RNA-seq and ChIP-seq as Complementary Approaches for Comprehension of Plant Transcriptional Regulatory Mechanism



Abstract
:1. Introduction
2. RNA-seq Platform Selections
3. RNA-seq Workflow (Wet Laboratory)
3.1. Total RNA Isolation
3.2. Library Preparation
4. RNA-seq Workflow (Data Analysis)
4.1. Quality Control
4.2. Transcriptome Reconstruction
4.3. Reference-Guided Assembly
4.4. De Novo Assembly
4.5. Expression Quantification and Normalization for Differential Expression Analysis
4.6. Functional Annotation and Pathway Analysis
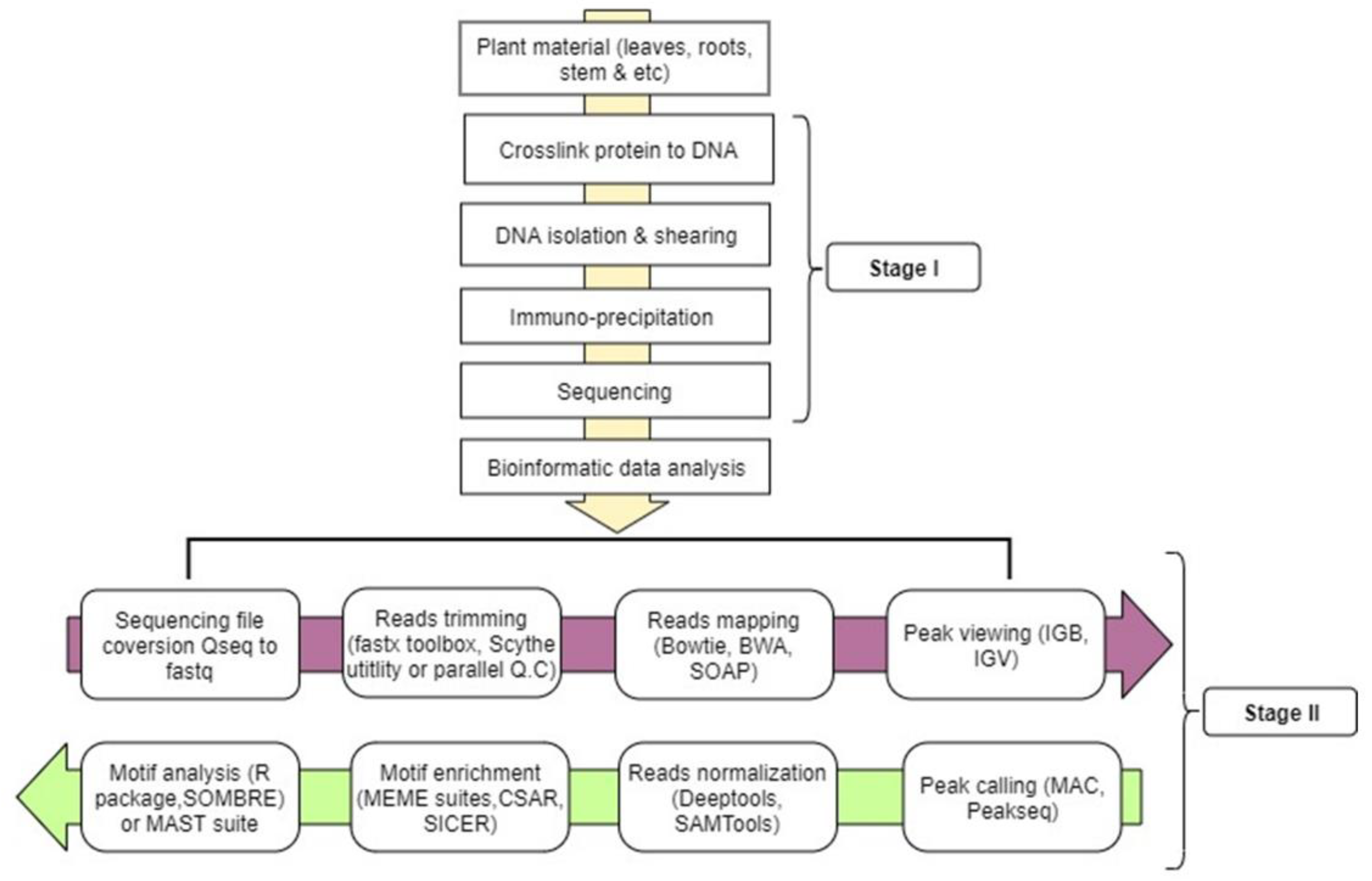
5. ChIP-seq Workflow (Wet Laboratory)
5.1. Crosslinking in Plant Samples
5.2. Chromatin Isolation
5.3. ChIPped-DNA Purification
5.4. Library Construction
6. ChIP-seq Workflow (Data Analysis)
6.1. Reads Mapping
6.2. Enrichment of Genomic Region
7. Genome-Wide Identification of Transcription Factor Co-Regulated Genes by RNA-seq and ChIP-seq
8. Third Generation Sequencing
9. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next Generation Sequencing |
| RNA-seq | RNA Sequencing |
| RIN | RNA Integrity Number |
| rRNA | ribosomal RNA |
| mRNA | micro RNA |
| siRNA | small interfering RNA |
| piRNA | piwi-interacting RNA |
| cDNA | complementary DNA |
| dUTPs | deoxy-UTPs |
| UDG | Uracil-N-Glycosylase |
| SBS | Sequencing by synthesis |
| BAM | Binary of SAM |
| IGV | Integrative Genomic Viewer |
| RPKM | Reads per kilobase of transcript per million mapped reads |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
| TPM | Transcripts per million |
| TMM | Trimmed mean of M-values |
| DEG | Differentially Expressed Gene |
| ChIP-seq | Chromatin Immunoprecipitation and Sequencing |
| UV | Ultraviolet |
| ChIP qPCR | ChIP quantitative realtime PCR |
| ENCODE | Encyclopedia of DNA Elements |
| BWA | Burrow Wheelers alignment |
| GFP | Green fluorescence protein |
| YFP | Yellow fluorescence protein |
| SET | Single-ends tags |
| PET | Paired-ends tags |
| JA | Jasmonic acid |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
References
- Yang, I.; Kim, S. Analysis of whole transcriptome sequencing data: Workflow and software. Genom. Inform. 2015, 13, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.; Montgomery, S. RNA sequencing and analysis. Cold Spring Harb Protoc. 2015, 11, 951–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anamika, K.; Verma, S.; Jere, A.; Desai, A. Transcriptomic Profiling Using Next Generation Sequencing-Advances, Advantages, and Challenges. In Next Generation Sequencing-Advances, Applications and Challenges; IntechOpen: Rijeka, Croatia, 2015; pp. 7355–7365. [Google Scholar]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Bahari, M.N.A.; Sakeh, N.M.; Abdullah, S.N.A.; Ramli, R.R.; Kadkhodaei, S. Transciptome profiling at early infection of Elaeis guineensis by Ganoderma boninense provides novel insights on fungal transition from biotrophic to necrotrophic phase. BMC Plant Biol. 2018, 18, 377. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, W.A.M.; Bártfai, R.; Stunnenberg, H.G. Transcriptome Analysis Using RNA-Seq. In Malaria; Humana Press: Totowa, NJ, USA, 2012; pp. 221–239. [Google Scholar]
- Agarwal, A.; Koppstein, D.; Rozowsky, J.; Sboner, A.; Habegger, L.; Hillier, L.D.W.; Sasidharan, R.; Reinke, V.; Waterston, R.H.; Gerstein, M. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genom. 2010, 11, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kröger, C.; Colgan, A.; Srikumar, S.; Händler, K.; Sivasankaran, S.K.; Hammarlöf, D.L.; Canals, R.; Grissom, J.E.; Conway, T.; Hokamp, K.; et al. An infection-relevant transcriptomic compendium for Salmonella enterica Serovar Typhimurium. Cell Host Microbe 2013, 14, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Hoffmann, S.; Darfeuille, F.; Nature, J.R. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250. [Google Scholar] [CrossRef]
