Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer


Abstract
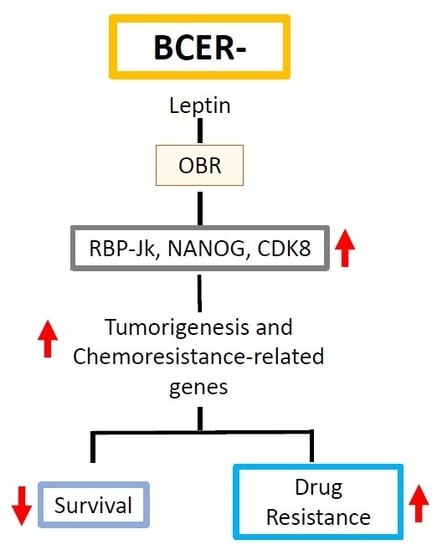
:
1. Introduction
2. Results
2.1. OBR and Leptin-Targeted Gene Co-Expression Correlates with Low Survival of BCER− Patients
2.2. Leptin Impaired Chemotherapeutic Effects on TNBC Cells
2.3. Leptin Increased the Expression of Key Proteins Associated with Survival and Motility in TNBC Cells Treated with Paclitaxel
2.4. Leptin Induced the Expression of Proliferation and Chemoresistance-Related Genes in TNBC Cells
3. Discussion
4. Materials and Methods
4.1. Antibodies, Hormones, and Reagents
4.2. Leptin Peptide Antagonists
4.3. Cell Cultures
4.4. CellTiter-Glo Assay
4.5. Cell Cycle Progression Assay
4.6. MTT Assay
4.7. Western Blot
4.8. RNA Silencing of OBR in TNBC Cells
4.8.1. Optimizing Leptin Receptor Knockdown in MDA-MB231 Cell Line
4.8.2. Detection of Leptin Signaling in OBR shRNA Knockdown Cells
4.9. Combined Leptin Peptide Antagonist-Chemotherapy Treatment
4.10. RNA Extraction and RNA-Seq
4.11. Breast Cancer Patient Kaplan–Meier Survival Curves
4.12. Protein-Protein Interaction (PPI) Analysis of Differentially Expressed Genes (DEG)
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO Fact Sheets in Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 11 September 2019).
- For, D.; Benefit, P. AACR Cancer Progress Report; AACR: Philadelphia, PA, USA, 2018. [Google Scholar]
- Lauby-secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 8, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Enhanced Expression of Leptin and Leptin Receptor (OB-R) in Human Breast Cancer. Clin. Cancer Res. 2004, 10, 4325–4332. [Google Scholar] [CrossRef] [Green Version]
- Mendonsa, A.M.; Chalfant, M.C.; Gorden, L.D.; VanSaun, M.N. Modulation of the leptin receptor mediates tumor growth and migration of pancreatic cancer cells. PLoS ONE 2015, 10, e0126686. [Google Scholar] [CrossRef]
- Daley-Brown, D.; Harbuzariu, A.; Kurian, A.A.; Oprea-Ilies, G.; Gonzalez-Perez, R.R. Leptin-induced Notch and IL-1 signaling crosstalk in endometrial adenocarcinoma is associated with invasiveness and chemoresistance. World J. Clin. Oncol. 2019, 10, 222–233. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; Mckee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: Possible role of obesity-related stimuli. Clin. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Vaisse, C.; Halaas, J.L.; Horvath, C.M.; Dernell, J.; Stoffel, M.; Friedman, J.M. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat. Genet. 1996, 14, 95–97. [Google Scholar] [CrossRef]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J. Methodol. March. 2016, 26, 43–55. [Google Scholar] [CrossRef]
- Guo, S.; Gonzalez-Perez, R.R. Notch, IL-1 and Leptin Crosstalk Outcome (NILCO) Is Critical for Leptin-Induced Proliferation, Migration and VEGF/VEGFR-2 Expression in Breast Cancer. PLoS ONE 2011, 6, e21467. [Google Scholar] [CrossRef] [Green Version]
- Candelaria, P.V.; Rampoldi, A.; Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin signaling and cancer chemoresistance: Perspectives. World J. Clin. Oncol. 2017, 8, 106–117. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, leptin and breast cancer: Epidemiological evidence and proposed mechanisms. Cancers 2019, 11, 1–27. [Google Scholar]
