Synergistic Activation of Toll-Like and NOD Receptors by Complementary Antigens as Facilitators of Autoimmune Disease: Review, Model and Novel Predictions


Abstract
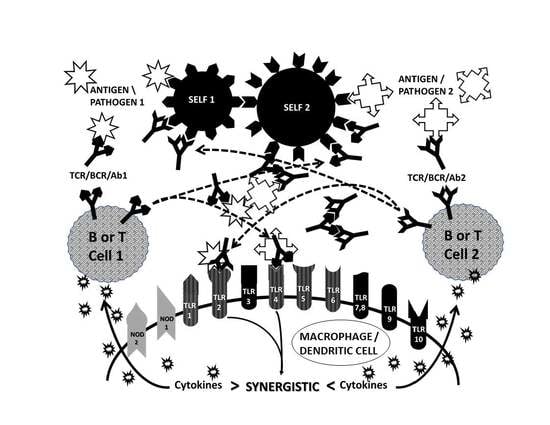
:
1. Introduction
2. Toll-Like Receptor Activation in Autoimmune Disease
2.1. Normal Innate Immune Function
2.2. Evidence for an Essential Role of Innate Immunity in Autoimmune Disease
2.3. Limited Substitutability of One Adjuvant for Another in AD Induction Related to TLR/NOD Activation Profiles
2.4. Difficulties Explaining the Roles of Innate Immunity in Autoimmune Disease
2.5. Complementary Antigen Theory of Autoimmunity
2.6. Synergisms with the TLR and NOD Signaling Pathways
2.7. Complementarity of Antigens in the Induction of AD
2.8. Complementary Antigen Theory Integrated with TLR/NOD Synergisms
2.9. Antigenic Complementarity Drives Ongoing Innate Activation Throughout the Course of AD
2.10. Implications of Synergistic TLR Activation for Therapeutics
2.11. Unresolved Problems
3. Summary with Tests and Predictions that Differentiate CAT from other AD Theories
Funding
Conflicts of Interest
References
- Frizinsky, S.; Haj-Yahia, S.; Machnes Maayan, D.; Lifshitz, Y.; Maoz-Segal, R.; Offengenden, I.; Kidon, M.; Agmon-Levin, N. The innate immune perspective of autoimmune and autoinflammatory conditions. Rheumatology 2019, 58, vi1–vi8. [Google Scholar] [CrossRef] [Green Version]
- Correa, R.G.; Milutinovic, S.; Reed, J.C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep. 2012, 32(6), 597–608. [Google Scholar] [CrossRef]
- Mohammad Hosseini, A.; Majidi, J.; Baradaran, B.; Yousefi, M. Toll-like receptors in the pathogenesis of autoimmune diseases. Adv. Pharm. Bull. 2015, 5 (Suppl. 1), 605. [Google Scholar] [CrossRef]
- Dabbagh, K.; Lewis, D.B. Toll-like Receptors and T-helper-1/T-helper-2 Responses. Curr. Opin. Infect. Dis. 2003, 16, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Tukhvatulin, A.; Gitlin, I.; Shcheblyakov, D.; Artemicheva, N.; Burdelya, L.; Shmarov, M.; Naroditsky, B.; Gudkov, A.; Gintsburg, A.; Logunov, D. Combined stimulation of Toll-Like receptor 5 and NOD1 strongly potentiates activity of NF-B, resulting in enhanced innate immune reactions and resistance to Salmonella enterica Serovar Typhimurium infection. Infect. Immun. 2013, 81, 3855–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, L.O.; Zamboni, D.S. NOD1 and NOD2 signaling in infection and inflammation. Front. Immunol. 2012, 3, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapetanovic, R.; Cavaillon, J.-M. Early events in innate immunity in recognition of microbial pathogens. Expert Opin. Biol. Ther. 2007, 7, 907–918. [Google Scholar] [CrossRef]
- Papadimitraki, E.D.; Bertsias, G.K.; Boumpas, D.T. Toll like receptors and autoimmunity: A critical appraisal. J. Autoimmun. 2007, 29, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Waldner, H. The role of innate immune responses in autoimmune disease development. Autoimmun. Rev. 2009, 8, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, Y.; Feng, G.; Su, S.B. The critical role of Toll-like receptor signaling pathways in the induction and progression of autoimmune diseases. Curr. Mol. Med. 2009, 9, 365–374. [Google Scholar] [CrossRef]
- Tomer, Y. Genetic susceptibility to autoimmune thyroid disease: Past; present; and future. Thyroid 2010, 20, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Duffy, L.; O’Reilly, S.C. Toll-like receptors in the pathogenesis of autoimmune diseases: Recent and emerging translational developments. Immuno Targets Ther. 2016, 5, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Hänninen, A. Infections in MS: An innate immunity perspective. Acta Neurol. Scand. 2017, 136 (Suppl. 201), 10–14. [Google Scholar] [CrossRef]
- Kiripolsky, J.; McCabe, L.G.; Kramer, J.M. Innate immunity in Sjögren’s syndrome. Clin. Immunol. 2017, 182, 4–13. [Google Scholar] [CrossRef]
- Sozzani, S.; Del Prete, A.; Bosisio, D. Dendritic cell recruitment and activation in autoimmunity. J. Autoimmun. 2017, 85, 126–140. [Google Scholar] [CrossRef]
- Matta, B.; Song, S.; Li, D.; Barnes, B.J. Interferon regulatory factor signaling in autoimmune disease. Cytokine. 2017, 98, 15–26. [Google Scholar] [CrossRef]
- Brown, M.A.; Weinberg, R.B. Mast cells and innate lymphoid cells: Underappreciated players in CNS autoimmune demyelinating disease. Front. Immunol. 2018, 9, 514. [Google Scholar] [CrossRef] [Green Version]
- Xiong, T.; Turner, J.E. Innate lymphoid cells in autoimmunity and chronic inflammatory diseases. Semin. Immunopathol. 2018, 40, 393–406. [Google Scholar] [CrossRef]
- Vignesh, P.; Rawat, A.; Sharma, M.; Singh, S. Complement in autoimmune diseases. Clin. Chim. Acta 2017, 465, 123–130. [Google Scholar] [CrossRef]
- Cojocaru, M.; Serbănescu, A.; Cojocaru, I.M. Changes of serum complement and of circulating immune complexes in patients with multiple sclerosis. Rom. J. Intern. Med. 1993, 31, 131. [Google Scholar]