- Saliba, A.; Santos, S.; Vogel, J. New RNA-seq approaches for the study of bacterial pathogens. Curr. Opin. Microbiol. 2017, 35, 78–87. [Google Scholar] [CrossRef]
- Liu, G.; Cheng, H.; You, W.; Song, E.L.; Liu, X.M.; Wan, F.C. Transcriptome profiling of muscle by RNA-Seq reveals significant differences in digital gene expression profiling between Angus and Luxi cattle. Anim. Prod. Sci. 2015, 55, 1172–1178. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Han, R.; Wang, Y.; Li, G.; Liu, X.; Tian, Y.; Kang, X.; Li, Z. Transcriptome analysis of the pectoral muscles of local chickens and commercial broilers using Ribo-Zero ribonucleic acid sequencing. PLoS ONE 2017, 12, e0184115. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Sodhi, S.S.; Song, K.D.; Kim, J.H.; Mongre, R.K.; Sharma, N.; Singh, N.K.; Kim, S.W.; Lee, H.K.; Jeong, D.K. Evaluation of body growth and immunity-related differentially expressed genes through deep RNA sequencing in the piglets of Jeju native pig and Berkshire. Anim. Genet. 2015, 46, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Guo, Y.; Du, W.; Zhang, X.; Li, A.; Miao, X. Global transcriptome analysis identifies differentially expressed genes related to lipid metabolism in Wagyu and Holstein cattle. Sci. Rep. 2017, 7, 5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Su, Z.Y.; Zhang, C.; Gaspar, J.M.; Wang, R.; Hart, R.P.; Verzi, M.P.; Kong, A.N.T. Mechanisms of colitis-accelerated colon carcinogenesis and its prevention with the combination of aspirin and curcumin: Transcriptomic analysis using RNA-seq. Biochem. Pharmacol. 2017, 135, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Jason, D.G.; Joshua, F.K.; Rina, L.D.; Todd, G.R.; Masahiro, O.; John, H.M.; Bruce, S.M. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry 2017, 22, 296. [Google Scholar] [CrossRef]
- Siede, D.; Rapti, K.; Gorska, A.; Katus, H.; Altmüller, J.; Boeckel, J.; Meder, B.; Maack, C.; Völkers, M.; Müller, O.; et al. Identification of circular RNAs with host gene-independent expression in human model systems for cardiac differentiation and disease. J. Mol. Cell. Cardiol. 2017, 109, 48–56. [Google Scholar] [CrossRef]
- Mironova, V.V.; Weinholdt, C.; Grosse, I. RNA-seq data analysis for studying abiotic stress in horticultural plants. In Abiotic Stress Biology in Horticultural Plants; Springer: Tokyo, Japan, 2015; pp. 197–220. ISBN 9784431552512. [Google Scholar]
- Schlüter, U.; Denton, A.K.; Brä utigam, A. Understanding metabolite transport and metabolism in C 4 plants through RNA-seq. Curr. Opin. Plant Biol. 2016, 31, 83–90. [Google Scholar] [CrossRef]
- Ma, W.; Chen, C.; Liu, Y.; Zeng, M.; Meyers, B.C.; Li, J.; Xia, R. Coupling of microRNA-directed phased small interfering RNA generation from long noncoding genes with alternative splicing and alternative polyadenylation in small RNA-mediated gene silencing. New Phytol. 2018, 217, 1535–1550. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, J.; Song, X.; Zhang, Z.; Jiang, Y.; Zhu, Y.; Zhao, H.; Ni, D. An RNA-Seq transcriptome analysis revealing novel insights into aluminum tolerance and accumulation in tea plant. Planta 2017, 246, 91–103. [Google Scholar] [CrossRef]
- Tian, B.; Wang, S.; Todd, T.C.; Johnson, C.D.; Tang, G.; Trick, H.N. Genome-wide identification of soybean microRNA responsive to soybean cyst nematodes infection by deep sequencing. BMC Genom. 2017, 18, 165–183. [Google Scholar] [CrossRef]
- Chotewutmontri, P.; Stiffler, N.; Watkins, K.P.; Barkan, A. Ribosome profiling in Maize. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1676, pp. 165–183. [Google Scholar]
