Genome-Wide Identification of Epigenetic Regulators in Quercus suber L.

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
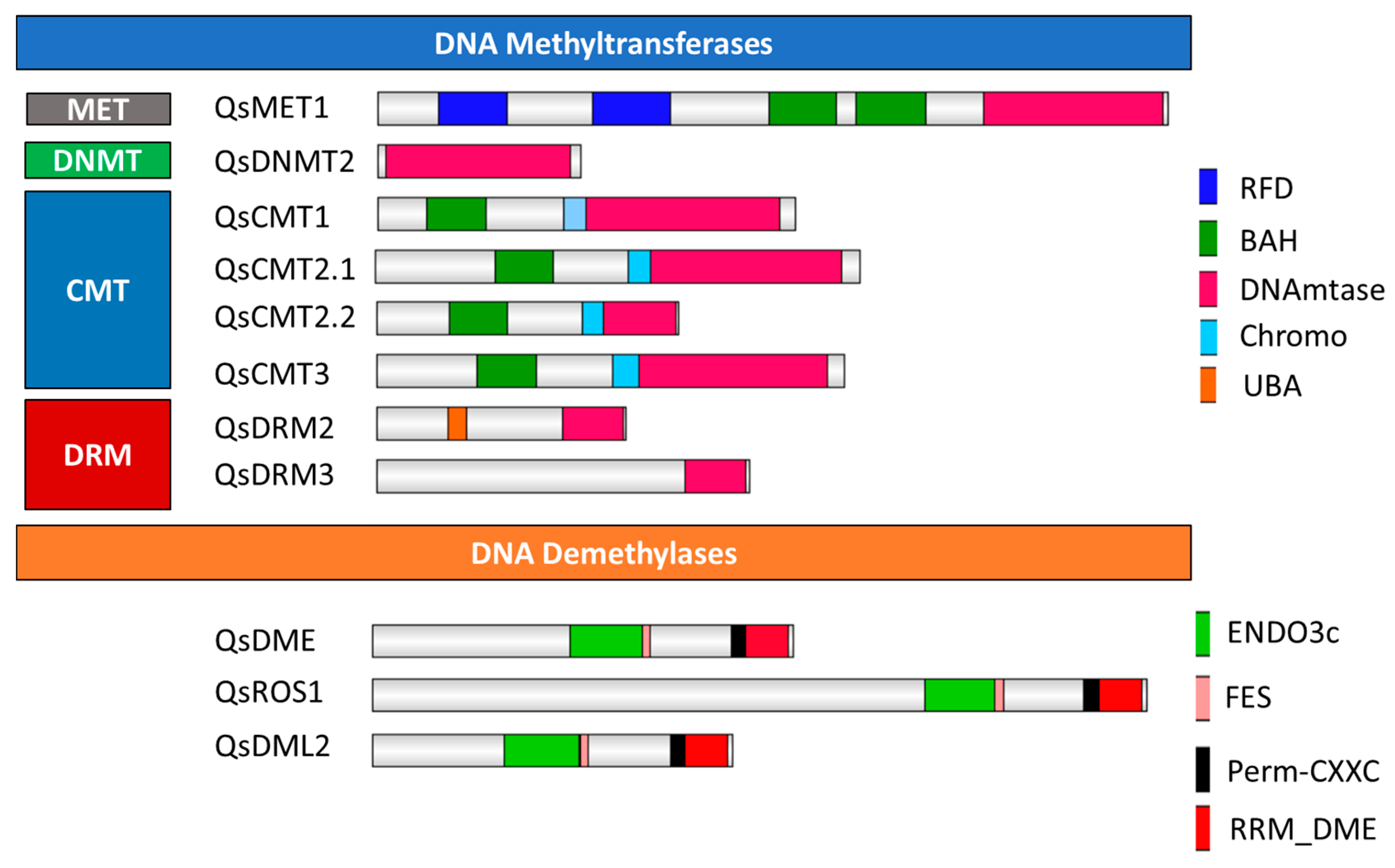
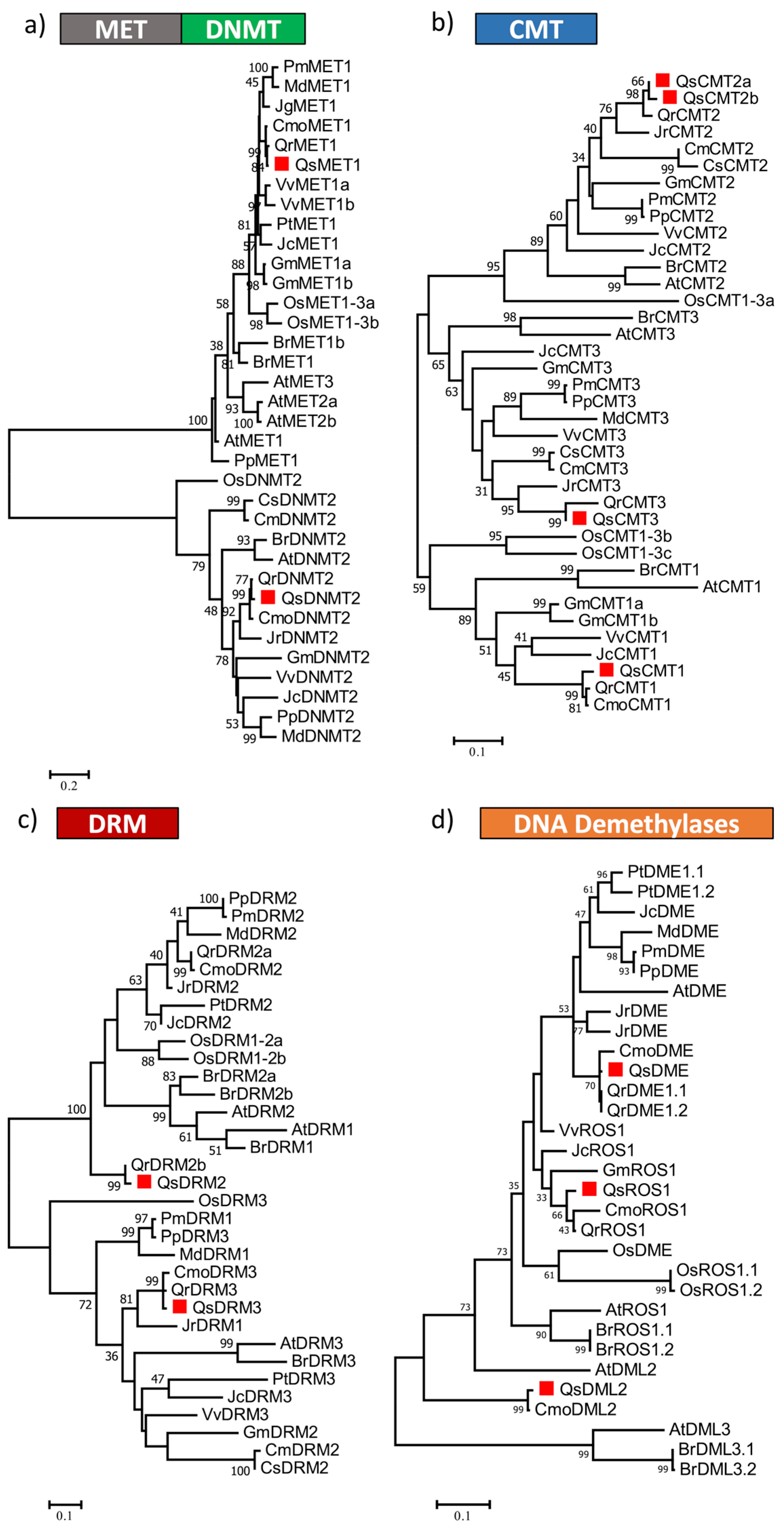
2.1. Identification and Classification of Quercus suber DNA Methyltransferases
2.2. Identification and Classification of Quercus suber DNA Demethylases
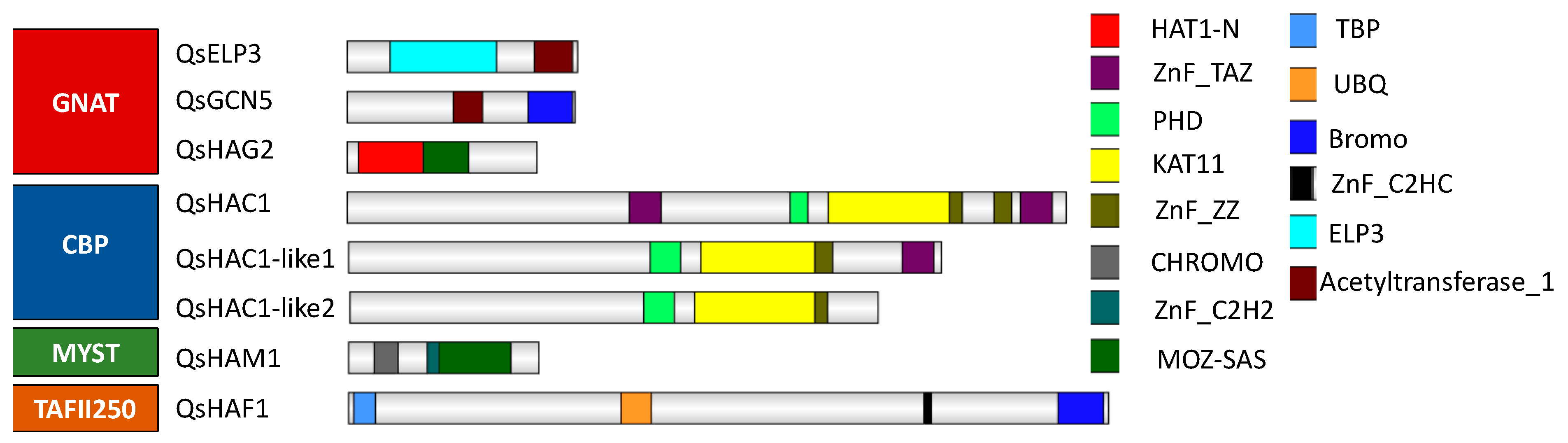
2.3. Identification and Classification of Quercus suber Histone Acetyltransferases
2.4. Identification and Classification of Quercus suber Histone Methyltransferases
2.5. Identification and Classification of Quercus suber Histone Demethylases
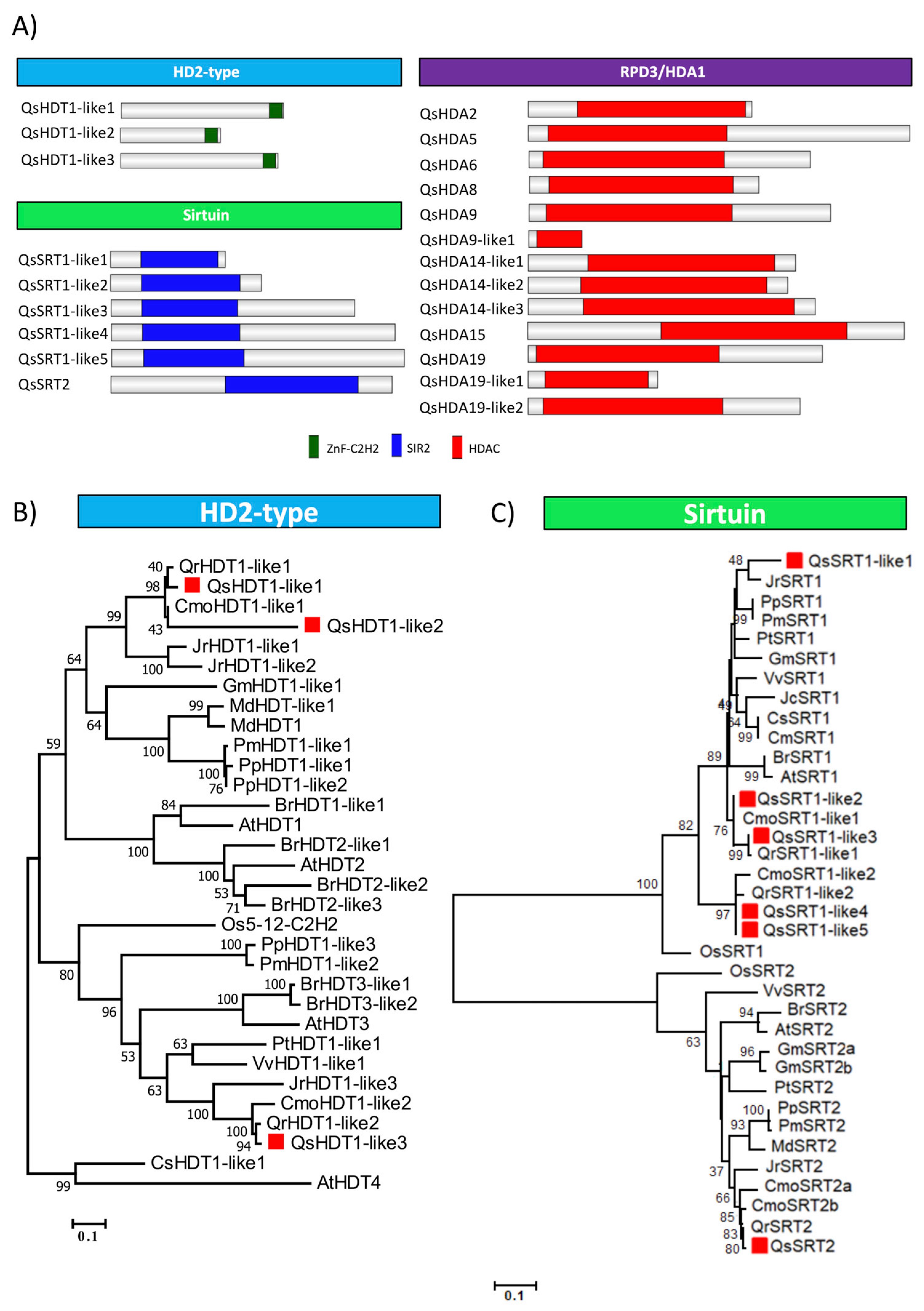
2.6. Identification and Classification of Quercus suber Histone Deacetylases
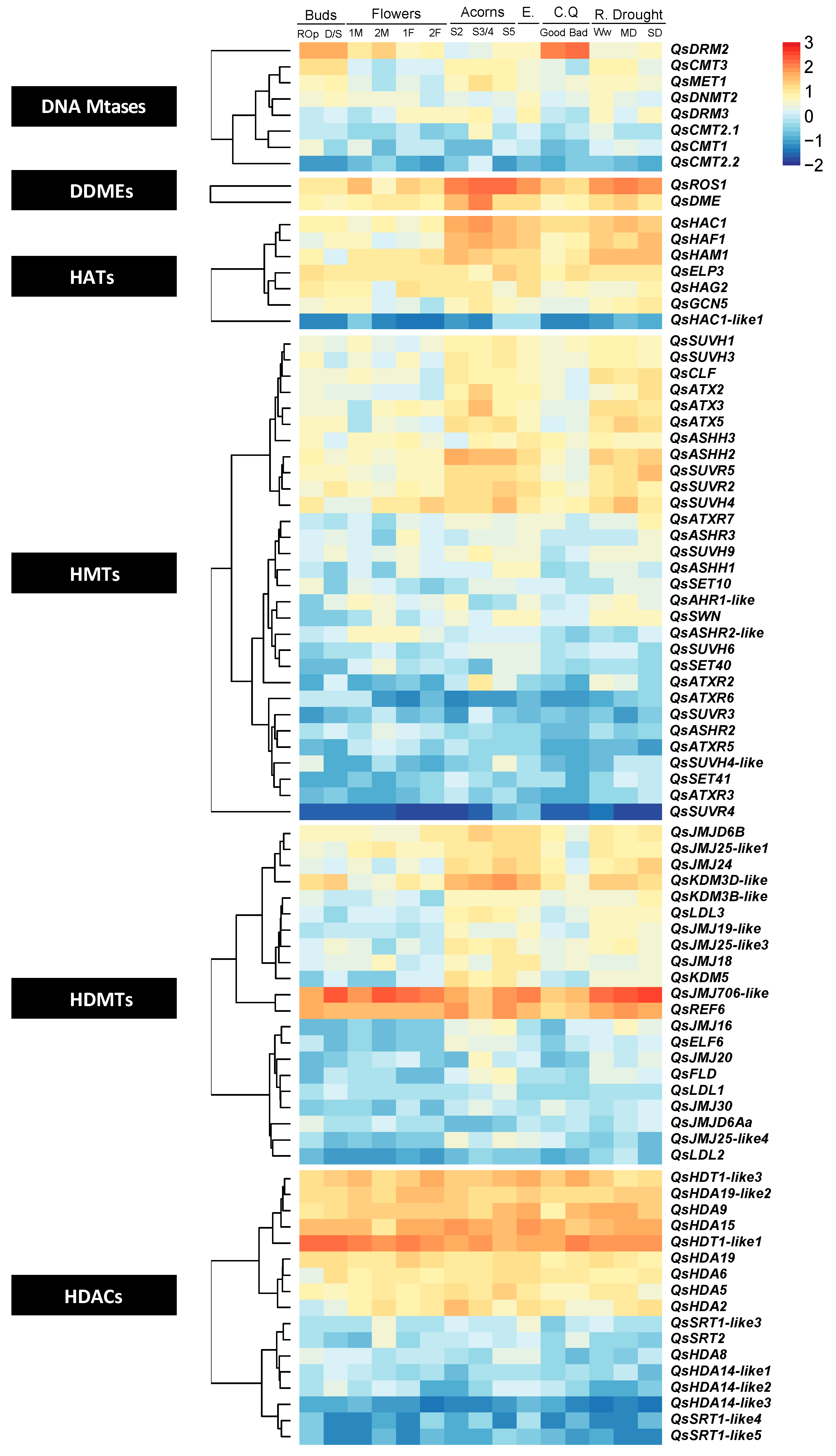
2.7. Gene Expression of Epigenetic Regulators during Quercus suber Plant Development
3. Discussion
4. Materials and Methods
4.1. Identification of Quercus suber DNA (De)Methyltransferases and Histone Modifiers
4.2. Prediction of Protein Domains
4.3. Phylogenetic Analysis
4.4. Transcriptome Data Analysis
4.5. Differential Expression Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gruenbaum, Y.; Naveh-Many, T.; Cedar, H.; Razin, A. Sequence specificity of methylation in higher plant DNA. Nature 1981, 292, 860–862. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Li, E. Structure and Function of Eukaryotic DNA Methyltransferases. Curr. Top. Dev. Biol. 2004, 60, 55–89. [Google Scholar]
- Su, C.; Wang, C.; He, L.; Yang, C.; Wang, Y. Shotgun Bisulfite Sequencing of the Betula platyphylla Genome Reveals the Tree’s DNA Methylation Patterning. Int. J. Mol. Sci. 2014, 15, 22874–22886. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Omalley, R.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Naumann, U.; Daxinger, L.; Kanno, T.; Eun, C.; Long, Q.; Lorkovic, Z.; Matzke, M.; Matzke, A.J.M. Genetic evidence that DNA methyltransferase DRM2 has a direct catalytic role in RNA-directed DNA methylation in Arabidopsis thaliana. Genetics 2011, 187, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldan-Arjona, T.; David, L.; Zhu, J. ROS1, a Repressor of Transcriptional Gene Silencing in Arabidopsis, Encodes a DNA Glycosylase/Lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Gehring, M.; Choi, Y.; Margossian, L.; Pu, H.; Harada, J.J.; Goldberg, R.B.; Pennell, R.I.; Fischer, R.L. Imprinting of the MEA Polycomb Gene Is Controlled by Antagonism between MET1 Methyltransferase and DME Glycosylase. Dev. Cell 2003, 5, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M.; Huh, J.H.; Hsieh, T.-F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, I. Zur Bestimmung der Isocitronensäure neben Citronensäure. Anal. Bioanal. Chem. 1966, 219, 207–208. [Google Scholar] [CrossRef]