- Sakurai, T.; Fujita, T.; Kono, I.; Kabashima, T.; Yamane, K.; Tamura, N.; Kashiwagi, H. Complement-mediated solubilization of immune complexes in systemic lupus erythematosus. Clin. Exp. Immunol. 1982, 48, 37–42. [Google Scholar]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by coxsackie B Viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Heidecker, B.; Kittleson, M.M.; Kasper, E.K.; Wittstein, I.S.; Champion, H.C.; Russell, S.D.; Hruban, R.H.; Rodriguez, E.R.; Baughman, K.L.; Hare, J.M. Transcriptomic biomarkers for the accurate diagnosis of myocarditis. Circulation 2011, 123, 1174–1184. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, U.; Kurrer, M.O.; Sonderegger, I.; Iezzi, G.; Tafuri, A.; Hunziker, L.; Suzuki, S.; Bachmaier, K.; Bingisser, R.M.; Penninger, J.M.; et al. Activation of dendritic cells through the Interleukin 1 Receptor 1 is critical for the induction of autoimmune myocarditis. J. Exp. Med. 2003, 197, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Roberts, B.J.; Dragon, J.A.; Moussawi, M.; Huber, S.A. Sex-specific signaling through Toll-Like Receptors 2 and 4 contributes to survival outcome of coxsackievirus B3 Infection in C57Bl/6 Mice. Biol. Sex. Differ. 2012, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Fairweather, D.; Fisancho-Kiss, S.; Rose, N.R. Viruses as adjuvants for autoimmunity: Evidence from coxsackievirus-induced myocarditis. Rev. Med. Virol. 2005, 15, 17–27. [Google Scholar] [CrossRef]
- Zhang, P.; Cox, C.J.; Alvarez, K.M.; Cunningham, M.W. Cutting edge: Cardiac myosin activates innate immune responses through Toll-like receptors. J. Immunol. 2009, 183, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Pagni, P.P.; Traub, S.; Demaria, O.; Chasson, L.; Alexopoulou, L. Contribution of TLR7and TLR9 signaling to the susceptibility of MyD88-deficient mice to myocarditis. Autoimmunity 2010, 43, 275–287. [Google Scholar] [CrossRef]
- Pahlman, L.I.; Morgelin, M.; Eckert, J.; Johansson, L.; Russell, W.; Riesbeck, K.; Soehnlein, O.; Lindbom, L.; Norrby-Teglund, A.; Schumann, R.R.; et al. Streptococcal M protein: A multipotent and powerful inducer of inflammation. J. Immunol. 2006, 177, 1221–1228. [Google Scholar] [CrossRef] [Green Version]
- Rose, N.R. The adjuvant effect in infection and autoimmunity. Clin. Rev. Allergy Immunol. 2008, 34, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef]
- Bao, J.; Sun, T.; Yue, Y.; Xiong, S. Macrophage NLRP3 inflammasome activated by CVB3 capsid proteins contributes to the development of viral myocarditis. Mol. Immunol. 2019, 114, 41–48. [Google Scholar] [CrossRef]
- Tschöpe, C.; Müller, I.; Xia, Y.; Savvatis, K.; Pappritz, K.; Pinkert, S.; Lassner, D.; Heimesaat, M.M.; Spillmann, F.; Miteva, K.; et al. NOD2 (Nucleotide-Binding Oligomerization Domain 2) is a major pathogenic mediator of coxsackievirus B3-induced myocarditis. Circ. Heart Fail. 2017, 10, e003870. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, Y.; Wang, C.; Jiang, B. Rotavirus and coxsackievirus infection activated different profiles of Toll-like receptors and chemokines in intestinal epithelial cells. Inflamm. Res. 2009, 58, 585–592. [Google Scholar] [CrossRef]
- Robinet, M.; Maillard, S.; Cron, M.A.; Berrih-Aknin, S.; Le Panse, R. Review on Toll-like receptor activation in myasthenia gravis: Application to the development of new experimental models. Clin. Rev. Allergy Immunol. 2017, 52, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Robinet, M.; Villeret, B.; Maillard, S.; Cron, M.A.; Berrih-Aknin, S.; Le Panse, R. Use of Toll-like receptor agonists to induce ectopic lymphoid structures in myasthenia gravis mouse models. Front. Immunol. 2017, 8, 1029. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhao, C.; Chen, X.; Jiang, L.; Su, X. Monocytes primed with GTS-21/α7 nAChR (nicotinic acetylcholine receptor) agonist develop anti-inflammatory memory. QJM: Inter. J. Med. 2017, 110, 437–445. [Google Scholar] [CrossRef]
- Cavalcante, P.; Galbardi, B.; Franzi, S.; Marcuzzo, S.; Barzago, C.; Bonanno, S.; Camera, G.; Maggi, L.; Kapetis, D.; Andreetta, F.; et al. Increased expression of Toll-like receptors 7 and 9 in myasthenia gravis thymus characterized by active Epstein-Barr virus infection. Immunobiology 2016, 221, 516–527. [Google Scholar] [CrossRef]
- Deerhake, M.E.; Biswas, D.D.; Barclay, W.E.; Shinohara, M.L. Pattern recognition receptors in multiple sclerosis and its animal models. Front. Immunol. 2019, 10, 2644. [Google Scholar] [CrossRef] [Green Version]
- Wolf, N.A.; Amouzegar, T.K.; Swanborg, R.H. Synergistic interaction between Toll-like receptor agonists is required for induction of experimental autoimmune encephalomyelitis in Lewis rats. J. Neuroimmunol. 2007, 185, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Lenz, D.C.; Lu, L.; Conant, S.B.; Wolf, N.A.; Gérard, H.C.; Whittum-Hudson, J.A.; Hudson, A.P.; Swanborg, R.H. A Chlamydia pneumoniae-specific peptide induces experimental autoimmune encephalomyelitis in rats. J. Immunol. 2001, 167, 1803–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, C.; Liu, Y.-J.; Kobayashi, K.S. Muramyl dipeptide and its derivatives: Peptide adjuvant in immunological disorders and cancer therapy. Curr. Bioact. Compd. 2011, 7, 180–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehori, J.; Fukase, K.; Akazawa, T.; Uematsu, S.; Akira, S.; Funami, K.; Singai, M.; Matsumoto, M.; Azuma, I.; Toyoshima, K.; et al. Dendritic cell maturation induced by muramyl dipeptide (MDP) derivatives: Monoacylated MDP confers TLR2/TLR4 activation. J. Immunol. 2005, 174, 7096–7103. [Google Scholar] [CrossRef]
- Gelbart, T.; West, C. Synergy between TLR2 and TLR4: A safety mechanism. Blood Cells Mol. Dis. 2001, 27, 728–730. [Google Scholar]
- Deng, Y.N.; Zhou, W.B. Expression of TLR4 and TLR9 mRNA in Lewis rats with experimental allergic neuritis. Neuroimmunomodulation 2007, 14, 337–343. [Google Scholar] [CrossRef]
- Marta, M.; Andersson, A.; Isaksson, M.; Kämpe, O.; Lobell, A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2008, 38, 565–575. [Google Scholar] [CrossRef]