- Zhang, Y.; Gu, L.; Hou, Y.; Wang, L.; Deng, X.; Hang, R.; Chen, D.; Zhang, X.; Zhang, Y.; Liu, C.; et al. Integrative genome-wide analysis reveals HLP1, a novel RNA-binding protein, regulates plant flowering by targeting alternative polyadenylation. Cell Res. 2015, 25, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Duttke, S.; Hetzel, J.; Groth, M.; Feng, S. RNA-directed DNA methylation involves co-transcriptional small-RNA-guided slicing of polymerase V transcripts in Arabidopsis. Nat. Plants 2018, 4, 181. [Google Scholar] [CrossRef] [PubMed]
- Hellman, L.; Fried, M. Electrophoretic Mobility Shift Assay (EMSA) for Detecting Protein-Nucleic Acid Interactions. Nat. Protoc. 2007, 2, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Azzeme, A.M.; Abdullah, S.N.A.; Aziz, M.A.; Wahab, P.E.M. Oil palm drought inducible DREB1 induced expression of DRE/CRT-and non-DRE/CRT-containing genes in lowland transgenic tomato under cold and PEG. Plant Physiol. Biochem. 2017, 112, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.V. Marker proteins for gene expression. Curr. Opin. Biotechnol. 1995, 6, 50–58. [Google Scholar] [CrossRef]
- Feng, S.Y.; Ota, K.; Yamada, Y.; Sawabu, N.; Ito, T. A yeast one-hybrid system to detect methylation-dependent DNA-protein interactions. Biochem. Biophys. Res. Commun. 2004, 313, 922–925. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, M.; Abdullah, S.N.A.; Aziz, M.A.; Namasivayam, P. A novel CBF that regulates abiotic stress response and the ripening process in oil palm (Elaeis guineensis) fruits. Tree Genet. Genomes 2015, 11, 56. [Google Scholar] [CrossRef]
- Aparicio, O.; Geisberg, J.V.; Sekinger, E.; Yang, A.; Moqtaderi, Z.; Struhl, K. Chromatin Immunoprecipitation for Determining the Association of Proteins with Specific Genomic Sequences In Vivo. In Current Protocols in Molecular Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Gilmour, D.S.; Lis, J.T. Detecting protein-DNA interactions in vivo: Distribution of RNA polymerase on specific bacterial genes (UV cross-linking/gene regulation/leucine operon/attenuation). Proc. Natl. Acad. Sci. USA 1984, 81, 4275–4279. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Koga, Y.; Ecker, J.R. Profiling of Transcription Factor Binding Events by Chromatin Immunoprecipitation Sequencing (ChIP-seq). Curr. Protoc. Plant Biol. 2016, 1, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Haring, M.; Offermann, S.; Danker, T.; Horst, I.; Peterhansel, C.; Stam, M. Chromatin immunoprecipitation: Optimization, quantitative analysis and data normalization. Plant Methods 2007, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Adli, M.; Bernstein, B.E. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat. Protoc. 2011, 6, 1656–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Sherwood, B.; Ji, H. Computational Prediction of the Global Functional Genomic Landscape: Applications, Methods, and Challenges. Hum. Hered. 2016, 81, 88–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamimuzzaman, M.; Vodkin, L. Genome-wide identification of binding sites for NAC and YABBY transcription factors and co-regulated genes during soybean seedling development by ChIP-Seq and RNA-Seq. BMC Genom. 2013, 14, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Shi, S.; Hao, Z.; Xiong, W.; Luo, M. OsbZIP81, A Homologue of Arabidopsis VIP1, May Positively Regulate JA Levels by Directly Targetting the Genes in JA Signaling and Metabolism Pathway in Rice. Int. J. Mol. Sci. 2019, 20, 2360. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Zhao, J.; Tzeng, D.T.W.; Liu, Y.; Deng, L.; Yang, T.; Zhai, Q.; Wu, F.; Huang, Z.; Zhou, M.; et al. MYC2 Orchestrates a Hierarchical Transcriptional Cascade that Regulates Jasmonate-Mediated Plant Immunity in Tomato. Plant Cell 2017, 29, 1883–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Basturea, G.; Corney, D.C. RNA-seq Using Next Generation Sequencing A comprehensive review of RNA-seq methodologies RNA-seq Using Next Generation Sequencing. Mater Methods 2016, 3, 203. [Google Scholar]
