomic Alterations, Gene Expression Profiles, and Immune Microenvironment of Their Human Counterparts. Cancer Res. 2020, 80, 877–889. [Google Scholar] [CrossRef]
- Lawrenson, K.; Fonseca, M.A.; Segato, F.; Lee, J.M.; Corona, R.I.; Seo, J.H.; Coetzee, S.; Lin, Y.G.; Pejovic, T.; Mhawech-Fauceglia, P.; et al. Integrated Molecular Profiling Studies to Characterize the Cellular Origins of High-Grade Serous Ovarian Cancer. bioRxiv 2018, 330597. [Google Scholar] [CrossRef] [Green Version]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular Analysis of High-Grade Serous Ovarian Carcinoma with and without Associated Serous Tubal Intra-Epithelial Carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative Proteomic Profiling of Ovarian Cancer Cell Lines Reveals Precursor Cell Associated Proteins and Functional Status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Hao, D.; Li, J.; Jia, S.; Meng, Y.; Zhang, C.; Wang, L.; Di, L.J. Integrated Analysis Reveals Tubal- and Ovarian-Originated Serous Ovarian Cancer and Predicts Differential Therapeutic Responses. Clin. Cancer Res. 2017, 23, 7400–7411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, A.; Tan, S.; Singh, G.; Rizk, P.; Swathi, Y.; Tan, T.Z.; Huang, R.Y.J.; Leushacke, M.; Barker, N. Lgr5 Marks Stem/Progenitor Cells in Ovary and Tubal Epithelia. Nat. Cell Biol. 2014, 16, 745–757. [Google Scholar] [CrossRef]
- Connolly, D.C.; Bao, R.; Nikitin, A.Y.; Stephens, K.C.; Poole, T.W.; Hua, X.; Harris, S.S.; Vanderhyden, B.C.; Hamilton, T.C. Female Mice Chimeric for Expression of the Simian Virus 40 Tag under Control of the Misiir Promoter Develop Epithelial Ovarian Cancer. Cancer Res. 2003, 63, 1389–1397. [Google Scholar]
- Hensley, H.; Quinn, B.A.; Wolf, R.L.; Litwin, S.L.; Mabuchi, S.; Williams, S.J.; Williams, C.; Hamilton, T.C.; Connolly, D.C. Magnetic Resonance Imaging for Detection and Determination of Tumor Volume in a Genetically Engineered Mouse Model of Ovarian Cancer. Cancer Biol. Ther. 2007, 6, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, S.; Altomare, D.A.; Connolly, D.C.; Klein-Szanto, A.; Litwin, S.; Hoelzle, M.K.; Hensley, H.H.; Hamilton, T.C.; Testa, J.R. Rad001 (Everolimus) Delays Tumor Onset and Progression in a Transgenic Mouse Model of Ovarian Cancer. Cancer Res. 2007, 67, 2408–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.J.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, P53, and Brca1 or Brca2 Cooperate in Inducing Metastatic Serous Epithelial Ovarian Cancer. Cancer Res. 2012, 72, 4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laviolette, L.A.; Garson, K.; Macdonald, E.A.; Senterman, M.K.; Courville, K.; Crane, C.A.; Vanderhyden, B.C. 17β-Estradiol Accelerates Tumor Onset and Decreases Survival in a Transgenic Mouse Model of Ovarian Cancer. Endocrinology 2010, 151, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; LaBarge, M.A. Context, Tissue Plasticity, and Cancer: Are Tumor Stem Cells Also Regulated by the Microenvironment? Cancer Cell 2005, 7, 17–23. [Google Scholar]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal Fibroblasts in Cancer Initiation and Progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Bissell, M.J.; Kenny, P.A.; Radisky, D.C. Microenvironmental Regulators of Tissue Structure and Function Also Regulate Tumor Induction and Progression: The Role of Extracellular Matrix and Its Degrading Enzymes. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2005. [Google Scholar]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. Tgf-Β Modulates Ovarian Cancer Invasion by Upregulating Caf-Derived Versican in the Tumor Microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A Collagen-Remodeling Gene Signature Regulated by Tgf-Β Signaling Is Associated with Metastasis and Poor Survival in Serous Ovarian Cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, D.C.; Hensley, H.H. Xenograft and Transgenic Mouse Models of Epithelial Ovarian Cancer and Non-Invasive Imaging Modalities to Monitor Ovarian Tumor Growth in Situ: Applications in Evaluating Novel Therapeutic Agents. Curr. Protoc. Pharmacol. 2009, 45, Unit14.12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.M.; Annunziata, C.M. Characterization of Ovarian Cancer Cell Lines as in Vivo Models for Preclinical Studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. Crispr/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef] [Green Version]
- Walton, J.B.; Farquharson, M.; Mason, S.; Port, J.; Kruspig, B.; Dowson, S.; Stevenson, D.; Murphy, D.; Matzuk, M.; Kim, J.; et al. Crispr/Cas9-Derived Models of Ovarian High Grade Serous Carcinoma Targeting Brca1, Pten and Nf1, and Correlation with Platinum Sensitivity. Sci. Rep. 2017, 7, 16827. [Google Scholar] [CrossRef]
- Maniati, E.; Berlato, C.; Gopinathan, G.; Heath, O.; Kotantaki, P.; Lakhani, A.; McDermott, J.; Pegrum, C.; Delaine-Smith, R.M.; Pearce, O.M.; et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020, 30, 525–540. [Google Scholar] [CrossRef] [Green Version]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a Syngeneic Mouse Model for Events Related to Ovarian Cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An Organoid Platform for Ovarian Cancer Captures Intra- and Interpatient Heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with Crispr–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Öllinger, R.; Friedrich, M.; Ehmer, U.; Barenboim, M.; Steiger, K.; Heid, I.; Mueller, S.; Maresch, R.; Engleitner, T.; et al. Crispr/Cas9 Somatic Multiplex-Mutagenesis for High-Throughput Functional Cancer Genomics in Mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13982–13987. [Google Scholar] [CrossRef] [Green Version]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed Pancreatic Genome Engineering and Cancer Induction by Transfection-Based Crispr/Cas9 Delivery in Mice. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic Crispr/Cas9-Mediated Tumour Suppressor Disruption Enables Versatile Brain Tumour Modelling. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; de Stanchina, E.; et al. In Vivo Engineering of Oncogenic Chromosomal Rearrangements with the Crispr/Cas9 System. Nature 2014, 516, 423–427. [Google Scholar] [CrossRef] [Green Version]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.J.; Mitros, T.; Muñoz, D.P.; Boffelli, D.; et al. Selection-Free Genome Editing of the Sickle Mutation in Human Adult Hematopoietic Stem/Progenitor Cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome Editing with Cas9 in Adult Mice Corrects a Disease Mutation and Phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [Green Version]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. Crispr–Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 Inhibits Crispr–Cas9 Engineering in Human Pluripotent Stem Cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, D.; MHattersley, S.; DStafford, N.; JHaswell, S.; Greenman, J. Development of Microfluidic-Based Analytical Methodology for Studying the Effects of Chemotherapy Agents on Cancer Tissue. Curr. Anal. Chem. 2013, 9, 2–8. [Google Scholar] [CrossRef]