- Miranda-Hernandez, S.; Gerlach, N.; Fletcher, J.M.; Biros, E.; Mack, M.; Körner, H.; Baxter, A.G. Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Segal, B.M.; Chang, J.T.; Shevach, E.M. CpG oligonucleotides are potent adjuvants for the activation of autoreactive encephalitogenic T cells in vivo. J. Immunol. 2000, 164, 5683–5688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelofs, M.; Joosten, L.; Abdollahi-Roodsaz, S. Expression of Toll-like receptor (TLR) 2, TLR3, TLR4 and TLR7 is increased in rheumatoid arthritis synovium and regulates cytokine production by dendritic cells upon stimulation of TLR specific pathways. Arthritis Res. Ther. 2005, 7, 70. [Google Scholar] [CrossRef]
- Kim, S.J.; Chen, Z.; Chamberlain, N.D.; Essani, A.B.; Volin, M.V.; Amin, M.A.; Volkov, S.; Gravallese, E.M.; Arami, S.; Swedler, W.; et al. Ligation of TLR5 promotes myeloid cell infiltration and differentiation into mature osteoclasts in RA and experimental arthritis. J. Immunol. 2014, 193, 3902–3913. [Google Scholar] [CrossRef]
- Franca, R.; Vieira, S.M.; Talbot, J.; Peres, R.S.; Pinto, L.G.; Zamboni, D.S.; Louzada-Junior, P.; Cunha, F.Q.; Cunha, T.M. Expression and activity of NOD1 and NOD2/RIPK2 signalling in mononuclear cells from patients with rheumatoid arthritis. Scand. J. Rheumatol. 2016, 45, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi-Roodsaz, S.; Joosten, L.A.; Helsen, M.M.; Walgreen, B.; van Lent, P.L.; van den Bersselaar, L.A.; Koenders, M.I.; van den Berg, W.B. Shift from toll-like receptor 2 (TLR-2) toward TLR-4 dependency in the erosive stage of chronic streptococcal cell wall arthritis coincident with TLR-4-mediated interleukin-17 production. Arthritis Rheum. 2008, 58, 3753–3764. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Heinhuis, B.; Abdollahi-Roodsaz, S.; Ferwerda, G.; Lebourhis, L.; Philpott, D.J.; Nahori, M.A.; Popa, C.; Morre, S.A.; van der Meer, J.W.; et al. Differential Function of the NACHT-LRR (NLR) Members Nod1 and Nod2 in Arthritis. Proc. Natl. Acad. Sci. USA 2008, 105, 9017–9022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terato, K.; Ye, X.J.; Miyahara, H.; Cremer, M.A.; Griffiths, M.M. Induction by chronic autoimmune arthritis in DBA/1 mice by oral administration of type II collagen and Escherichia coli lipopolysaccharide. Br. J. Rheumatol. 1996, 35, 828–838. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi-Roodsaz, S.; Joosten, L.A.; Roelofs, M.F.; Radstake, T.R.; Matera, G.; Popa, C.; van der Meer, J.W.; Netea, M.G.; van den Berg, W.B. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007, 56, 2957–2967. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, M.; Liu, S. Collagen-induced arthritis models. Methods Mol. Biol. 2018, 1868, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Gorton, D.; Blyth, S.; Gorton, J.G.; Govan, B.; Ketheesan, N. An alternative technique for the induction of autoimmune valvulitis in a rat model of rheumatic heart disease. J. Immunol. Methods 2010, 355, 80–85. [Google Scholar] [CrossRef]
- Tifrea, D.F.; Pal, S.; le Bon, C.; Cocco, M.J.; Zoonens, M.; de la Maza, L.M. Improved protection against Chlamydia muridarum using the native major outer membrane protein trapped in Resiquimod-carrying amphipols and effects in protection with addition of a Th1 (CpG-1826) and a Th2 (Montanide ISA 720) adjuvant. Vaccine 2020, 30, 4412–4422. [Google Scholar] [CrossRef]
- Greenfield, E.A. Preparing and using adjuvants. Cold Spring Harb. Protoc. 2019, 2019. [Google Scholar] [CrossRef]
- Leuthard, D.S.; Duda, A.; Freiberger, S.N.; Weiss, S.; Dommann, I.; Fenini, G.; Contassot, E.; Kramer, M.F.; Skinner, M.A.; Kündig, T.M.; et al. Microcrystalline tyrosine and aluminum as adjuvants in allergen-specific immunotherapy protect from IgE-mediated reactivity in mouse models and act independently of inflammasome and TLR signaling. J. Immunol. 2018, 200, 3151–3159. [Google Scholar] [CrossRef] [Green Version]
- Hansen, B.S.; Hussain, R.Z.; Lovett-Racke, A.E.; Thomas, J.A.; Racke, M.K. Multiple toll-like receptor agonists act as potent adjuvants in the induction of autoimmunity. J. Neuroimmunol. 2006, 172, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Fairweather, D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J. Theor. Biol. 2015, 375, 101–123. [Google Scholar] [CrossRef] [Green Version]
- Root-Bernstein, R.; Fairweather, D. Complexities in the relationship between infection and autoimmunity. Curr. Allergy Asthma Rep. 2014, 14, 407. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, R.S.; Oldstone, M.B.; Wroblewska, Z.; Frankel, M.E.; Koprowski, H. Molecular mimicry in virus infection: Cross-reaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad Sci. USA 1983, 80, 2346–2350. [Google Scholar] [CrossRef] [Green Version]
- Donati, D. Viral infections and multiple sclerosis. Drug Discov. Today Dis. Models 2020. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W. T cell mimicry in inflammatory heart disease. Mol. Immunol. 2004, 40, 1121–1127. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Vonck, J.; Podufaly, A. Antigenic complementarity between coxsackie virus and streptococcus in the induction of rheumatic heart disease and autoimmune myocarditis. Autoimmunity 2009, 42, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gorska, A.; Urban, M.; Glowinska, B.; Kowalewski, M. Czy zakazenie aciorkowcem grupy A jest wylaczna infekcyjna przyczyna goraczki reumatycznej?—Opis przebiegu goraczki reumatycznej u 11-letniego chlopca ze wspolistniejaca infekcja wirusem Coxackie B1. [Is infection with group A streptococcus the only reason for rheumatic fever?—A case report of rheumatic fever coexisting with Coxsackie B1 virus infection]. Przegl. Lek. 1998, 55, 418–419. [Google Scholar] [PubMed]