- Lahens, N.F.; Ricciotti, E.; Smirnova, O.; Toorens, E.; Kim, E.J.; Baruzzo, G.; Hayer, K.E.; Ganguly, T.; Schug, J.; Grant, G.R. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression. BMC Genom. 2017, 18, 602. [Google Scholar] [CrossRef]
- Dündar, F.; Skrabanek, L.; Zumbo, P. Introduction to Differential Gene Expression Analysis Using RNA-Seq; Applied Bioinformatics Core/Weill Cornell Medical College: New York, NY, USA, September 2015. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Corey, D.R. RNA sequencing: Platform selection, experimental design, and data interpretation. Nucleic Acid Ther. 2012, 22, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and quality management in next-generation sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Zhang, Y.; Gordon, W.; Quan, J.; Xi, H.; Du, S.; von Schack, D.; Zhang, B. Comparison of stranded and non-stranded RNA-seq transcriptome profiling and investigation of gene overlap. BMC Genom. 2015, 16, 675. [Google Scholar] [CrossRef] [Green Version]
- Van Nieuwerburgh, F.; Soetaert, S.; Podshivalova, K.; Ay-Lin Wang, E.; Schaffer, L.; Deforce, D.; Salomon, D.R.; Head, S.R.; Ordoukhanian, P. Quantitative Bias in Illumina TruSeq and a Novel Post Amplification Barcoding Strategy for Multiplexed DNA and Small RNA Deep Sequencing. PLoS ONE 2011, 6, e26969. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef]
- Pajoro, A.; Severing, E.; Angenent, G.C.; Immink, R.G.H. Histone H3 lysine 36 methylation affects temperature-induced alternative splicing and flowering in plants. Genome Biol. 2017, 18, 102. [Google Scholar] [CrossRef]
- Piya, S.; Liu, J.; Burch-Smith, T.; Baum, T.J.; Hewezi, T. The roles of Arabidopsis Growth-Regulating Factors 1 and 3 in growth-stress antagonism. J. Exp. Bot. 2019. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatic 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Nature Methods Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Zeng, Z.; Shi, T. Comparative study of de novo assembly and genome-guided assembly strategies for transcriptome reconstruction based on RNA-Seq. Sci. China Life Sci. 2013, 56, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Van Verk, M.C.; Hickman, R.; Pieterse, C.M.J.; Van Wees, S.C.M. RNA-Seq: Revelation of the messengers. Trends Plant Sci. 2013, 18, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Zyprych-Walczak, J.; Szabelska, A.; Handschuh, L.; Górczak, K.; Klamecka, K.; Figlerowicz, M.; Siatkowski, I. The Impact of Normalization Methods on RNA-Seq Data Analysis. BioMed Res. Int. 2015, 2015, 621690. [Google Scholar] [CrossRef]
- Finotello, F.; Camillo, B. Di Measuring differential gene expression with RNA-seq: Challenges and strategies for data analysis. Brief. Funct. Genom. 2014, 14, 130–142. [Google Scholar] [CrossRef]
- Dillies, M.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 2012, 14, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Hendrickson, D.; Sauvageau, M.; Goff, L.; Rinn, J.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562. [Google Scholar] [CrossRef] [Green Version]
- Hardcastle, T.J.; Kelly, K.A. BaySeq: Empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinform. 2010, 11, 422. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; Furió-Tarí, P.; Turra, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucl. Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McCarthy, D.; Robinson, M.; Smyth, G. edgeR: Differential Expression Analysis of Digital Gene Expression Data User’s Guide. Bioconductor User’s Guide. Available online: http://www.bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf (accessed on 17 September 2008).
