AMPK Profiling in Rodent and Human Pancreatic Beta-Cells under Nutrient-Rich Metabolic Stress

 ,
,  ,
,
Abstract
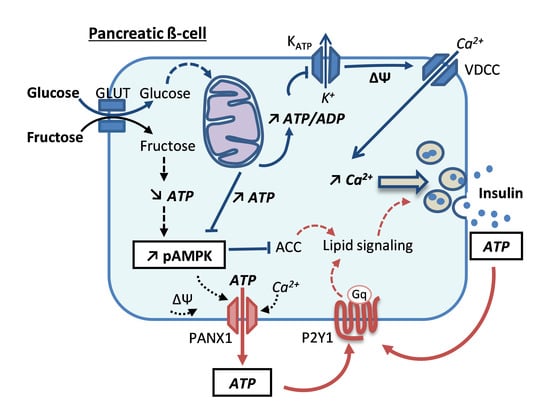
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. AMPK and Glucose Homeostasis
1.2. Function of the Insulin-Secreting Cell
1.3. AMPK in the β-Cell
1.4. Effects of Metabolic Stresses on AMPK in the β-Cell
2. Results
2.1. AMPK Profiling in β-Cells
2.2. Diabetogenic Conditions do not Alter AMPK Gene Expression in INS-1E β-Cells and Human Islets
2.3. Glucotoxic Conditions and Chronic Fructose Exposure do not Alter AMPK Protein Levels in INS-1E β-Cells and Human Islets, While Prolonged Treatment with Fructose Activates AMPK
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture, Human Islets and Treatments
4.3. RNA-Seq
4.4. Animals and Islet Isolation
4.5. Isolation of RNA and Quantitative RT-PCR
4.6. Immunoblotting
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prentki, M.; Peyot, M.L.; Masiello, P.; Madiraju, S.R.M. Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic β-Cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M. Insulin resistance and pancreatic beta cell failure. J. Clin. Investig. 2006, 116, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Hu, Q.; Screaton, R.A. AMPK and Friends: Central Regulators of β Cell Biology. Trends Endocrinol. Metab. 2018, 29, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Eberhard, C.E.; Screaton, R.A. Role of AMPK in pancreatic β cell function. Mol. Cell. Endocrinol. 2013, 366, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T.; Szkudelska, K. The relevance of AMP-activated protein kinase in insulin-secreting β cells: A potential target for improving β cell function? J. Physiol. Biochem. 2019, 75, 423–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maechler, P. Mitochondrial function and insulin secretion. Mol. Cell. Endocrinol. 2013, 379, 12–18. [Google Scholar] [CrossRef]
- Maechler, P. Glutamate pathways of the β-cell and the control of insulin secretion. Diabetes Res. Clin. Pract. 2017, 131, 149–153. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef] [Green Version]
- Henquin, J.C. Do pancreatic β cells “taste” nutrients to secrete insulin? Sci. Signal. 2012, 5, pe36. [Google Scholar] [CrossRef]
- Malaisse, W.J. Insulin release: The receptor hypothesis. Diabetologia 2014, 57, 1287–1290. [Google Scholar] [CrossRef]
- Wollheim, C.B.; Maechler, P. β cell glutamate receptor antagonists: Novel oral antidiabetic drugs? Nat Med. 2015, 21, 310–311. [Google Scholar] [CrossRef]
- Bartley, C.; Brun, T.; Oberhauser, L.; Grimaldi, M.; Molica, F.; Kwak, B.R.; Bosco, D.; Chanson, M.; Maechler, O. Chronic fructose renders pancreatic β-cells hyper-responsive to glucose-stimulated insulin secretion through extracellular ATP signaling. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E25–E41. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salt, I.P.; Johnson, G.; Ashcroft, S.J.; Hardie, D.G. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic β cells, and may regulate insulin release. Biochem. J. 1998, 335 Pt 3, 533–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva Xavier, G.; Leclerc, I.; Salt, I.; Doiron, B.; Hardie, D.G.; Kahn, A.; Rutter, G.A. Role of AMP-activated protein kinase in the regulation by glucose of islet β cell gene expression. Proc. Natl. Acad. Sci. USA 2000, 97, 4023–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.J.; Partridge, C.J.; Asipu, A.; Mair, L.A.; Hunter, M.; Sivaprasadarao, A. Increased ATP-sensitive K+ channel expression during acute glucose deprivation. Biochem. Biophys. Res. Commun. 2006, 348, 1123–1131. [Google Scholar] [CrossRef]
- Lim, A.; Park, S.H.; Sohn, J.W.; Jeon, J.H.; Park, J.H.; Song, D.K.; Lee, S.H.; Ho, W.K. Glucose deprivation regulates KATP channel trafficking via AMP-activated protein kinase in pancreatic β-cells. Diabetes 2009, 58, 2813–2819. [Google Scholar] [CrossRef] [Green Version]
- da Silva Xavier, G.; Leclerc, I.; Varadi, A.; Tsuboi, T.; Moule, S.K.; Rutter, G.A. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem. J. 2003, 371 Pt 3, 761–774. [Google Scholar] [CrossRef]
- Rutter, G.A.; Da Silva Xavier, G.; Leclerc, I. Roles of 5’-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem. J. 2003, 375 Pt 1, 1–16. [Google Scholar]