- Root-Bernstein, R. Autoimmunity and the microbiome: T-cell receptor mimicry of "self" and microbial antigens mediates self tolerance in holobionts. BioEssays 2016, 38, 1068–1083. [Google Scholar] [CrossRef]
- De Groot, A.S.; Moise, L.; Liu, R.; Gutierrez, A.H.; Tassone, R.; Bailey-Kellogg, C.; Martin, W. Immune camouflage: Relevance to vaccines and human immunology. Hum. Vaccin Immunother. 2014, 10, 3570–3575. [Google Scholar] [CrossRef] [Green Version]
- Plotz, P.H. Autoantibodies are anti-idiotype antibodies to antiviral antibodies. Lancet 1983, 2, 824–826. [Google Scholar] [CrossRef]
- Erlanger, B.F.; Cleveland, W.L.; Wassermann, N.H.; Ku, H.H.; Hill, B.L.; Sarangarajan, R.; Rajagopalan, R.; Cayanis, E.; Edelman, I.S.; Penn, A.S. Auto-anti-idiotype: A basis for autoimmunity and a strategy for anti-receptor antibodies. Immunol. Rev. 1986, 94, 23–37. [Google Scholar] [CrossRef]
- Weremeichik, H.; Moraska, A.; Herzum, M.; Weller, A.; Huber, S.A. Naturally occurring anti-idiotypic antibodies--mechanisms for autoimmunity and immunoregulation? Heart J. 1991, 12 (Suppl. D), 154–157. [Google Scholar] [CrossRef] [PubMed]
- Paque, R.E. Role of anti-idiotypic antibodies in induction.; regulation.; and expression of coxsackievirus-induced myocarditis. Prog. Med. Virol. 1992, 39, 204–227. [Google Scholar]
- Lu, N.Q. A speculative view of immune recognition. Immunol. Invest. 1994, 23, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Sindhwani, R.; Rojkind, M. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J. Exp. Med. 1995, 181, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Scheerder, I.; Vandekerckhove, J.; Robbrecht, J.; Algoed, L.; De Buyzere, M.; De Langhe, J.; De Schrijver, G.; Clement, D. Post-cardiac injury syndrome and an increased humoral immune response against the major contractile proteins (actin and myosin). Am. J. Cardiol. 1985, 56, 631–633. [Google Scholar] [CrossRef]
- Grosman-Rimon, L.; Ajrawat, P.; Lioe, J.; Tumiati, L.C.; Rao, V.; Billia, F.; Chruscinski, A. Increases in serum autoantibodies after left ventricular assist device implantation. J. Card. Fail. 2019, 25, 301–306. [Google Scholar] [CrossRef]
- Vargas, M.E.; Watanabe, J.; Singh, S.J.; Robinson, W.H.; Barres, B.A. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc. Natl. Acad. Sci. USA 2010, 107, 11993–11998. [Google Scholar] [CrossRef] [Green Version]
- Olsson, T.; Sun, J.B.; Solders, G.; Xiao, B.G.; Höjeberg, B.; Ekre, H.P.; Link, H. Autoreactive T and B cell responses to myelin antigens after diagnostic sural nerve biopsy. J. Neurol. Sci. 1993, 117, 130–139. [Google Scholar] [CrossRef]
- Lehmann, P.V.; Forsthuber, T.; Miller, A.; Sercarz, E.E. Spreading of T cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992, 358, 155–157. [Google Scholar] [CrossRef]
- Powell, A.M.; Black, M.M. Epitope spreading: Protection from pathogens. but propagation of autoimmunity? Clin. Exp. Dermatol. 2001, 26, 427–432. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Rethinking molecular mimicry in rheumatic heart disease and autoimmune myocarditis: Laminin, collagen IV, CAR, and B1AR as initial targets of disease. Front. Pediatr. 2014, 2, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serreze, D.V.; Ottendorfer, E.W.; Ellis, T.M.; Gauntt, C.J.; Atkinson, M.A. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes 2000, 49, 708–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Herrath, M.G.; Fujinami, R.S.; Whitton, J.L. Microorganisms and autoimmunity: Making the barren field fertile? Nat. Rev. Microbiol. 2003, 1, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Kappler, J.; Marrack, P. Bystander virus infection prolongs activated T cell survival. J. Immunol. 1999, 162, 4527–4535. [Google Scholar]
- McCoy, L.; Tsunoda, I.; Fujinami, R.S. Multiple sclerosis and virus induced immune responses: Autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 2006, 39, 9–19. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krah, D.L.; Choppin, P.W. Mice immunized with measles virus develop antibodies to a cell surface receptor for binding virus. J. Virol. 1988, 62, 1565–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, L.M.; Goldman, M.; Lambert, P.H. The production of anti-idiotypic antibodies and of idiotype-anti-idiotype immune complexes after polyclonal activation induced by bacterial LPS. J. Immunol. 1982, 128, 2126–2233. [Google Scholar] [PubMed]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, L.Y.E.; Too, H.K.I.; Tan, E.L.; Chow, T.-K.V.; Shek, P.-C.L.; Tham, E.; Alonso, S. Antiviral activity of Lactobacillus reuteri Protectis against Coxsackievirus A and Enterovirus 71 infection in human skeletal muscle and colon cell lines. Virol. J. 2016, 13, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantú-Bernal, S.L.; Domínguez-Gámez, M.; Medina-Peraza, I.; Aros-Uzarraga, E.; Ontiveros, N.; Flores-Mendoza, L.; Gomez-Flores, R.; Tamez-Guerra, P.; González-Ochoa, G. Enhanced Viability and Anti-rotavirus Effect of Bifidobacterium longum and Lactobacillus plantarum in Combination With Chlorella sorokiniana in a Dairy Product. Front. Microbiol. 2020, 11, 875. [Google Scholar] [CrossRef] [PubMed]