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.; Blake, J.; Botstein, D.; Butler, H. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucl. Acids Res. 2012, 49, D109–D114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A Refined Protein Classification and Annotation Framework Applicable to Multi-Omics Data Analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nuceic Acids Res. 2017, 45, gkw937. [Google Scholar] [CrossRef] [PubMed]
- Strickler, S.R.; Bombarely, A.; Mueller, L.A. Designing a transcriptome next-generation sequencing project for a nonmodel plant species. Am. J. Bot. 2012, 99, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [Green Version]
- Quievryn, G.; Zhitkovich, A. Loss of DNA–protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 2000, 21, 1573–1580. [Google Scholar] [CrossRef]
- McGhee, J.D.; Von Hippel, P.H. Formaldehyde as a Probe of DNA Structure. I. Reaction with Exocyclic Amino Groups of DNA Bases. Biochemistry 1975, 14, 1281–1296. [Google Scholar] [CrossRef]
- Hoffman, E.A.; Frey, B.L.; Smith, L.M.; Auble, D.T. Formaldehyde crosslinking: A tool for the study of chromatin complexes. J. Biol. Chem. 2015, 290, 26404–26411. [Google Scholar] [CrossRef] [Green Version]
- Orlando, V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem. Sci. 2000, 25, 99–104. [Google Scholar] [CrossRef]
- Saleh, A.; Alvarez-Venegas, R.; Avramova, Z. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat. Protoc. 2008, 3, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Muiño, J.M.; Østerås, M.; Farinelli, L.; Krajewski, P.; Angenent, G.C. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat. Protoc. 2010, 5, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Duś, K.; Mann, M. Proteomic workflow for analysis of archival formalin-fixed and paraffin-embedded clinical samples to a depth of 10000 proteins. Proteom. Clin. Appl. 2013, 7, 225–233. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [Green Version]
- Hills, P.N.; Van Staden, J.; Scott, P. An improved DNA extraction procedure for plant tissues with a high phenolic content. S. Afr. J. Bot. 2005, 68, 549–550. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Winter, C.M.; Wu, M.-F.; Kwon, C.S.; William, D.A.; Wagner, D. PROTOCOL: Chromatin Immunoprecipitation from Arabidopsis Tissues. Arab. Book Am. Soc. Plant Biol. 2014, 12, e0170. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Ye, Z.; Lenz, S.W.; Clark, C.R.; Bharucha, A.; Farrugia, G.; Robertson, K.D.; Zhang, Z.; Ordog, T.; Lee, J.H. Purification of nanogram-range immunoprecipitated DNA in ChIP-seq application. BMC Genom. 2017, 18, 985. [Google Scholar] [CrossRef] [Green Version]
- De Folter, S.; Urbanus, S.L.; van Zuijlen, L.G.; Kaufmann, K.; Angenent, G.C. Tagging of MADS domain proteins for chromatin immunoprecipitation. BMC Plant Biol. 2007, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente, L.; Conesa, A.; Lloret, A.; Badenes, M.L.; Ríos, G. Genome-wide changes in histone H3 lysine 27 trimethylation associated with bud dormancy release in peach. Tree Genet. Genomes 2015, 11, 45. [Google Scholar] [CrossRef]
- Hussey, S.G.; Mizrachi, E.; Groover, A.; Berger, D.K.; Myburg, A.A. Genome-wide mapping of histone H3 lysine 4 trimethylation in Eucalyptus grandis developing xylem. BMC Plant Biol. 2015, 15, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brind’Amour, J.; Liu, S.; Hudson, M.; Chen, C.; Karimi, M.M.; Lorincz, M.C. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 2015, 6, 6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, D.K.; Rohozinski, D.; Song, Q.; Taylor, S.H.; Juenger, T.E.; Harmon, F.G.; Chen, Z.J. Temporal Shift of Circadian-Mediated Gene Expression and Carbon Fixation Contributes to Biomass Heterosis in Maize Hybrids. PLoS Genet. 2016, 12, e1006197. [Google Scholar] [CrossRef] [PubMed]
- Posé, D.; Verhage, L.; Ott, F.; Yant, L.; Mathieu, J.; Angenent, G.C.; Immink, R.G.H.; Schmid, M. Temperature-dependent regulation of flowering by antagonistic FLM variants. Nature 2013, 503, 414–417. [Google Scholar] [CrossRef]
