Cdc42-Dependent Transfer of mir301 from Breast Cancer-Derived Extracellular Vesicles Regulates the Matrix Modulating Ability of Astrocytes at the Blood–Brain Barrier

 , , ,
, , ,
Abstract
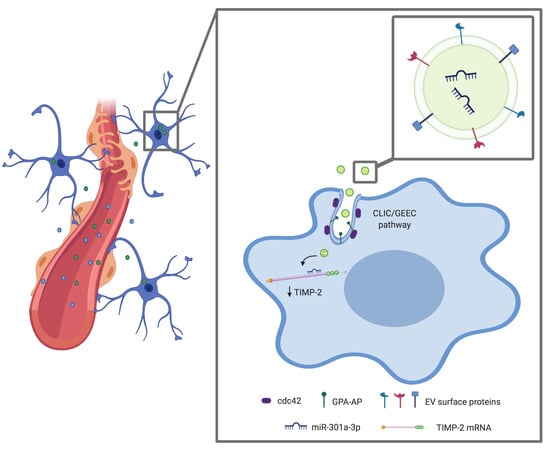
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Astrocyte Uptake of Breast Cancer-Derived EVs Is Mediated through the CLIC/GEEC Pathway
2.2. Br-EVs Are Enriched in Interacting Partners of the CLIC/GEEC Cargo
2.3. Br-EVs Decrease the Astrocyte Expression of TIMP-2
2.4. miR-301a-3p Transferred by Breast Cancer-Derived EVs Downregulate TIMP-2 in Astrocytes
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. EV Isolation and Characterization
4.3. In Vitro EV Uptake Studies
4.4. In Vitro EV Functional Studies
4.5. Validation of EV Surface Proteins
4.6. Proteomics Analysis
4.7. In Vivo Experiments
4.8. miRNA Target Validation
4.9. RNA Isolation and Analysis
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cagney, D.N.; Martin, A.M.; Catalano, P.J.; Redig, A.J.; Lin, N.U.; Lee, E.Q.; Wen, P.Y.; Dunn, I.F.; Bi, W.L.; Weiss, S.E.; et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: A population-based study. Neuro Oncol. 2017, 19, 1511–1521. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.M.; Cagney, D.N.; Catalano, P.J.; Warren, L.E.; Bellon, J.R.; Punglia, R.S.; Claus, E.B.; Lee, E.Q.; Wen, P.Y.; Haas-Kogan, D.A.; et al. Brain Metastases in Newly Diagnosed Breast Cancer: A Population-Based Study. JAMA Oncol. 2017, 3, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE 2009, 4, e5857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [Green Version]
- Morad, G.; Moses, M.A. Brainwashed by extracellular vesicles: The role of extracellular vesicles in primary and metastatic brain tumour microenvironment. J. Extracell. Vesicles 2019, 8, 1627164. [Google Scholar] [CrossRef] [Green Version]
- Fais, S.; O′Driscoll, L.; Borras, F.E.; Buzas, E.; Camussi, G.; Cappello, F.; Carvalho, J.; Cordeiro da Silva, A.; Del Portillo, H.; El Andaloussi, S.; et al. Evidence-Based Clinical Use of Nanoscale Extracellular Vesicles in Nanomedicine. ACS Nano 2016, 10, 3886–3899. [Google Scholar] [CrossRef]
- Morad, G.; Carman, C.V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-Derived Extracellular Vesicles Breach the Intact Blood-Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J.K. Exosomal Annexin II Promotes Angiogenesis and Breast Cancer Metastasis. Mol. Cancer Res. 2017, 15, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lotvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, G.; Hoshino, A.; Kenific, C.M.; Matei, I.R.; Steiner, L.; Freitas, D.; Kim, H.S.; Oxley, P.R.; Scandariato, I.; Casanova-Salas, I.; et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat. Cell Biol. 2019, 21, 1403–1412. [Google Scholar] [CrossRef]
- Yoneda, T.; Williams, P.J.; Hiraga, T.; Niewolna, M.; Nishimura, R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res. 2001, 16, 1486–1495. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.; Wang, S.; Huang, Y.; Stamnes, M.; Chen, J.L. Roles of rho GTPases in intracellular transport and cellular transformation. Int. J. Mol. Sci. 2013, 14, 7089–7108. [Google Scholar] [CrossRef]