- Al Kassaa, I. Antiviral Probiotics: A New Concept in Medical Sciences. In New Insights Antiviral Probiotics; Springer: Cham, Switzerland, 2016; pp. 1–46. [Google Scholar] [CrossRef]
- Fijan, S.; Frauwallner, A.; Langerholc, T.; Krebs, B.; ter Haar, J.A.; Heschl, A.; Turk, D.M.; Rogeli, I. Efficacy of Using Probiotics with Antagonistic Activity against Pathogens of Wound Infections: An Integrative Review of Literature. Biomed. Res. Int. 2019, 2019, 7585486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westall, F.C.; Root-Bernstein, R. Cause and prevention of postinfectious and postvaccinal neuropathies in light of a new theory of autoimmunity. Lancet 1986, 2, 251–252. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Self, Nonself, and the Paradoxes of Autoimmunity. In Organism and the Development of Self; Root-Bernstein, R.S., Ed.; Kluwer: Boston, MA, USA, 1991; pp. 159–209. [Google Scholar]
- Root-Bernstein, R.; Rallo, A. Antigenic complementarity resulting in idiotype-antiidiotype immune complexes: Possible contributor to AIDS pathogenesis and autoimmunity. Autoimmunity 2004, 37, 203–210. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Couturier, J. Antigenic complementarity in the origins of autoimmunity: A general theory illustrated with a case study of idiopathic thrombocytopenia purpura. Clin. Dev. Immunol. 2006, 13, 49–65. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Antigenic complementarity in the induction of autoimmunity: A general theory and review. Autoimmun. Rev. 2007, 6, 272–277. [Google Scholar] [CrossRef]
- Khetsuriani, N.; LaMonte-Fowlkes, A.; Oberste, M.S.S.; Pallansch, M.A. Centers for Disease Control. Enterovirus Surveillance-United States, 1970—2005. MMWR Surveill. Summaries 2006, 55, 1–20. [Google Scholar]
- Carapetis, J.R.; Steer, A.C.; Mulholland, E.K.; Weber, M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 2005, 5, 685–694. [Google Scholar] [CrossRef]
- Sims Sanyahumbi, A.; Colquhoun, S.; Wyber, R. Global Disease Burden of Group A Streptococcus. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations [Internet]; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar]
- Guilherme, L.; Köhler, K.F.; Kalil, J. Rheumatic heart disease: Mediation by complex immune events. Adv. Clin. Chem. 2011, 53, 31–50. [Google Scholar] [PubMed]
- Land, M.A.; Bisno, A.L. Acute rheumatic fever: A vanishing disease in suburbia. JAMA 1983, 249, 895–898. [Google Scholar] [CrossRef]
- Stockmann, C.; Ampofo, K.; Hersh, A.L. Evolving epidemiologic characteristics of invasive group A streptococcal disease in Utah, 2002–2010. Clin. Infect. Dis. 2012, 55, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Higgins, P.M. Splenomegaly in acute infections due to group A streptococci and viruses. Epidemiol. Infect. 1992, 109, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, N.R. The role of infection in the pathogenesis of autoimmune disease. Semin. Immunol. 1998, 10, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nomura, F.; Kawai, T.; Takeuchi, O.; Mühlradt, P.F.; Takeda, K.; Akira, S. Synergy and cross-tolerance between toll-like receptor (TLR) 2- and TLR4-mediated signaling pathways. J. Immunol. 2000, 165, 7096–7101. [Google Scholar] [CrossRef]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Mickelson, D.J.; Solberg, J.C.; Lipson, K.E.; Inglefield, J.R.; Alkan, S.S. TLR-TLR cross talk in human PBMC resulting in synergistic and antagonistic regulation of type-1 and 2 interferons, IL-12 and TNF-alpha. Int. Immun. 2007, 7, 1111–1121. [Google Scholar] [CrossRef]
- Krumbiegel, D.; Zepp, F.; Meyer, C.U. Combined Toll-like receptor agonists synergistically increase production of inflammatory cytokines in human neonatal dendritic cells. Hum. Immunol. 2007, 68, 813–822. [Google Scholar] [CrossRef]
- Vanhoutte, F.; Paget, C.; Breuilh, L.; Fontaine, J.; Vendeville, C.; Goriely, S.; Ryffel, B.; Faveeuw, C.; Trottein, F. Toll-like receptor (TLR)2 and TLR3 synergy and cross-inhibition in murine myeloid dendritic cells. Immunol. Lett. 2008, 116, 86–94. [Google Scholar] [CrossRef]
- Mäkeläm, S.M.; Strengell, M.; Pietilä, T.E.; Osterlund, P.; Julkunen, I. Multiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cells. J. Leukoc. Biol. 2009, 85, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.R.; Jeong, S.K.; Ahn, B.C.; Lee, B.J.; Shin, S.J.; Yum, J.S.; Ha, S.J. Combination of TLR1/2 and TLR3 ligands enhances CD4(+) T cell longevity and antibody responses by modulating type I IFN production. Sci. Rep. 2016, 6, 32526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischetti, L.; Zhong, Z.; Pinder, C.L.; Tregoning, J.S.; Shattock, R.J. The synergistic effects of combining TLR ligand based adjuvants on the cytokine response are dependent upon p38/JNK signalling. Cytokine 2017, 99, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.P.; Carstens, M.R.; Lewis, J.S.; Dolgova, N.; Xia, C.Q.; Clare-Salzler, M.J.; Keselowsky, B.G. A cell-based microarray to investigate combinatorial effects of microparticle-encapsulated adjuvants on dendritic cell activation. J. Mater. Chem. B 2016, 4, 1672–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouri-Shirazi, M.; Tamjidi, S.; Nourishirazi, E.; Guinet, E. TLR8 combined withTLR3 or TLR4 agonists enhances DC-NK driven effector Tc1 cells. Immunol. Lett. 2017, 193, 58–66. [Google Scholar] [CrossRef]
- Jung, Y.O.; Cho, M.L.; Kang, C.M.; Jhun, J.Y.; Park, J.S.; Oh, H.J.; Min, J.K.; Park, S.H.; Kim, H.Y. Toll-like receptor 2 and 4 combination engagement upregulate IL-15 synergistically in human rheumatoid synovial fibroblasts. Immunol. Lett. 2007, 109, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in autoimmune diseases: A comprehensive review. Clin. Rev. Allergy Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef]