- Fan, M.; Bai, M.Y.; Kim, J.G.; Wang, T.; Oh, E.; Chen, L.; Park, C.H.; Son, S.H.; Kim, S.K.; Mudgett, M.B.; et al. The bHLH transcription factor HBI1 mediates the trade-off between growth and pathogen-associated molecular pattern–triggered immunity in Arabidopsis. Plant Cell 2014, 26, 828–841. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Chen, X.; Qian, S.; Zhong, X. The plant-specific histone residue Phe41 is important for genome-wide H3.1 distribution. Nat. Commun. 2018, 9, 630. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, W.; Singh, R.; Zheng, Y.; Cao, Z.; Li, M.; Lunde, C.; Hake, S.; Zhang, Z. GRF-interacting factor1 Regulates Shoot Architecture and Meristem Determinacy in Maize. Plant Cell 2018, 30, 360–374. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.; Park, D.; Myers, K.; Grass, J.; Kiley, P.; Landick, R.; Keleş, S. dPeak: High Resolution Identification of Transcription Factor Binding Sites from PET and SET ChIP-Seq Data. PLoS Comput. Biol. 2013, 9, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.; Krajewski, P.; Ladunga, I.; Lefebvre, C.; Li, Q.; Liu, T.; Madrigal, P.; Taslim, C.; Zhang, J. Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLoS Comput. Biol. 2013, 9, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Qiao, Z.; Qi, W.; Wang, Q.; Yuan, Y.; Yang, X.; Tang, Y.; Mei, B.; Lv, Y.; Zhao, H.; et al. Genome-Wide Characterization of cis -Acting DNA Targets Reveals the Transcriptional Regulatory Framework of Opaque2 in Maize. Plant Cell 2015, 27, 532–545. [Google Scholar] [CrossRef] [Green Version]
- Yant, L.; Ott, F.; Keller, H.; Weigel, D.; Schmid, M. 20th International of Conference on Arabidopsis Research, Scotland, United Kingdom. In Proceedings of the Design and Analysis of ChIP-Seq Experiments in Plants: A Systematic Comparison of ChIP-Seq and ChIP-chip for APETALA2 (AP2), FD, and SCHLAFMÜTZE (SMZ), Edinburgh, UK, 30 June–4 July 2009. [Google Scholar]
- Nakato, R.; Shirahige, K. Recent advances in ChIP-seq analysis: From quality management to whole-genome annotation. Brief. Bioinform. 2017, 18, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepke, S.; Wold, B.; Mortazavi, A. Computation for ChIP-seq and RNA-seq studies. Nat. Methods 2009, 6, S22. [Google Scholar] [CrossRef] [PubMed]
- Furey, T.S. ChIP–seq and beyond: New and improved methodologies to detect and characterize protein–DNA interactions. Nat. Rev. Genet. 2012, 13, 840–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, L.R.; DeModena, J.; Sternberg, P.W. RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 2009, 324, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Rozowsky, J.; Euskirchen, G.; Auerbach, R.K.; Zhang, Z.D.; Gibson, T.; Bjornson, R.; Carriero, N.; Snyder, M.; Gerstein, M.B. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 2009, 27, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Muiño, J.M.; Kaufmann, K.; van Ham, R.C.; Angenent, G.C.; Krajewski, P. ChIP-seq Analysis in R (CSAR): An R package for the statistical detection of protein-bound genomic regions. Plant Methods 2011, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Taleb, H.A.; AL-Dherasi, A.; Al-Mosaib, S.; Alnoud, M.; Vilaphong, S. Peak Calling Algorithms and Their Applications for Next-Generation Sequencing Technologies. Indian J. Nat. Sci. 2019, 9, 16659–16670. [Google Scholar]
- Johannes, F.; Wardenaar, R.; Colomé-Tatché, M.; Mousson, F.; de Graaf, P.; Mokry, M.; Guryev, V.; Timmers, H.T.M.; Cuppen, E.; Jansen, R.C. Comparing genome-wide chromatin profiles using ChIP-chip or ChIP-seq. Bioinformatics 2010, 26, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Burrows, M.; Wheeler, D. A Block-Sorting Lossless Data Compression Algorithm; Technical Report for Digital Equipment Corporation: Maynard, MA, USA, May 1994. [Google Scholar]
- Ferragina, P.; Manzini, G. An Experimental Study of an Opportunistic Index. In Proceedings of the Twelfth Annual ACM-SIAM Symposium on Discrete Algorithms, Washington, DC, USA, 7–9 January 2001; pp. 269–278. [Google Scholar]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Xi, Y.; Pan, X.; Li, Z.; Kaestner, K.; Tyler, J.; Dent, S.; He, X.; Li, W. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013, 23, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latrasse, D.; Jégu, T.; Li, H.; de Zelicourt, A.; Raynaud, C.; Legras, S.; Gust, A.; Samajova, O.; Veluchamy, A.; Rayapuram, N.; et al. MAPK-triggered chromatin reprogramming by histone deacetylase in plant innate immunity. Genome Biol. 2017, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Ricardi, M.M.; González, R.M.; Zhong, S.; Domínguez, P.G.; Duffy, T.; Turjanski, P.G.; Salgado Salter, J.D.; Alleva, K.; Carrari, F.; Giovannoni, J.J.; et al. Genome-wide data (ChIP-seq) enabled identification of cell wall-related and aquaporin genes as targets of tomato ASR1, a drought stress-responsive transcription factor. BMC Plant Biol. 2014, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Yu, H.; Xiong, G.; Wang, J.; Jiao, Y.; Liu, G.; Jing, Y.; Meng, X.; Hu, X.; Qian, Q.; et al. Genome-Wide Binding Analysis of the Transcription Activator IDEAL PLANT ARCHITECTURE1 Reveals a Complex Network Regulating Rice Plant Architecture. Plant Cell 2013, 25, 3743–3759. [Google Scholar] [CrossRef] [Green Version]