- Hebbes, T.R.; Thorne, A.W.; Crane-Robinson, C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988, 7, 1395–1402. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, D.W.-K.; Wang, T.; Chandrasekharan, M.B.; Aramayo, R.; Kertbundit, S.; Hall, T.C. Plant SET domain-containing proteins: Structure, function and regulation. Biochim. Biophys. Acta (BBA) Bioenerg. 2007, 1769, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1 Instance, Histone H3 K9 (H3-K9) Methylation Is Associ-Ated with Heterochromatin Formation (Nakayama et Al and Also. Sidney Kimmel Compr. Cancer Cent. 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, Y.I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone Demethylation by a Family of JmjC Domain-Containing Proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- Kumar, S.; Cheng, X.; Klimašauskas, S.; Sha, M.; Pósfai, J.; Roberts, R.J.; Wilson, G.G. The DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 1994, 22, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pósfai, J.; Bhagwat, A.S.; Pósfai, G.; Roberts, R.J. Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res. 1989, 17, 2421–2435. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulou, A.; Kossida, S. Plant Cytosine-5 DNA Methyltransferases: Structure, Function, and Molecular Evolution. Curr. Top. Dev. Biol. 2007, 90, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Ju, Z.; Gao, C.; Mei, X.; Fu, D.; Zhu, H.; Luo, Y.; Zhu, B. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in Solanum lycopersicum. Gene 2014, 550, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhang, Z.; Han, B.; Li, H.; Dai, H.; He, P.; Tian, H. Isolation of DNA-methyltransferase genes from strawberry (Fragaria × ananassa Duch.) and their expression in relation to micropropagation. Plant Cell Rep. 2009, 28, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Kumari, R.; Tiwari, S.; Goyal, S. Genomic Survey, Gene Expression Analysis and Structural Modeling Suggest Diverse Roles of DNA Methyltransferases in Legumes. PLoS ONE 2014, 9, e88947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianoglio, S.; Moglia, A.; Acquadro, A.; Comino, C.; Portis, E. The genome-wide identification and transcriptional levels of DNA methyltransferases and demethylases in globe artichoke. PLoS ONE 2017, 12, e0181669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Singh, R.K.M.; Malik, G.; Deveshwar, P.; Tyagi, A.K.; Kapoor, S.; Kapoor, M. Rice cytosine DNA methyltransferases-gene expression profiling during reproductive development and abiotic stress. FEBS J. 2009, 276, 6301–6311. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, H.; Xu, Q.; Deng, X. Characterization of DNA Methylation Variations during Fruit Development and Ripening of Sweet Orange. Plant Mol. Biol. Rep. 2014, 33, 1–11. [Google Scholar] [CrossRef]
- Eissenberg, J.C. Molecular biology of the chromo domain: An ancient chromatin module comes of age. Gene 2001, 275, 19–29. [Google Scholar] [CrossRef]
- Callebaut, I.; Courvalin, J.-C.; Mornon, J.-P. The BAH (bromo-adjacent homology) domain: A link between DNA methylation, replication and transcriptional regulation. FEBS Lett. 1999, 446, 189–193. [Google Scholar] [CrossRef]