- Richards, S.K.; Parton, L.E.; Leclerc, I.; Rutter, G.A.; Smith, R.M. Over-expression of AMP-activated protein kinase impairs pancreatic β-cell function in vivo. J. Endocrinol. 2005, 187, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Yavari, A.; Stocker, C.J.; Ghaffari, S.; Wargent, E.T.; Steeples, V.; Czibik, G.; Pinter, K.; Bellahcene, M.; Woods, A.; de Morentin, P.B.M.; et al. Chronic Activation of γ2 AMPK Induces Obesity and Reduces β Cell Function. Cell Metab. 2016, 23, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryer, L.G.; Parbu-Patel, A.; Carling, D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, R.; Nie, A.; Jian, F.; Liu, Y.; Tang, H.; Zhang, J.; Zhang, Y.; Shao, L.; Li, F.; Zhou, L.; et al. Acute exposure of β-cells to troglitazone decreases insulin hypersecretion via activating AMPK. Biochim. Biophys. Acta 2014, 1840, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.C. The molecular mechanisms of pancreatic β-cell glucotoxicity: Recent findings and future research directions. Mol. Cell. Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C. Glucolipotoxicity, β-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Oberhauser, L.; Granziera, S.; Colom, A.; Goujon, A.; Lavallard, V.; Matile, S.; Roux, A.; Brun, T.; Maechler, P. Palmitate and oleate modify membrane fluidity and kinase activities of INS-1E β-cells alongside altered metabolism-secretion coupling. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118619. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Han, D.; Kim, Y.; Jin, J.; Ho, W.K.; Kim, Y. Interactome analysis of AMP-activated protein kinase (AMPK)-α1 and -β1 in INS-1 pancreatic β-cells by affinity purification-mass spectrometry. Sci. Rep. 2014, 4, 4376. [Google Scholar] [CrossRef] [Green Version]
- Merglen, A.; Theander, S.; Rubi, B.; Chaffard, G.; Wollheim, C.B.; Maechler, P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 2004, 145, 667–678. [Google Scholar] [CrossRef]
- Roche, E.; Farfari, S.; Witters, L.A.; Assimacopoulos-Jeannet, F.; Thumelin, S.; Brun, T.; Corkey, B.; Saha, A.; Prentki, M. Long-term exposure of β-INS cells to high glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene expression. Diabetes 1998, 47, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Maechler, P. β-cell mitochondrial carriers and the diabetogenic stress response. Biochim. Biophys. Acta 2016, 1863, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Scarcia, P.; Li, N.; Gaudet, P.; Duhamel, D.; Palmieri, F.; Maechler, P. Changes in mitochondrial carriers exhibit stress-specific signatures in INS-1Eβ-cells exposed to glucose versus fatty acids. PLoS ONE 2013, 8, e82364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravnskjaer, K.; Boergesen, M.; Dalgaard, L.T.; Mandrup, S. Glucose-induced repression of PPARα gene expression in pancreatic β-cells involves PP2A activation and AMPK inactivation. J. Mol. Endocrinol. 2006, 36, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Holloway, G.P.; Steinberg, G.R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 2013, 366, 135–151. [Google Scholar] [CrossRef]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.-Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.; Kieffer, T.J.; et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Vetterli, L.; Carobbio, S.; Frigerio, F.; Karaca, M.; Maechler, P. The Amplifying Pathway of the β-Cell Contributes to Diet-induced Obesity. J. Biol. Chem. 2016, 291, 13063–13075. [Google Scholar] [CrossRef] [Green Version]
- Pournourmohammadi, S.; Grimaldi, M.; Stridh, M.H.; Lavallard, V.; Waagepetersen, H.S.; Wollheim, C.B.; Maechler, P. Epigallocatechin-3-gallate (EGCG) activates AMPK through the inhibition of glutamate dehydrogenase in muscle and pancreatic ss-cells: A potential beneficial effect in the pre-diabetic state? Int. J. Biochem. Cell Biol. 2017, 88, 220–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Sanchez, K.; Leyva, M.J.; Wu, M.; Betts, N.M.; Aston, C.E.; Lyons, T.J. Green tea supplementation affects body weight, lipids, and lipid peroxidation in obese subjects with metabolic syndrome. J. Am. Coll. Nutr. 2010, 29, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Sae-tan, S.; Grove, K.A.; Lambert, J.D. Weight control and prevention of metabolic syndrome by green tea. Pharmacol. Res. 2011, 64, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Moustaid-Moussa, N.; Chen, L.; Mo, H.; Shastri, A.; Su, R.; Bapat, P.; Kwun, I.; Shen, C.