- Mateos, J.L.; Madrigal, P.; Tsuda, K.; Rawat, V.; Richter, R.; Romera-Branchat, M.; Fornara, F.; Schneeberger, K.; Krajewski, P.; Coupland, G. Combinatorial activities of SHORT VEGETATIVE PHASE and FLOWERING LOCUS C define distinct modes of flowering regulation in Arabidopsis. Genome Biol. 2015, 16, 31. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, L.; Qi, B.; Zhao, B.; Ko, E.E.; Riggan, N.D.; Chin, K.; Qiao, H. EIN2 mediates direct regulation of histone acetylation in the ethylene response. Proc. Natl. Acad. Sci. USA 2017, 114, 201707937. [Google Scholar] [CrossRef] [Green Version]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2011, 27, 1696–1697. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Bailey, T.L. DREME: Motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011, 27, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas-Chollier, M.; Darbo, E.; Herrmann, C.; Defrance, M.; Thieffry, D.; van Helden, J. A complete workflow for the analysis of full-size ChIP-seq (and similar) data sets using peak-motifs. Nat. Protoc. 2012, 7, 1551–1568. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenhart, R.A.; Bai, Y.; Valter De Oliveira, L.F.; Bucker Neto, L.; Schunemann, M.; Maraschin, F.D.S.; Mariath, J.; Silverio, A.; Sachetto-Martins, G.; Margis, R.; et al. New insights into aluminum tolerance in rice: The ASR5 protein binds the STAR1 promoter and other aluminum-responsive genes. Mol. Plant 2014, 7, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xie, S.; Dong, X.; Zhao, X.; Zeng, B.; Chen, J.; Li, H.; Yang, W.; Zhao, H.; Wang, G.; et al. Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res. 2014, 24, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Lei, K.J.; Lin, Y.M.; Ren, J.; Bai, L.; Miao, Y.C.; An, G.Y.; Song, C.P. Modulation of the phosphate-deficient responses by MicroRNA156 and its targeted SQUAMOSA PROMOTER BINDING PROTEIN-LIKE 3 in arabidopsis. Plant Cell Physiol. 2016, 57, 192–203. [Google Scholar] [CrossRef]
- Pedmale, U.V.; Huang, S.S.C.; Zander, M.; Cole, B.J.; Hetzel, J.; Ljung, K.; Reis, P.A.B.; Sridevi, P.; Nito, K.; Nery, J.R.; et al. Cryptochromes Interact Directly with PIFs to Control Plant Growth in Limiting Blue Light. Cell 2016, 164, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.; Cheng, Y.-J.; Hu, B.; Ma, X.; Wu, R.; Wang, J.-W.; Liu, C. Nonrandom domain organization of theArabidopsisgenome at the nuclear periphery. Genome Res. 2017, 27, 1162–1173. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Jia, J.; Zhao, X.; Zhang, M.Y.; Huang, X.; Ji, E.; Ni, L.; Jiang, M. The ascorbate peroxidase APX1 is a direct target of a zinc finger transcription factor ZFP36 and a late embryogenesis abundant protein OsLEA5 interacts with ZFP36 to co-regulate OsAPX1 in seed germination in rice. Biochem. Biophys. Res. Commun. 2018, 495, 339–345. [Google Scholar] [CrossRef]
- Balanzà, V.; Martínez-Fernández, I.; Sato, S.; Yanofsky, M.F.; Kaufmann, K.; Angenent, G.C.; Bemer, M.; Ferrándiz, C. Genetic control of meristem arrest and life span in Arabidopsis by a FRUITFULL-APETALA2 pathway. Nat. Commun. 2018, 9, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.C.; Vining, K.; Dombrowski, J.E. Genome-wide (ChIP-seq) identification of target genes regulated by BdbZIP10 during paraquat-induced oxidative stress. BMC Plant Biol. 2018, 18, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth–metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.W.; Lee, D.K.; Jung, H.; Chung, P.J.; Kim, Y.S.; Choi, Y.D.; Suh, J.W.; Kim, J.K. Overexpression of OsTF1L, a rice HD-Zip transcription factor, promotes lignin biosynthesis and stomatal closure that improves drought tolerance. Plant Biotechnol. J. 2019, 17, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Cheng, L.; Gao, Y.; Zhang, B.; Zheng, X.; Wang, L.; Li, P.; Sun, Q.; Li, H. Plant HP1 protein ADCP1 links multivalent H3K9 methylation readout to heterochromatin formation. Cell Res. 2019, 29, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Chen, W.; Wang, B.; Yao, Q.M.; Valliyodan, B.; Bai, M.Y.; Zhao, M.Z.; Ye, H.; Wang, Z.Y.; Nguyen, H.T. GmBZL3 acts as a major BR signaling regulator through crosstalk with multiple pathways in Glycine max. BMC Plant Biol. 2019, 19, 86. [Google Scholar] [CrossRef] [Green Version]