- Rhee, J.M.; Pirity, M.K.; Lackan, C.S.; Long, J.Z.; Kondoh, G.; Takeda, J.; Hadjantonakis, A.K. In vivo imaging and differential localization of lipid-modified GFP-variant fusions in embryonic stem cells and mice. Genesis 2006, 44, 202–218. [Google Scholar] [CrossRef] [Green Version]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.S.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, M.T.; Kirkham, M.; Riches, J.; Cortese, K.; Walser, P.J.; Simpson, F.; Hill, M.M.; Jones, A.; Lundmark, R.; Lindsay, M.R.; et al. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J. Cell Biol. 2010, 190, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado-Baez, L.; Williamson, C.; Donaldson, J.G. Clathrin-independent endocytosis: A cargo-centric view. Exp. Cell Res. 2013, 319, 2759–2769. [Google Scholar] [PubMed] [Green Version]
- Lakshminarayan, R.; Wunder, C.; Becken, U.; Howes, M.T.; Benzing, C.; Arumugam, S.; Sales, S.; Ariotti, N.; Chambon, V.; Lamaze, C.; et al. Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat. Cell Biol. 2014, 16, 595–606. [Google Scholar] [CrossRef]
- Strater, N. Ecto-5′-nucleotidase: Structure function relationships. Purinergic Signal. 2006, 2, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Lakhan, S.E.; Sabharanjak, S.; De, A. Endocytosis of glycosylphosphatidylinositol-anchored proteins. J. Biomed. Sci. 2009, 16, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berditchevski, F. Complexes of tetraspanins with integrins: More than meets the eye. J. Cell Sci. 2001, 114 Pt 23, 4143–4151. [Google Scholar]
- Fang, J.; Shing, Y.; Wiederschain, D.; Yan, L.; Butterfield, C.; Jackson, G.; Harper, J.; Tamvakopoulos, G.; Moses, M.A. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc. Natl. Acad. Sci. USA 2000, 97, 3884–3889. [Google Scholar] [PubMed] [Green Version]
- Harper, J.; Moses, M.A. Molecular regulation of tumor angiogenesis: Mechanisms and therapeutic implications. EXS 2006, 96, 223–268. [Google Scholar]
- Overall, C.M.; Kleifeld, O. Tumour microenvironment-opinion: Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar]
- Roy, R.; Morad, G.; Jedinak, A.; Moses, M.A. Metalloproteinases and their Roles in Human Cancer. Anat. Rec. 2019, in press. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Mendes, O.; Kim, H.T.; Stoica, G. Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin. Exp. Metastasis 2005, 22, 237–246. [Google Scholar] [CrossRef]
- Mendes, O.; Kim, H.T.; Lungu, G.; Stoica, G. MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin. Exp. Metastasis 2007, 24, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kato, Y.; Erzinger, S.A.; Kiriakova, G.M.; Qian, Y.; Palmieri, D.; Steeg, P.S.; Price, J.E. The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer 2012, 12, 583. [Google Scholar]
- Wu, K.; Fukuda, K.; Xing, F.; Zhang, Y.; Sharma, S.; Liu, Y.; Chan, M.D.; Zhou, X.; Qasem, S.A.; Pochampally, R.; et al. Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J. Biol. Chem. 2015, 290, 9842–9854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojiani, M.V.; Ghoshal-Gupta, S.; Kutiyanawalla, A.; Mathur, S.; Rojiani, A.M. TIMP-1 overexpression in lung carcinoma enhances tumor kinetics and angiogenesis in brain metastasis. J. Neuropathol. Exp. Neurol. 2015, 74, 293–304. [Google Scholar]
- Wang, L.; Cossette, S.M.; Rarick, K.R.; Gershan, J.; Dwinell, M.B.; Harder, D.R.; Ramchandran, R. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLoS ONE 2013, 8, e80933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar]