-L. Novel insights of dietary polyphenols and obesity. J. Nutr. Biochem. 2014, 25, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortsater, H.; Grankvist, N.; Wolfram, S.; Kuehn, N.; Sjoholm, A. Diet supplementation with green tea extract epigallocatechin gallate prevents progression to glucose intolerance in db/db mice. Nutr. Metab. (Lond.) 2012, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.; Kang, D.-H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [Green Version]
- Kinote, A.; A Faria, J.; A Roman, E.; Solon, C.; Razolli, D.S.; Ignacio-Souza, L.; Sollon, C.S.; Nascimento, L.F.; De Araújo, T.M.; Barbosa, A.P.L.; et al. Fructose-induced hypothalamic AMPK activation stimulates hepatic PEPCK and gluconeogenesis due to increased corticosterone levels. Endocrinology 2012, 153, 3633–3645. [Google Scholar] [CrossRef] [Green Version]
- Burmeister, M.A.; Ayala, J.; Drucker, D.J.; Ayala, J.E. Central glucagon-like peptide 1 receptor-induced anorexia requires glucose metabolism-mediated suppression of AMPK and is impaired by central fructose. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E677–E685. [Google Scholar] [CrossRef] [Green Version]
- Cha, S.H.; Wolfgang, M.; Tokutake, Y.; Chohnan, S.; Lane, M.D. Differential effects of central fructose and glucose on hypothalamic malonyl-CoA and food intake. Proc. Natl. Acad. Sci. USA 2008, 105, 16871–16875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.J.; Kow, L.M.; Funabashi, T.; Mobbs, C.V. Hypothalamic glucose sensor: Similarities to and differences from pancreatic β-cell mechanisms. Diabetes 1999, 48, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Brun, T.; Bartley, C.; Usardi, A.; Bosco, D.; Ravnskjær, K.; Mandrup, S.; Maechler, P. Peroxisome proliferator-activated receptor alpha (PPARα) protects against oleate-induced INS-1E β cell dysfunction by preserving carbohydrate metabolism. Diabetologia 2010, 53, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Brun, T.; Li, N.; Jourdain, A.A.; Gaudet, P.; Duhamel, D.; Meyer, J.; Bosco, D.; Maechler, P. Diabetogenic milieus induce specific changes in mitochondrial transcriptome and differentiation of human pancreatic islets. Hum. Mol. Genet. 2015, 24, 5270–5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.F.; Madsen, J.G.; Frafjord, K.O.; Poulsen, L.; Salo, S.; Boergesen, M.; Loft, A.; Larsen, B.D.; Madsen, M.S.; Holst, J.J.; et al. Integrative Genomics Outlines a Biphasic Glucose Response and a ChREBP-RORγ Axis Regulating Proliferation in β Cells. Cell Rep. 2016, 16, 2359–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouiller, D.G.; Cirulli, V.; Halban, P.A. Uvomorulin mediates calcium-dependent aggregation of islet cells, whereas calcium-independent cell adhesion molecules distinguish between islet cell types. Dev. Biol. 1991, 148, 233–242. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brun, T.; Jiménez-Sánchez, C.; Madsen, J.G.S.; Hadadi, N.; Duhamel, D.; Bartley, C.; Oberhauser, L.; Trajkovski, M.; Mandrup, S.; Maechler, P. AMPK Profiling in Rodent and Human Pancreatic Beta-Cells under Nutrient-Rich Metabolic Stress. Int. J. Mol. Sci. 2020, 21, 3982. https://doi.org/10.3390/ijms21113982
Brun T, Jiménez-Sánchez C, Madsen JGS, Hadadi N, Duhamel D, Bartley C, Oberhauser L, Trajkovski M, Mandrup S, Maechler P. AMPK Profiling in Rodent and Human Pancreatic Beta-Cells under Nutrient-Rich Metabolic Stress. International Journal of Molecular Sciences. 2020; 21(11):3982. https://doi.org/10.3390/ijms21113982
Chicago/Turabian StyleBrun, Thierry, Cecilia Jiménez-Sánchez, Jesper Grud Skat Madsen, Noushin Hadadi, Dominique Duhamel, Clarissa Bartley, Lucie Oberhauser, Mirko Trajkovski, Susanne Mandrup, and Pierre Maechler. 2020. "AMPK Profiling in Rodent and Human Pancreatic Beta-Cells under Nutrient-Rich Metabolic Stress" International Journal of Molecular Sciences 21, no. 11: 3982. https://doi.org/10.3390/ijms21113982