- Barone, I.; Brusco, L.; Fuqua, S.A.W. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin. Cancer Res. 2010, 16, 2702–2708. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, A.; Castoria, G.; De Falco, A.; Bilancio, A.; Giovannelli, P.; Di Donato, M.; Marino, I.; Yamaguchi, H.; Appella, E.; Auricchio, F. Polyproline and Tat transduction peptides in the study of the rapid actions of steroid receptors. Steroids 2012, 77, 974–978. [Google Scholar] [CrossRef]
- Rene Gonzalez, R.; Watters, A.; Xu, Y.; Singh, U.P.; Mann, D.R.; Rueda, B.R.; Penichet, M.L. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 2009, 11, R36. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Á.; Lánczky, A.; Menyhárt, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Mihály, Z.; Kormos, M.; Lánczky, A.; Dank, M.; Budczies, J.; Szász, M.A.; Gyorffy, B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 219–232. [Google Scholar] [CrossRef]
- Mishra, P.; Tang, W.; Ambs, S. ADHFE1 is a MYC-linked oncogene that induces metabolic reprogramming and cellular de-differentiation in breast cancer. Mol. Cell. Oncol. 2018, 5, e1432260. [Google Scholar] [CrossRef] [Green Version]
- Wainszelbaum, M.J.; Liu, J.; Kong, C.; Srikanth, P.; Samovski, D.; Su, X.; Stahl, P.D. TBC1D3, a hominoid-specific gene, delays IRS-1 degradation and promotes insulin signaling by modulating p70 S6 kinase activity. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Jing, H.; Qu, X.; Liu, L.; Xia, H. A novel long noncoding rna (LncRNA), LL22NC03-N64E9.1, promotes the proliferation of lung cancer cells and is a potential prognostic molecular biomarker for lung cancer. Med. Sci. Monit. 2018, 24, 4317–4323. [Google Scholar] [CrossRef]
- Sesé, M.; Fuentes, P.; Esteve-Codina, A.; Béjar, E.; McGrail, K.; Thomas, G.; Aasen, T.; Ramón y Cajal, S. Hypoxia-mediated translational activation of ITGB3 in breast cancer cells enhances TGF-β signaling and malignant features in vitro and in vivo. Oncotarget 2017, 8, 114856–114876. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta Rev. Cancer 2011, 1815, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Parish, C.R.; Wong, M.-L.; Licinio, J.; Blackburn, A.C. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE 2017. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, D.; Parsyan, A.; Petroulakis, E.; Hershey, J.; Sonenberg, N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle 2010, 9, 4106–4109. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Qin-Yun, M.; Pang, L.W.; Chen, Z.M.; Zhu, Y.J.; Chen, G.; Chen, J. The significance of MAGED4 expression in non small cell lung cancer as analyzed by real-time fluorescence quantitative PCR. Oncol. Lett. 2012, 4, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Kasamatsu, A.; Miyamoto, I.; Saito, T.; Higo, M.; Endo-Sakamoto, Y.; Shiiba, M.; Tanzawa, H.; Uzawa, K. Overexpression of TMOD1 is associated with enhanced regional lymph node metastasis in human oral cancer. Int. J. Oncol. 2016, 48, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.D.; Hum, N.R.; Thomas, C.B.; Kohlgruberl, A.; Sebastian, A.; Collette, N.M.; Coleman, M.A.; Christiansen, B.A.; Loots, G.G. SOST inhibits prostate cancer invasion. PLoS ONE 2015, 10, e0142058. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.X.; Musco, S.; Lisitsina, N.M.; Yaklichkin, S.Y.; Lisitsyn, N.A. Genomic organization of a new candidate tumor suppressor gene, LRP1B. Genomics 2000, 69, 271–274. [Google Scholar] [CrossRef]
- Flamini, M.I.; Gauna, G.V.; Sottile, M.L.; Nadin, B.S.; Sanchez, A.M.; Vargas-Roig, L.M. Retinoic acid reduces migration of human breast cancer cells: Role of retinoic acid receptor beta. J. Cell. Mol. Med. 2014, 18, 1113–1123. [Google Scholar] [CrossRef]