- Trujillo-de Santiago, G.; Flores-Garza, B.G.; Tavares-Negrete, J.A.; Lara-Mayorga, I.M.; González-Gamboa, I.; Zhang, Y.S.; Rojas-Martínez, A.; Ortiz-López, R.; Álvarez, M.M. The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors. Materials 2019, 12, 2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, L.; Zhu, Z.; Xu, Z.; He, T.; Li, E.; Guo, Z.; Liu, F.; Jiang, C.; Wang, Q. Cancer Associated Fibroblast-Derived Hepatocyte Growth Factor Inhibits the Paclitaxel-Induced Apoptosis of Lung Cancer A549 Cells by up-Regulating the Pi3k/Akt and Grp78 Signaling on a Microfluidic Platform. PLoS ONE 2015, 10, e0129593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Tu, T.Y.; Kim, C.; Thiery, J.P.; Kamm, R.D. Identification of Drugs as Single Agents or in Combination to Prevent Carcinoma Dissemination in a Microfluidic 3d Environment. Oncotarget 2015, 6, 36603–36614. [Google Scholar] [CrossRef] [Green Version]
- de Almeida Monteiro Melo Ferraz, M.; Nagashima, J.B.; Venzac, B.; Le Gac, S.; Songsasen, N. A Dog Oviduct-on-a-Chip Model of Serous Tubal Intraepithelial Carcinoma. Sci. Rep. 2020, 10, 1575. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Subtype | Frequency | Molecular Features | GEMMs |
|---|---|---|---|
| Mucinous | <5% | CDKN2A copy number alterations [12] KRAS, TP53, RNF43, BRAF, PIK3CA, ARID1A [12,13] mutations ERBB2 [12], HER2 [14], HOXD9 [15] amplification | Amhr2-Cre; LSL-KrasG12D/+PtenloxP/loxP; Trp53R172H/+ [16] |
| Clear cell | ~10% | ARID1A [17] and PIK3CA [18] mutations Ubiquitous HNF1β expression [19] Loss of PTEN expression [20] | No GEMMs |
| Endometrioid | ~10% | ARID1a [17] and PPP2R1A [21] mutations PTEN alterations [22] | (ADCRE) - LSL-K-rasG12D/+PtenloxP/loxP [23] (ADCRE) - ApcloxP/loxP;PtenloxP/loxP [24] Ovgp1-iCreERT2;ApcloxP/loxP; PtenloxP [25] |
| Low-grade serous | <5% | KRAS, BRAF, ERBB2 mutations [26,27] | Amhr2-Cre; LSL-KrasG12D/+; PtenloxP/loxP [28] |
| High-grade serous | ~70% | TP53 [29,30] and BRCA1/2 [30,31] mutations CCNE1 and RB1 [32] aberrations | Older GEMMs reviewed in detail (Howell) Most recent HGSC GEMMS: Pax8-rtTA; TetO-Cre; Brca1loxP/loxP; Trp53mut; PtenloxP/loxP [33] Ovgp1-iCreERT2; Brca1loxP/loxP; Trp53loxP/loxP; Rb1loxP/loxP; Nf1 loxP/loxP [34] Lgr5-Cre; Trp53R172H/+; T121 [35] |
| Syngeneic Cell Line | Engraftment Location | Median Survival (days) |
|---|---|---|
| ID8 | i.p. | 101 * |
| ID8Trp53-/- | i.p. | 47 * |
| ID8Trp53-/-;Brca2-/- | i.p. | 57 * |
| ID8Trp53-/-;Brca1-/- | i.p. | 46 ^ |
| ID8Trp53-/-;Pten-/- | i.p. | 34 ^ |
| ID8Trp53-/-;Pten+/- | i.p. | 40.5 ^ |
| ID8Trp53-/-;Nf1-/- | i.p. | 36.5 ^ |
| ID8Trp53-/-;Brca2-/-;Pten-/- | i.p. | 40 ^ |
| 60577: p53-/-;Brca1-/- | i.p. | 36 # |
| 30200: p53-/-;Brca1-/- | i.p. | 77 # |
| HGS1: Pax8-Cre;p53-/-;Pten-/-;Brca2-/- | i.p. | 91 # |
| HGS2: Pax8-Cre;p53-/-;Pten-/-;Brca2-/- | i.p. | 80.5 # |
| HGS3: Pax8-Cre;p53-/-;Pten-/-;Brca2+/- | i.p. | 87.5 # |
| HGS4: Pax8-Cre;p53-/-;Pten-/-;Brca2+/- | i.p. | 80.5 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakarya, R.; Howell, V.M.; Colvin, E.K. Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches. Int. J. Mol. Sci. 2020, 21, 4806. https://doi.org/10.3390/ijms21134806
Zakarya R, Howell VM, Colvin EK. Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches. International Journal of Molecular Sciences. 2020; 21(13):4806. https://doi.org/10.3390/ijms21134806
Chicago/Turabian StyleZakarya, Razia, Viive M. Howell, and Emily K. Colvin. 2020. "Modelling Epithelial Ovarian Cancer in Mice: Classical and Emerging Approaches" International Journal of Molecular Sciences 21, no. 13: 4806. https://doi.org/10.3390/ijms21134806