- Cao, X.; Aufsatz, W.; Zilberman, D.; Mette, M.; Huang, M.S.; Matzke, M.; Jacobsen, S.E. Role of the DRM and CMT3 Methyltransferases in RNA-Directed DNA Methylation. Curr. Biol. 2003, 13, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jacobsen, S.E. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Gao, C.; Bian, X.; Zhao, S.; Zhao, C.; Xia, H.; Song, H.; Hou, L.; Wan, S.; Wang, X.-J. Genome-Wide Identification and Comparative Analysis of Cytosine-5 DNA Methyltransferase and Demethylase Families in Wild and Cultivated Peanut. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cigliano, R.A.; Sanseverino, W.; Cremona, G.; Ercolano, M.R.; Conicella, C.; Consiglio, F.M. Genome-wide analysis of histone modifiers in tomato: Gaining an insight into their developmental roles. BMC Genom. 2013, 14, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Ying, P.; Liu, X.; Li, C.; Xia, R.; Li, J.; Zhao, M. Genome-Wide Identification of Histone Modifiers and Their Expression Patterns during Fruit Abscission in Litchi. Front. Plant Sci. 2017, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Latrasse, D.; Benhamed, M.; Henry, Y.; Domenichini, S.; Kim, W.; Zhou, D.-X.; Delarue, M. The MYST histone acetyltransferases are essential for gametophyte development in Arabidopsis. BMC Plant Biol. 2008, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- De La Cruz, X.; Lois, S.; Sánchez-Molina, S.; Martínez-Balbás, M.A. Do protein motifs read the histone code? BioEssays 2005, 27, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Luo, M.; Zhang, W.; Zhao, J.; Zhang, J.; Wu, K.; Tian, L.; Duan, J. Histone acetyltransferases in rice (Oryza sativa L.): Phylogenetic analysis, subcellular localization and expression. BMC Plant Biol. 2012, 12, 145. [Google Scholar] [CrossRef] [Green Version]
- Ghasroddashti, E.; Pantarotto, J.; Macrae, R.; Gerig, L. SU-FF-T-412: The Reliability of Surrogates in Predicting Tumour Motion: A Comparison of Surrogate Based and Non-Surrogate Based Approach. Med. Phys. 2007, 34, 2496. [Google Scholar] [CrossRef]
- Pontivianne, F.; Blevins, T.; Pikaard, C.S. Arabidopsis Histone Lysine Methyltransferases. Adv. Bot. Res. 2010, 2296, 1–18. [Google Scholar] [CrossRef]
- Emmanuel, A.; Kamm, M.; Cohen, R. Preface. Best Pr. Res. Clin. Gastroenterol. 2002, 16, 527. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, C.; Shen, W.; Ruan, Y. Phylogenetic analysis and classification of the Brassica rapa SET-domain protein family. BMC Plant Biol. 2011, 11, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppmann, V.; Thorstensen, T.; Kristiansen, P.E.; Veiseth, S.V.; Rahman, M.A.; Finne, K.; Aalen, R.B.; Aasland, R. The CW domain, a new histone recognition module in chromatin proteins. EMBO J. 2011, 30, 1939–1952. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Huang, Y. Uncovering the mechanistic basis for specific recognition of monomethylated H3K4 by the CW domain ofArabidopsishistone methyltransferase SDG8. J. Biol. Chem. 2018, 293, 6470–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorstensen, T.; Fischer, A.; Sandvik, S.V.; Johnsen, S.S.; Grini, P.; Reuter, G.; Aalen, R.B. The Arabidopsis SUVR4 protein is a nucleolar histone methyltransferase with preference for monomethylated H3K9. Nucleic Acids Res. 2006, 34, 5461–5470. [Google Scholar] [CrossRef] [Green Version]