- Zhang, C.; Zhou, S.; Zhu, Y.; Mashrah, M.; Zhang, X.; He, Z.; Yao, Z.; Zhang, C.; Guo, F.; Hu, Y. Expression pattern of DKK3, dickkopf WNT signaling pathway inhibitor 3, in the malignant progression of oral submucous fibrosis. Oncol. Rep. 2017, 37, 979–985. [Google Scholar] [CrossRef]
- Jette, C.; Peterson, P.W.; Sandoval, I.T.; Manos, E.J.; Hadley, E.; Ireland, C.M.; Jones, D.A. The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. J. Biol. Chem. 2004, 279, 34397–34405. [Google Scholar] [CrossRef] [Green Version]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Schulten, H.J.; Bangash, M.; Karim, S.; Dallol, A.; Hussein, D.; Merdad, A.; Al-Thoubaity, F.K.; Al-Maghrabi, J.; Jamal, A.; Al-Ghamdi, F.; et al. Comprehensive molecular biomarker identification in breast cancer brain metastases. J. Transl. Med. 2017, 15, 269. [Google Scholar] [CrossRef] [Green Version]
- Lage, H.; Perlitz, C.; Abele, R.; Tampé, R.; Dietel, M.; Schadendorf, D.; Sinha, P. Enhanced expression of human ABC-transporter tap is associated with cellular resistance to mitoxantrone. FEBS Lett. 2001, 503, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Siegel, R.L.; Rosenberg, P.S.; Jemal, A. Emerging cancer trends among young adults in the USA: Analysis of a population-based cancer registry. Lancet Public Health 2019, 4, e137–e147. [Google Scholar] [CrossRef] [Green Version]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interf. Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.C.; Huang, K.W.; Liu, C.W.; Chang, Y.C.; Lin, W.M.; Yang, T.Y.; Hsiao, M. Leptin signaling axis specifically associates with clinical prognosis and is multifunctional in regulating cancer progression. Oncotarget 2018, 9, 17210–17219. [Google Scholar] [CrossRef] [Green Version]
- Bilancio, A.; Migliaccio, A. Phosphoinositide 3-kinase assay in breast cancer cell extracts. Methods Mol. Biol. 2014, 1204, 145–153. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin-Notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget 2018, 9, 18239–18253. [Google Scholar] [CrossRef] [Green Version]
- Harmon, T.; Harbuzariu, A.; Lanier, V.; Lipsey, C.C.; Kirlin, W.; Yang, L.; Gonzalez-Perez, R.R. Nanoparticle-linked antagonist for leptin signaling inhibition in breast cancer. World J. Clin. Oncol. 2017, 8, 54–66. [Google Scholar] [CrossRef]
- Fusco, R.; Galgani, M.; Procaccini, C.; Franco, R.; Pirozzi, G.; Fucci, L.; Laccetti, P.; Matarese, G. Cellular and molecular crosstalk between leptin receptor and estrogen receptor-α in breast cancer: Molecular basis for a novel therapeutic setting. Endocr. Relat. Cancer 2010, 17, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Catalano, S.; Mauro, L.; Marsico, S.; Giordano, C.; Rizza, P.; Hago, V.; Montanaro, D.; Maggiolini, M.; Panno, M.L.; Andó, S. Leptin Induces, via ERK1/ERK2 Signal, Functional Activation of Estrogen Receptor α in MCF-7 Cells. J. Biol. Chem. 2004, 279, 19908–19915. [Google Scholar] [CrossRef] [Green Version]
- Catalano, S.; Marsico, S.; Giordano, C.; Mauro, L.; Rizza, P.; Panno, M.L.; Andò, S. Leptin Enhances, via AP-1, Expression of Aromatase in the MCF-7 Cell Line. J. Biol. Chem. 2003, 278, 28668–28676. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Shimomura, Y.; Nakanishi, Y.; Futawatari, T.; Ohtani, K.; Sato, N.; Mori, M. Estrogen increases in vivo leptin production in rats and human subjects. J. Endocrinol. 1997, 154, 285–292. [Google Scholar] [CrossRef]