- Wikén, M.; Grunewald, J.; Eklund, A.; Wahlström, J. Higher monocyte expression of TLR2 and TLR4, and enhanced pro-inflammatory synergy of TLR2 with NOD2 stimulation in sarcoidosis. J. Clin. Immunol. 2009, 9, 78–89. [Google Scholar] [CrossRef]
- Fritz, J.H.; Girardin, S.E.; Fitting, C.; Werts, C.; Mengin-Lecreulx, D.; Caroff, M.; Cavaillon, J.-M.; Philpott, D.J.; Adib-Conquy, M. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur. J. Immunol. 2005, 35, 2459–2470. [Google Scholar] [CrossRef]
- Farzi, A.; Reichmann, F.; Meinitzer, A.; Mayerhofer, R.; Jain, P.; Hassan, A.M.; Fröhlich, E.E.; Wagner, K.; Painsipp, E.; Rinner, B.; et al. Synergistic effects of NOD1 or NOD2 and TLR4 activation on mouse sickness behavior in relation to immune and brain activity markers. Brain Behav. Immun. 2015, 44, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Tada, H.; Aiba, S.; Shibata, K.; Ohteki, T.; Takada, H. Synergistic effect of NOD1 and NOD2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect. Immun. 2005, 73, 7967–7976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, H.; Uehara, A. Enhancement of TLR-mediated innate immune responses by peptidoglycans through NOD signaling. Curr. Pharm. Des. 2006, 12, 4163–4172. [Google Scholar] [CrossRef] [PubMed]
- Re, F.; Strominger, J.L. IL-10 released by concomitant TLR2 stimulation blocks the induction of a subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in human dendritic cells. J. Immunol. 2004, 173, 7548–7555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salk, J.; Romine, J.S.; Westall, F.C.; Wiederholt, W.C. Myelin basic protein studies in experimental allergic encephalomyelitis and multiple sclerosis: A summary with theoretical considerations of multiple sclerosis etiology. In Experimental Allergic Encephalomyelitis and Multiple Sclerosis; Davison, E.N., Cuzner, M.L., Eds.; Academic Press: London, UK, 1980; pp. 141–156. [Google Scholar]
- Root-Bernstein, R. The structure of a serotonin and LSD binding site of myelin basic protein. J. Theor. Biol. 1983, 100, 373–378. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Westall, F.C. Sleep factors: Do muramyl peptides activate serotonin binding sites? Lancet 1983, 321, 653. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Westall, F.C. Serotonin binding sites. II. Muramyl dipeptide binds to serotonin binding sites on myelin basic protein, LHRH, and MSH-ACTH 4-10. Brain Res. Bull. 1990, 25, 827–841. [Google Scholar] [CrossRef]
- Hess, K.L.; Andorko, J.I.; Tostanoski, L.H.; Jewell, C.M. Polyplexes assembled from self-peptides and regulatory nucleic acids blunt toll-like receptor signaling to combat autoimmunity. Biomaterials 2017, 118, 51–62. [Google Scholar] [CrossRef]
- Hayman, W.A.; Toth, I.; Flinn, N.; Scanlon, M.; Good, M.F. Enhancing the immunogenicity and modulating the fine epitope recognition of antisera to a helical group A streptococcal peptide vaccine candidate from the M protein using lipid-core peptide technology. Immunol. Cell. Biol. 2002, 80, 178–187. [Google Scholar] [CrossRef]
- Olive, C.; Batzloff, M.R.; Horváth, A.; Wong, A.; Clair, T.; Yarwood, P.; Toth, I.; Good, M.F. A lipid core peptide construct containing a conserved region determinant of the group A streptococcal M protein elicits heterologous opsonic antibodies. Infect. Immun. 2002, 70, 2734–2738. [Google Scholar] [CrossRef] [Green Version]
- Dörnerk, A.; Grunert, H.P.; Lindig, V. Treatment of coxsackievirus-B3-infected BALB/c mice with the soluble coxsackie adenovirus receptor CAR4/7 aggravates cardiac injury. J. Mol. Med. 2006, 84, 842–851. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Park, I.K.; Kohyama, K. B-cell epitope spreading is a critical step for the switch from C-protein-induced myocarditis to dilated cardiomyopathy. Am. J. Pathol. 2007, 170, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root-Bernstein, R. How to make a non-antigenic protein (auto) antigenic: Molecular complementarity alters antigen processing and activates adaptive-innate immunity synergy. Anticancer. Agents Med. Chem. 2015, 15, 1242–1259. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.F.; Gilbert, J.G. Axonal degeneration in experimental diphtheritic polyneuritis. Trans. Am. Neurol. Assoc. 1974, 99, 62–66. [Google Scholar]
- McDonald, W.I.; Gilman, S. Demyelination and muscle spindle function. Effect of diphtheritic polyneuritis on nerve conduction and muscle spindle function in the cat. Arch. Neurol. 1968, 18, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Waksman, B.H. Experimental study of diphtheritic polyneuritis in the rabbit and guinea pig. III. The bloodnerve barrier in the rabbit. J. Neuropathol. Exp. Neurol. 1961, 20, 35–77. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Dobbelstein, C. Insulin binds to glucagon forming a complex that is hyper-antigenic and inducing complementary antibodies having an idiotype-antiidiotype relationship. Autoimmunity 2001, 33, 153–169. [Google Scholar] [CrossRef]
- Kogut, E.P.; Livashova, N.V.; Bondarenk, A.P.; Zherdeva, A.I.; Shuvalova, I.A. Eksperimental’noe izuchenie koksaki-streptokokkovoi infektsii [Experimental study of coxsackie-streptococcal infection]. Vopr. Virusol. 1978, 6, 690–695. (In Russian) [Google Scholar]