- Baumbusch, L.O. The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res. 2001, 29, 4319–4333. [Google Scholar] [CrossRef]
- Zhou, X.; Ma, H. Evolutionary history of histone demethylase families: Distinct evolutionary patterns suggest functional divergence. BMC Evol. Biol. 2008, 8, 294. [Google Scholar] [CrossRef] [Green Version]
- Miguel, A.; De Vega, J.J.; Marum, L.; Chaves, I.; Santo, T.; Leitão, J.; Varela, M.C.; Miguel, C.M. Characterization of the cork oak transcriptome dynamics during acorn development. BMC Plant Biol. 2015, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, R.T.; Fortes, A.M.; Pinheiro, C.; Pereira, H. Comparison of good- and bad-quality cork: Application of high-throughput sequencing of phellogenic tissue. J. Exp. Bot. 2014, 65, 4887–4905. [Google Scholar] [CrossRef] [Green Version]
- Rocheta, M.; Sobral, R.; Magalhães, J.; Amorim, M.I.; Ribeiro, A.T.; Pinheiro, M.; Egas, C.; Morais-Cecã, L.; Costa, M.M.R.; Sobral, R.; et al. Comparative transcriptomic analysis of male and female flowers of monoecious Quercus suber. Front. Plant Sci. 2014, 5, 599. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, A.; Verde, N.; Reis, F.; Martins, I.; Costa, D.; Lino-Neto, T.; Castro, P.H.; Tavares, R.; Azevedo, H. RNA-Seq and Gene Network Analysis Uncover Activation of an ABA-Dependent Signalosome During the Cork Oak Root Response to Drought. Front. Plant Sci. 2016, 6, 3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usié, A.; Simões, F.; Barbosa, P.; Meireles, B.; Chaves, I.; Alves, E.; Folgado, A.; Almeida, M.H.; Matos, J.; Ramos, A.M. Comprehensive Analysis of the Cork Oak (Quercus suber) Transcriptome Involved in the Regulation of Bud Sprouting. Forests 2017, 8, 486. [Google Scholar] [CrossRef] [Green Version]
- Lesur, I.; Le Provost, G.; Bento, P.; Da Silva, C.; Leple, J.-C.; Murat, F.; Ueno, S.; Bartholomé, J.; Lalanne, C.; Ehrenmann, F.; et al. The oak gene expression atlas: Insights into Fagaceae genome evolution and the discovery of genes regulated during bud dormancy release. BMC Genom. 2015, 16, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, A.P.; Usié, A.; Barbosa, P.; Barros, P.M.; Capote, T.; Chaves, I.; Simões, F.; Abreu, I.A.; Carrasquinho, I.; Faro, C.; et al. The draft genome sequence of cork oak. Sci. Data 2018, 5, 180069. [Google Scholar] [CrossRef]
- Aquea, F.; Timmermann, T.; Arce-Johnson, P. Analysis of histone acetyltransferase and deacetylase families of Vitis vinifera. Plant Physiol. Biochem. 2010, 48, 194–199. [Google Scholar] [CrossRef]
- Liew, L.C.; Singh, M.B.; Bhalla, P.L. An RNA-Seq Transcriptome Analysis of Histone Modifiers and RNA Silencing Genes in Soybean during Floral Initiation Process. PLoS ONE 2013, 8, e77502. [Google Scholar] [CrossRef] [Green Version]