- Zheng, Q.; Banaszak, L.; Fracci, S.; Basali, D.; Dunlap, S.M.; Hursting, S.D.; Rich, J.N.; Hjlemeland, A.B.; Vasanji, A.; Berger, N.A.; et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr. Relat. Cancer 2013, 20, 797–808. [Google Scholar] [CrossRef]
- Harbuzariu, A.; Oprea-Ilies, G.; Gonzalez-Perez, R. The Role of Notch Signaling and Leptin-Notch Crosstalk in Pancreatic Cancer. Medicines 2018, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Harbuzariu, A.; Rampoldi, A.; Daley-Brown, D.S.; Candelaria, P.; Harmon, T.L.; Lipsey, C.C.; Beech, D.J.; Quarshie, A.; Ilies, G.O.; Gonzalez-Perez, R.R. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 2017, 8, 7740–7752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacy, A.E.; Jansson, P.J.; Richardson, D.R. Molecular Pharmacology of ABCG2 and its role in chemoresistance. Mol. Pharmacol. 2013, 84, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R.R.; Leavis, P.C. A Peptide Derived from the Human Leptin Molecule Is a Potent Inhibitor of the Leptin Receptor Function in Rabbit Endometrial Cells. Endocrine 2003, 21, 185–195. [Google Scholar] [CrossRef]
- Huff, L.M.; Lee, J.S.; Robey, R.W.; Fojo, T. Characterization of gene rearrangements leading to activation of MDR-1. J. Biol. Chem. 2006, 281, 36501–36509. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Control vs. Leptin | Leptin vs. Leptin + mC6 | Function in Cancer | ||
|---|---|---|---|---|---|
| Log FC | p-Value | Log FC | p-Value | ||
| WNT4 | 3.45 | 0.00 | −2.88 | 0.03 | Oncogenesis, regulation of cell fate and development [26] |
| ADHFE1 | 2.37 | 0.03 | −2.34 | 0.04 | Oncogenesis and metabolic reprograming [22] |
| TBC1D3 | 3.99 | 0.03 | −4.66 | 0.04 | Oncogenesis [23] |
| ABCB1 | 2.09 | 0.01 | −1.56 | 0.13 | Multidrug resistance [29] |
| LL22NC03 | 6.35 | 0.01 | −1.47 | 0.08 | Proliferation [24] |
| MAGED4 | 5.20 | 0.13 | −8.13 | 0.05 | Lymph node BC metastasis [30] |
| SOST | −1.39 | 0.37 | 3.57 | 0.01 | Inhibition of WNT pathway [32] |
| LRP1B | −1.56 | 0.37 | 3.49 | 0.01 | Tumor suppressor activity in non-small cell lung cancer [33] |
| EIF4BP7 | −1.13 | 0.75 | 2.27 | 0.02 | Proliferation and overexpression of oncogenes [28] |
| RARB | −1.48 | 0.26 | 2.25 | 0.04 | Tumor suppressors [34] |
| DDK3 | 1.48 | 0.19 | −2.11 | 0.02 | Cancer cell growth, metastasis and chemoresistance [35] |
| RDH5 | 2.27 | 0.03 | 1.01 | 0.99 | Tumorigenesis [36] |
| NEED8 | −1.09 | 0.11 | −3.78 | 0.11 | Cancer cell growth, survival and activation of ubiquitin ligase complexes [37] |
| TMOD1 | − | − | −3.95 | 0.03 | Lymph node metastasis in oral cancer [31] |
| FOXD4L1 | − | − | −4.53 | 0.03 | Cell cycle regulation [38] |
| TAP2 | −1.01 | 0.75 | −5.79 | 0.01 | Multidrug resistance (ABC protein transporter) [39] |
| ITGB3 | 8.06 | 0.00 | 1.01 | 0.87 | Cell migration and invasion [25] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipsey, C.C.; Harbuzariu, A.; Robey, R.W.; Huff, L.M.; Gottesman, M.M.; Gonzalez-Perez, R.R. Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 3794. https://doi.org/10.3390/ijms21113794
Lipsey CC, Harbuzariu A, Robey RW, Huff LM, Gottesman MM, Gonzalez-Perez RR. Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer. International Journal of Molecular Sciences. 2020; 21(11):3794. https://doi.org/10.3390/ijms21113794
Chicago/Turabian StyleLipsey, Crystal C., Adriana Harbuzariu, Robert W. Robey, Lyn M. Huff, Michael M. Gottesman, and Ruben R. Gonzalez-Perez. 2020. "Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer" International Journal of Molecular Sciences 21, no. 11: 3794. https://doi.org/10.3390/ijms21113794