- Suresh, L.; Chandrasekar, S.; Rao, R.S.; Ravi, V.; Badrinath, S. Coxsackie virus and rheumatic fever, a correlative study. J. Assoc. Physicians India 1989, 37, 582–585. [Google Scholar]
- Zaher, S.R.; Kassem, A.S.; Hughes, J.J. Coxsackie virus infections in rheumatic fever. Indian J. Pediatr. 1993, 60, 289–298. [Google Scholar] [CrossRef]
- Vikerfors, T.; Stjerna, A.; Olcen, P.; Malmcrona, R.; Magnius, L. Acute myocarditis, serologic diagnosis, clinical findings and follow-up. Acta Med. Scand. 1988, 223, 45–52. [Google Scholar] [CrossRef]
- Novikov, I.O. Dignostike nervmaticheskikh miokarditov [Diagnosis of nonrheumatic myocarditis]. Kardiologiia 1983, 23, 50–55. (In Russian) [Google Scholar] [PubMed]
- Root-Bernstein, R. Autoreactive T-cell receptor (Vbeta/D/Jbeta) sequences in diabetes are homologous to insulin.; glucagon, the insulin receptor, and the glucagon receptor. J. Mol. Recognit. 2009, 22, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Podufaly, A. Autoreactive T-cell receptor (Vbeta/D/Jbeta) sequences in diabetes recognize insulin, the insulin receptor, and each other, and are targets of insulin antibodies. Open Autoimmun. J. 2012, 4, 10–22. [Google Scholar] [CrossRef]
- Low, H.Z.; Witte, T. Aspects of innate immunity in Sjögren’s syndrome. Arthritis Res Ther. 2011, 13, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-H.; Li, Z.; Cui, L.; Li, Y.; Yoon, H.-J.; Choi, W.; Lee, J.-B.; Liu, Z.; Yoon, K.-C. Expression of Nod-like Receptors and Clinical Correlations in Patients With Dry Eye Disease. Am. J. Ophthalmol. 2019, 200, 150–160. [Google Scholar] [CrossRef]
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339. [Google Scholar] [CrossRef]
- Benhamou, Y.; Bellien, J.; Armengol, G.; Brakenhielm, E.; Adriouch, S.; Iacob, M.; Remy-Jouet, I.; Le Cam-Duchez, V.; Monteil, C.; Renet, S.; et al. Role of Toll-like receptors 2 and 4 in mediating endothelial dysfunction and arterial remodeling in primary arterial antiphospholipid syndrome. Arthritis Rheumatol. 2014, 66, 3210–3220. [Google Scholar] [CrossRef] [Green Version]
- Dema, B.; Charles, N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Gilkeson, G. Sex Differences in monocytes and TLR4 associated immune responses, implications for systemic lupus erythematosus (SLE). J. Immunother Appl. 2014, 1, 1. [Google Scholar] [CrossRef]
- Marques, C.P.; Maor, Y.; de Andrade, M.S.; Rodrigues, V.P.; Benatti, B.B. Possible evidence of systemic lupus erythematosus and periodontal disease association mediated by Toll-like receptors 2 and 4. Clin. Exp. Immunol. 2016, 183, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, S.; Wang, M.R.; Wang, T.T.; Li, B.K.; Zhu, J.M. Potential roles of nucleotide-binding oligomerization domain 2 in the pathogenesis of systemic lupus erythematosus. Rheumatol. Int. 2014, 34, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Uccellini, M.B.; Avalos, A.M.; Mashak-Rothstein, A.; Viglianti, G.A. Toll-like Receptor-Dependent Immune Complex Activation of B Cells and Dendritic Cells. Methods Mol. Biol. 2009, 517, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Moody, K.L.; Uccellini, M.B.; Avalos, A.M.; Marshak-Rothstein, A.; Viglianti, G.A. Toll-like receptor-dependent immune complex activation of B cells and dendritic cells. Methods Mol. Biol. 2016, 1390, 249–272. [Google Scholar] [CrossRef] [PubMed]
- Leadbetter, E.A.; Rifkin, I.R.; Hohlbaum, A.M.; Beaudette, B.C.; Shlomchik, M.J.; Marshak-Rothstein, A. Chromatin-IgG Complexes Activate B cells by dual engagement of IgM and toll-like receptors. Nature 2002, 416, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Liu, J.; Zhang, T.; Shen, Q.; Yu, Y.; Cao, X. Immune complex/Ig negatively regulate TLR4-triggered inflammatory response in macrophages through Fc gamma RIIb-dependent PGE2 production. J. Immunol. 2009, 182, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollahi-Roodsaz, S.; Koenders, M.I.; Walgreen, B.; Bolscher, J.; Helsen, M.M.; van den Bersselaar, L.A.; van Lent, P.; van de Loo, F.A.J.; van den Berg, W.B. Toll-like receptor 2 controls acute immune complex–driven arthritis in mice by regulating the inhibitory FcReceptor IIB. Arthritis Rheumatism 2013, 65, 2583–2593. [Google Scholar] [CrossRef]
- Couser, W.G. TLR2, 3, 7 and 9 activation by immune complexes in autoimmune glomerular diseases. Basic and Translational Concepts of Immune-Mediated Glomerular Diseases. J. Am. Soc. Nephrol. 2012, 23, 381–399. [Google Scholar] [CrossRef] [Green Version]
- Raschi, E.; Chighizola, C.B.; Cesana, L.; Privitera, D.; Ingegnoli, F.; Mastaglio, C.; Meroni, P.L.; Borghi, M.O. Immune complexes containing scleroderma-specific autoantibodies induce a profibrotic and proinflammatory phenotype in skin fibroblasts. Arthritis Res. Ther. 2018, 20, 187. [Google Scholar] [CrossRef] [Green Version]
- Tukaj, S.; Kaminski, M. Heat shock proteins in the therapy of autoimmune diseases: Too simple to be true? Version 2. Cell. Stress Chaperones 2019, 24, 475–479. [Google Scholar] [CrossRef] [Green Version]
- Valentinis, B.; Capobianco, A.; Esposito, F.; Bianchi, A.; Rovere-Querini, P.; Manfredi, A.A.; Traversari, C. Human recombinant heat shock protein 70 affects the maturation pathways of dendritic cells in vitro and has an in vivo adjuvant activity. J. Leukoc. Biol. 2008, 84, 199–206. [Google Scholar] [CrossRef]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vabulas, R.M.; Wagner, H.; Schild, H. Heat shock proteins as ligands of toll like receptors. Curr. Top. Microbiol. Immunol. 2002, 270, 169–184. [Google Scholar] [PubMed]