- Papaefthimiou, D.; Likotrafiti, E.; Kapazoglou, A.; Bladenopoulos, K.; Tsaftaris, A. Epigenetic chromatin modifiers in barley: III. Isolation and characterization of the barley GNAT-MYST family of histone acetyltransferases and responses to exogenous ABA. Plant Physiol. Biochem. 2010, 48, 98–107. [Google Scholar] [CrossRef]
- Malik, G.; Dangwal, M.; Kapoor, S.; Kapoor, M. Role of DNA methylation in growth and differentiation inPhyscomitrella patensand characterization of cytosine DNA methyltransferases. FEBS J. 2012, 279, 4081–4094. [Google Scholar] [CrossRef]
- Horvath, D.; Sung, S.; Kim, D.; Chao, W.; Anderson, J. Characterization, expression and function of DORMANCY ASSOCIATED MADS-BOX genes from leafy spurge. Plant Mol. Biol. 2010, 73, 169–179. [Google Scholar] [CrossRef]
- Leida, C.; Conesa, A.; Llácer, G.; Badenes, M.L.; Ríos, G. Histone modifications and expression of DAM6 gene in peach are modulated during bud dormancy release in a cultivar-dependent manner. New Phytol. 2011, 193, 67–80. [Google Scholar] [CrossRef]
- Hao, X.; Chao, W.; Yang, Y.; Horvath, D. Coordinated Expression of Flowering Locus T and Dormancy Associated Mads-Box-Like Genes in Leafy Spurge. PLoS ONE 2015, 10, e0126030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, A.; Saito, T.; Sakamoto, D.; Sugiura, T.; Bai, S.; Moriguchi, T. Physiological differences between bud breaking and flowering after dormancy completion revealed byDAMandFT/TFL1expression in Japanese pear (Pyrus pyrifolia). Tree Physiol. 2015, 36, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Q.; Li, J.; Cai, D.; Qian, M.; Jia, H.; Bai, S.; Hussain, S.; Liu, G.; Teng, Y.; Zheng, X. Dormancy-associated MADS-box genes and microRNAs jointly control dormancy transition in pear (Pyrus pyrifolia white pear group) flower bud. J. Exp. Bot. 2015, 67, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.K.; Maurya, J.P.; Azeez, A.; Miskolczi, P.; Tylewicz, S.; Stojkovič, K.; Delhomme, N.; Busov, V.; Bhalerao, R.P. A genetic network mediating the control of bud break in hybrid aspen. Nat. Commun. 2018, 9, 4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomax, A.; Woods, D.P.; Dong, Y.; Bouché, F.; Rong, Y.; Mayer, K.; Zhong, X.; Amasino, R.M. An ortholog of Curly Leaf/Enhancer of Zeste like-1 is required for proper flowering in Brachypodium distachyon. Plant J. 2018, 93, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Ruttink, T.; Arend, M.; Morreel, K.; Storme, V.; Rombauts, S.; Fromm, J.; Bhalerao, R.P.; A Boerjan, W.; Rohde, A. A Molecular Timetable for Apical Bud Formation and Dormancy Induction in Poplar. Plant Cell 2007, 19, 2370–2390. [Google Scholar] [CrossRef] [Green Version]
- Jacob, Y.; Feng, S.; LeBlanc, C.; Bernatavichute, Y.; Stroud, H.; Cokus, S.; Johnson, L.; Pellegrini, M.; Jacobsen, S.; Michaels, S. ATXR5 and ATXR6 Are Novel H3K27 Monomethyltransferases Required for Chromatin Structure and Gene Silencing. Nat. Struct. Mol. Biol. 2009, 16, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Saze, H.; Scheid, O.M.; Paszkowski, J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003, 34, 65–69. [Google Scholar] [CrossRef]