- Fernandez, C.A.; Butterfield, C.; Jackson, G.; Moses, M.A. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): Loop 6 is a novel angiogenesis inhibitor. J. Biol. Chem. 2003, 278, 40989–40995. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Zhang, N.; He, S.; Lui, Y.; Lu, G.; Zhao, L. MicroRNA-106a targets TIMP2 to regulate invasion and metastasis of gastric cancer. FEBS Lett. 2014, 588, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, A.; Yang, C.; Chen, Z.; Li, C.; Cai, L. MiR-761 Promotes Progression and Metastasis of Non-Small Cell Lung Cancer by Targeting ING4 and TIMP2. Cell. Physiol. Biochem. 2015, 37, 55–66. [Google Scholar] [CrossRef]
- Liang, B.; Yin, J.J.; Zhan, X.R. MiR-301a promotes cell proliferation by directly targeting TIMP2 in multiple myeloma. Int. J. Clin. Exp. Pathol. 2015, 8, 9168–9174. [Google Scholar] [PubMed]
- Morad, G.; Moses, M.A. Breast cancer-derived extracellular vesicles modulate the endocytic pathway in brain endothelial cells. (unpublished, manuscript in preparation).
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKelvey, K.J.; Powell, K.L.; Ashton, A.W.; Morris, J.M.; McCracken, S.A. Exosomes: Mechanisms of Uptake. J. Circ. Biomark. 2015, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Nonnenmacher, M.; Weber, T. Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell Host Microbe 2011, 10, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Qiao, Y.; Li, X.; Chen, J.; Ding, J.; Bai, L.; Shen, F.; Shi, B.; Liu, J.; Peng, L.; et al. Exosomes Exploit the Virus Entry Machinery and Pathway to Transmit Alpha Interferon-Induced Antiviral Activity. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelke, G.V.; Lasser, C.; Gho, Y.S.; Lotvall, J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.P.; Kim, E.Y.; Badr, C.E.; Weissleder, R.; Mempel, T.R.; Tannous, B.A.; Breakefield, X.O. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun. 2015, 6, 7029. [Google Scholar] [CrossRef] [PubMed]
- Jedinak, A.; Curatolo, A.; Zurakowski, D.; Dillon, S.; Bhasin, M.K.; Libermann, T.A.; Roy, R.; Sachdev, M.; Loughlin, K.R.; Moses, M.A. Novel non-invasive biomarkers that distinguish between benign prostate hyperplasia and prostate cancer. BMC Cancer 2015, 15, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sneath, P. Numerical Taxonomy; the Principles and Practice of Numerical Classification; W. H. Freeman: San Francisco, CA, USA, 1973. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morad, G.; Daisy, C.C.; Otu, H.H.; Libermann, T.A.; Dillon, S.T.; Moses, M.A. Cdc42-Dependent Transfer of mir301 from Breast Cancer-Derived Extracellular Vesicles Regulates the Matrix Modulating Ability of Astrocytes at the Blood–Brain Barrier. Int. J. Mol. Sci. 2020, 21, 3851. https://doi.org/10.3390/ijms21113851
Morad G, Daisy CC, Otu HH, Libermann TA, Dillon ST, Moses MA. Cdc42-Dependent Transfer of mir301 from Breast Cancer-Derived Extracellular Vesicles Regulates the Matrix Modulating Ability of Astrocytes at the Blood–Brain Barrier. International Journal of Molecular Sciences. 2020; 21(11):3851. https://doi.org/10.3390/ijms21113851
Chicago/Turabian StyleMorad, Golnaz, Cassandra C. Daisy, Hasan H. Otu, Towia A. Libermann, Simon T. Dillon, and Marsha A. Moses. 2020. "Cdc42-Dependent Transfer of mir301 from Breast Cancer-Derived Extracellular Vesicles Regulates the Matrix Modulating Ability of Astrocytes at the Blood–Brain Barrier" International Journal of Molecular Sciences 21, no. 11: 3851. https://doi.org/10.3390/ijms21113851