- Lewis, M.J.; McAndrew, M.B.; Wheeler, C.; Workman, N.; Agashe, P.; Koopmann, J.; Uddin, E.; Morris, D.L.; Zou, L.; Stark, R.; et al. Autoantibodies targeting TLR and SMAD pathways define new subgroups in systemic lupus erythematosus. J. Autoimmun. 2018, 91, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, M.C.; Choi, S. Recent progress in the development of Toll-like receptor (TLR) antagonists. Expert. Opin. Ther. Pat. 2016, 26, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Keogh, B.; Parker, A.E. Toll-like receptors as targets for immune disorders. Trends Pharmacol. Sci. 2011, 32, 435–442. [Google Scholar] [CrossRef]
- Henderson, J.; Bhattacharyya, S.; Varga, J.; O’Reilly, S. Targeting TLRs and the inflammasome in systemic sclerosis. Pharmacol. Ther. 2018, 192, 163–169. [Google Scholar] [CrossRef]
- Rahmani, F.; Rezaei, N. Therapeutic targeting of Toll-like receptors: A review of Toll-like receptors and their signaling pathways in psoriasis. Expert Rev. Clin. Immunol. 2016, 12, 1289–1298. [Google Scholar] [CrossRef]
- Zhu, J.; Mohan, C. Toll-like receptor signaling pathways--therapeutic opportunities. Med. Inflamm. 2010, 2010, 781235. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.Y.; Su, Y.W.; Lin, K.I.; Hsu, L.C.; Chuang, T.H. Natural modulators of endosomal toll-like receptor-mediated psoriatic skin inflammation. J. Immunol. Res. 2017, 2017, 7807313. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Patinote, C.; Karroum, N.B.; Moarbess, G.; Cirnat, N.; Kassab, I.; Bonnet, P.A.; Deleuze-Masquéfa, C. Agonist and antagonist ligands of toll-like receptors 7 and 8: Ingenious tools for therapeutic purposes. Eur. J. Med. Chem. 2020, 193, 112238. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Payán, J.; Saqui-Salces, M.; Jeyanathan, M.; Alcántara-Vazquez, A.; Castañon-Arreola, M.; Rook, G.; Hernandez-Pando, R. Extrapulmonary Locations of Mycobacterium tuberculosis DNA During Latent Infection. J. Infect. Dis. 2012, 206, 1194–1205. [Google Scholar] [CrossRef] [Green Version]
- Molina, D.K.; DiMiao, V.J.M. Normal Organ Weights in Men: Part II-the Brain, Lungs, Liver, Spleen, and Kidneys. Am. J. Forensic. Med. Pathol. 2012, 33, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Rose, N.R. Coxsackievirus-induced myocarditis in mice: A model of autoimmune disease for studying immunotoxicity. Methods 2007, 41, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marugán, R.B.; Garzón, S.G. DNA-guided hepatitis B treatment, viral load is essential, but not sufficient. World J. Gastroenterol. 2009, 15, 423–430. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
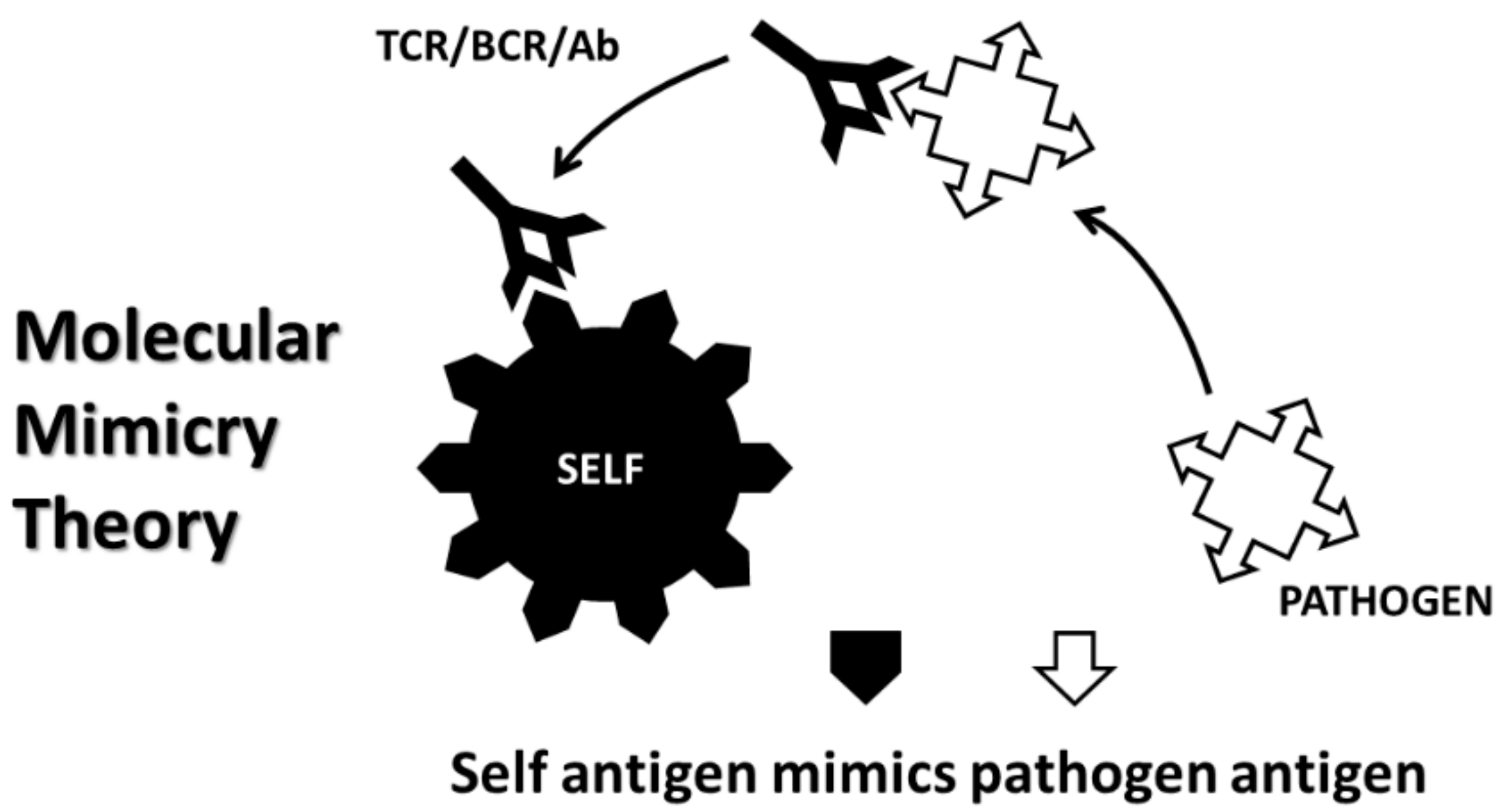{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TLR 1 | TLR 2 | TLR 3 | TLR 4 | TLR 5 | TLR 6 | TLR 7 | TLR 8 | TLR 9 | TLR 10 | NOD 1 | NOD 2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUTOTIMMUNE MYOCARDITIS | X | X | X | X | X | |||||||
| CX+myosin EAM | X | X | X | X | X | |||||||
| GAS M prot+CFA EAM | X | X | X | X | X | |||||||
| Cardiac myosin +CFA EAM | X | X | X | X | X | |||||||
| CX | X | X | X | X | ||||||||
| Cardiac myosin | X | X | ||||||||||
| GAS M protein | X | X | ||||||||||
| CFA | X | ? | X | X | X | X | ||||||
| MYESTHENIA GRAVIS | X | X | X | X | X | |||||||
| EAMG (CFA+AChR) | X | X | X | X | X | |||||||
| EAMG (I:C+LPS+AChR | X | X | X | X | ||||||||
| AChR | X | X | ||||||||||
| Poly I:C | X | |||||||||||
| LPS | X | |||||||||||
| CFA | X | ? | X | X | X | X | ||||||
| MULTIPLE SCLEROSIS | X | X | X | X | ||||||||
| MBP-CFA EAE | X | X | X | X | ||||||||
| MBP-CpG-LPS EAE | X | X | X | |||||||||
| MBP-MDP EAE | X | X | X | |||||||||
| MOG-CFA EAE | X | X | X | |||||||||
| MOG-CpG EAE | X | X | ||||||||||
| CpG ODN | X | |||||||||||
| MDP | X | X | X | |||||||||
| MBP | X | X | ||||||||||
| MOG | X | |||||||||||
| CFA | X | ? | X | X | X | X | ||||||
| RHEUMATOID ARTHRITIS | X | X | X | X | X | |||||||
| Strep cell wall RA | X | X | X | X | ||||||||
| Collagen II-CFA RA | X | X | X | X | X | |||||||
| Collagen II-LPS RA | X | X | ||||||||||
| Collagen II | X | |||||||||||
| LPS | X | |||||||||||
| CFA | X | ? | X | X | X | X |
| TLR & NOD SYNERGIES | TLR2-TLR3 | TLR2-TLR4 | TLR-TLR6 | TLR3-TLR4 | TLR3-TLR7, 8 | TLR4-TLR7, 8 | TLR4-TLR9 | TLR3-NOD1, 2 | TLR4-NOD1, 2 | TLR9-NOD1, 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Autoimmune myocarditis | X | X | X | |||||||
| Myasthenia gravis | X | X | X | |||||||
| Multiple sclerosis | X | X | X | X | ||||||
| Rheumatoid arthritis | X | X | ||||||||
| Sjogren’s syndrome | X | X | ? | ? | ? | |||||
| Anti-phospholipid syndrome | X | X | ||||||||
| Systemic lupus erythematosus | X | X | X | X | X |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R. Synergistic Activation of Toll-Like and NOD Receptors by Complementary Antigens as Facilitators of Autoimmune Disease: Review, Model and Novel Predictions. Int. J. Mol. Sci. 2020, 21, 4645. https://doi.org/10.3390/ijms21134645
Root-Bernstein R. Synergistic Activation of Toll-Like and NOD Receptors by Complementary Antigens as Facilitators of Autoimmune Disease: Review, Model and Novel Predictions. International Journal of Molecular Sciences. 2020; 21(13):4645. https://doi.org/10.3390/ijms21134645
Chicago/Turabian StyleRoot-Bernstein, Robert. 2020. "Synergistic Activation of Toll-Like and NOD Receptors by Complementary Antigens as Facilitators of Autoimmune Disease: Review, Model and Novel Predictions" International Journal of Molecular Sciences 21, no. 13: 4645. https://doi.org/10.3390/ijms21134645