- Ramos, M.J.N.; Rocheta, M.; Carvalho, L.C.; Inácio, V.; Graça, J.; Cecilio, L.M. Expression of DNA methyltransferases is involved in Quercus suber cork quality. Tree Genet. Genomes 2013, 9, 1481–1492. [Google Scholar] [CrossRef] [Green Version]
- Inácio, V.; Martins, M.T.; Graça, J.; Cecilio, L.M. Cork Oak Young and Traumatic Periderms Show PCD Typical Chromatin Patterns but Different Chromatin-Modifying Genes Expression. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Cao, X.; Jacobsen, S.E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 2002, 99, 16491–16498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.P.; Lindroth, A.M.; Cao, X.; Jacobsen, S.E. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 2002, 416, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Bostick, M.; Zhang, X.; Kraft, E.; Henderson, I.R.; Callis, J.; Jacobsen, S.E. The SRA Methyl-Cytosine-Binding Domain Links DNA and Histone Methylation. Curr. Biol. 2007, 17, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.M.; Law, J.A.; Khattar, A.; Henderson, I.R.; Jacobsen, S.E. SRA-Domain Proteins Required for DRM2-Mediated De Novo DNA Methylation. PLoS Genet. 2008, 4, e1000280. [Google Scholar] [CrossRef]
- Johnson, L.M.; Cao, X.; Jacobsen, S.E. Interplay between Two Epigenetic Marks. Curr. Biol. 2002, 12, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Malagnac, F.; Bartee, L.; Bender, J. An Arabidopsis SET domain protein required for maintenance but not establishment of DNA methylation. EMBO J. 2002, 21, 6842–6852. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Johnson, L.M.; Groth, M.; Feng, S.; Hale, C.J.; Li, S.; Vashisht, A.A.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Mechanism of DNA methylation-directed histone methylation by KRYPTONITE. Mol. Cell 2014, 55, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Inácio, V.; Barros, P.M.; Costa, A.; Roussado, C.; Gonçalves, E.M.F.; Costa, R.L.; Graça, J.; Oliveira, M.M.; Cecilio, L.M. Differential DNA Methylation Patterns Are Related to Phellogen Origin and Quality of Quercus suber Cork. PLoS ONE 2017, 12, e0169018. [Google Scholar] [CrossRef]
- Kim, Y.J.; Wang, R.; Gao, L.; Li, N.; Xu, C.; Mang, H.; Jeon, J.; Chen, X.; Zhong, X.; Kwak, J.M.; et al. POWERDRESS and HDA9 interact and promote histone H3 deacetylation at specific genomic sites in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, 14858–14863. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Tai, R.; Yu, C.-W.; Yang, S.; Chen, C.-Y.; Lin, W.-D.; Schmidt, W.; Wu, K. Regulation of flowering time by the histone deacetylase HDA 5 in A rabidopsis. Plant J. 2015, 82, 925–936. [Google Scholar] [CrossRef]
- Liu, X.; Chen, C.-Y.; Wang, K.-C.; Luo, M.; Tai, R.; Yuan, L.; Zhao, M.; Yang, S.; Tian, G.; Cui, Y.; et al. PHYTOCHROME INTERACTING FACTOR3 associates with the histone deacetylase HDA15 in repression of chlorophyll biosynthesis and photosynthesis in etiolated Arabidopsis seedlings. Plant Cell 2013, 25, 1258–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Zhao, Z.; Dong, A.; Soubigou-Taconnat, L.; Renou, J.-P.; Steinmetz, A.; Shen, W. Di- and Tri- but Not Monomethylation on Histone H3 Lysine 36 Marks Active Transcription of Genes Involved in Flowering Time Regulation and Other Processes in Arabidopsis thaliana. Mol. Cell. Biol. 2007, 28, 1348–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Yu, Y.; Meyer, D.; Wu, C.; Shen, W. Prevention of early flowering by expression of FLOWERING LOCUS C requires methylation of histone H3 K36. Nature 2005, 7, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; He, Y.; Jacob, Y.; Noh, Y.-S.; Michaels, S.; Amasino, R.M. Establishment of the Vernalization-Responsive, Winter-Annual Habit in Arabidopsis Requires a Putative Histone H3 Methyl Transferase. Plant Cell 2005, 17, 3301–3310. [Google Scholar] [CrossRef] [Green Version]
- Grini, P.; Thorstensen, T.; Alm, V.; Vizcay-Barrena, G.; Windju, S.S.; Jørstad, T.S.; A Wilson, Z.; Aalen, R.B. The ASH1 HOMOLOG 2 (ASHH2) Histone H3 Methyltransferase Is Required for Ovule and Anther Development in Arabidopsis. PLoS ONE 2009, 4, e7817. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.-Y.; Tamada, Y.; Kang, Y.E.; Amasino, R.M. Arabidopsis Trithorax-Related3/Set Domain Group2 Is Required for the Winter-Annual Habit of Arabidopsis Thaliana. Plant Cell Physiol. 2012, 53, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Napsucialy-Mendivil, S.; Alvarez-Venegas, R.; Shishkova, S.; Dubrovsky, J. Arabidopsis homolog of trithorax1 (ATX1) is required for cell production, patterning, and morphogenesis in root development. J. Exp. Bot. 2014, 65, 6373–6384. [Google Scholar] [CrossRef] [Green Version]
- Saleh, A.; Alvarez-Venegas, R.; Yilmaz, M.; Hou, G.; Sadder, M.; Al-Abdallat, A.; Xia, Y.; Lu, G.; Ladunga, I.; Avramova, Z. The Highly Similar Arabidopsis Homologs of Trithorax ATX1 and ATX2 Encode Proteins with Divergent Biochemical Functions. Plant Cell 2008, 20, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Feng, H.; Yu, Y.; Dong, A.; Shen, W. SDG2-Mediated H3K4 Methylation Is Required for Proper Arabidopsis Root Growth and Development. PLoS ONE 2013, 8, e56537. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Park, O.-S.; Seo, P.J. ATXR2 as a core regulator of de novo root organogenesis. Plant Signal. Behav. 2018, 13, e1449543–e1449605. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.; Wang, Y.; Ma, H.; Zhang, L. Expansion and Functional Divergence of Jumonji C-Containing Histone Demethylases: Significance of Duplications in Ancestral Angiosperms and Vertebrates. Plant Physiol. 2015, 168, 1321–1337. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.S.; Xu, Y.; Wong, J.-Y.; Goh, J.G.; Sun, B.; Wee, W.-Y.; Huang, J.; Ito, T. Jumonji demethylases moderate precocious flowering at elevated temperature via regulation of FLC in Arabidopsis. Nat. Commun. 2014, 5, 5098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wei, W.; Zhu, W.; Su, L.; Xiong, Z.; Zhou, M.; Zheng, Y.; Zhou, D.-X. Histone Deacetylase AtSRT1 Links Metabolic Flux and Stress Response in Arabidopsis. Mol. Plant 2017, 10, 1510–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Sun, Q.; Qin, F.; Li, C.; Zhao, Y.; Zhou, D.X. Down-Regulation of a Silent Information Regulator2-Related Histone Deacetylase Gene, OsSRT1, Induces DNA Fragmentation and Cell Death in Rice. Plant Physiol. 2007, 144, 1508–1519. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Hume Stroud, T.D.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. The Roles of Non-CG Methylation in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64. [Google Scholar] [CrossRef] [Green Version]
- Hartl, M.; Füßl, M.; Boersema, P.J.; Jost, J.O.; Kramer, K.; Bakirbas, A.; Sindlinger, J.; Plöchinger, M.; Leister, D.; Uhrig, R.G.; et al. Lysine acetylome profiling uncovers novel histone deacetylase substrate proteins in Arabidopsis. Mol. Syst. Biol. 2017, 13, 949. [Google Scholar] [CrossRef]
- Ebbs, M.L.; Bender, J. Locus-Specific Control of DNA Methylation by the Arabidopsis SUVH5 Histone Methyltransferase. Plant Cell 2006, 18, 1166–1176. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Leal, J.P.; Abreu, I.A.; Alabaça, C.S.; Almeida, M.H.; Almeida, P.; Almeida, T.; Amorim, M.I.; Araújo, S.; Azevedo, H.; Badia, A.; et al. A comprehensive assessment of the transcriptome of cork oak (Quercus suber) through EST sequencing. BMC Genom. 2014, 15, 371. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Brisse, S. AlienTrimmer: A tool to quickly and accurately trim off multiple short contaminant sequences from high-throughput sequencing reads. Genome 2013, 102, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, R.K.; Lubinsky, B.; Bansode, V.B.; Moinz, M.B.; McCormack, G.P.; Travers, S. QTrim: A novel tool for the quality trimming of sequence reads generated using the Roche/454 sequencing platform. BMC Bioinform. 2014, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Mass Genom. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 31. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, H.G.; Sobral, R.S.; Magalhães, A.P.; Morais-Cecílio, L.; Costa, M.M.R. Genome-Wide Identification of Epigenetic Regulators in Quercus suber L. Int. J. Mol. Sci. 2020, 21, 3783. https://doi.org/10.3390/ijms21113783
Silva HG, Sobral RS, Magalhães AP, Morais-Cecílio L, Costa MMR. Genome-Wide Identification of Epigenetic Regulators in Quercus suber L. International Journal of Molecular Sciences. 2020; 21(11):3783. https://doi.org/10.3390/ijms21113783
Chicago/Turabian StyleSilva, Helena G., Rómulo S. Sobral, Alexandre P. Magalhães, Leonor Morais-Cecílio, and M. Manuela R. Costa. 2020. "Genome-Wide Identification of Epigenetic Regulators in Quercus suber L." International Journal of Molecular Sciences 21, no. 11: 3783. https://doi.org/10.3390/ijms21113783