- Collani, S.; Neumann, M.; Yant, L.; Schmid, M. FT Modulates Genome-Wide DNA-Binding of the bZIP Transcription Factor FD. Plant Physiol. 2019, 180, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Ye, T.; Krebs, A.R.; Choukrallah, M.-A.; Keime, C.; Plewniak, F.; Davidson, I.; Tora, L. seqMINER: An integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011, 39, e35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 165, 1280–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, Y.; Shi, Z.; Jin, Y.; Sun, H.; Xie, F.; Zhang, L. Biosynthesis and Signal Transduction of ABA, JA, and BRs in Response to Drought Stress of Kentucky Bluegrass. Int. J. Mol. Sci. 2019, 20, 1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Wang, Y.; Fang, Y.; Zeng, L.; Xu, J.; Yu, H.; Shi, Z.; Pan, J.; Zhang, D.; Kang, S.; et al. A Rare Allele of GS2 Enhances Grain Size and Grain Yield in Rice. Mol. Plant 2015, 8, 1455–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapham, R.; Lee, L.Y.; Tsugama, D.; Lee, S.; Mengiste, T.; Gelvin, S.B. VIP1 and its homologs are not required for agrobacterium-mediated transformation, but play a role in botrytis and salt stress responses. Front. Plant Sci. 2018, 9, 749. [Google Scholar] [CrossRef]
- Tsugama, D.; Liu, S.; Takano, T. A bZIP Protein, VIP1, Is a Regulator of Osmosensory Signaling in Arabidopsis 1[W]. Plant Physiol. 2012, 159, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Jenior, M.L.; Koumpouras, C.C.; Westcott, S.L.; Highlander, S.K. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 2016, 4, e1869. [Google Scholar] [CrossRef] [Green Version]
- Gueidan, C.; Elix, J.A.; McCarthy, P.M.; Roux, C.; Mallen-Cooper, M.; Kantvilas, G. PacBio amplicon sequencing for metabarcoding of mixed DNA samples from lichen herbarium specimens. MycoKeys 2019, 53, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameur, A.; Kloosterman, W.P.; Hestand, M.S. Single-Molecule Sequencing: Towards Clinical Applications. Trends Biotechnol. 2019, 37, 72–85. [Google Scholar] [CrossRef]
- Laboratories, K. Partek: How to Integrate ChIP-Seq and RNA-Seq Data. Available online: https://www.partek.com/how-to-integrate-chip-seq-and-rna-seq-data/ (accessed on 9 December 2019).
- Wang, S.; Sun, H.; Ma, J.; Zang, C.; Wang, C.; Wang, J.; Tang, Q.; Meyer, C.A.; Zhang, Y.; Liu, X.S. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013, 8, 2502–2515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhang, B.; Lin, L.-L.; Zhao, S. Evaluation and comparison of computational tools for RNA-seq isoform quantification. BMC Genom. 2017, 18, 583. [Google Scholar] [CrossRef]
- O’Geen, H.; Henry, I.M.; Bhakta, M.S.; Meckler, J.F.; Segal, D.J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015, 43, 3389–3404. [Google Scholar] [CrossRef]
- Partridge, E.C.; Watkins, T.A.; Mendenhall, E.M. Every transcription factor deserves its map: Scaling up epitope tagging of proteins to bypass antibody problems. BioEssays 2016, 38, 801–811. [Google Scholar] [CrossRef]
- Savic, D.; Partridge, E.C.; Newberry, K.M.; Smith, S.B.; Meadows, S.K.; Roberts, B.S.; Mackiewicz, M.; Mendenhall, E.M.; Myers, R.M. CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. Genome Res. 2015, 25, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Moradpour, M.; Abdulah, S.N.A. CRISPR/dCas9 platforms in plants: Strategies and applications beyond genome editing. Plant Biotechnol. J. 2019. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chen, C.; Chen, H.; Wang, S.; Chen, X.; Cui, Y. Verification of DNA motifs in Arabidopsis using CRISPR/Cas9-mediated mutagenesis. Plant Biotechnol. J. 2018, 16, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Findings | Antibody Type | Refernce |
|---|---|---|---|
| Abiotic factor | Abscisic acid stress (ASR5) a TF binds Sensitive To Aluminum Rhizotoxicity 1 (STAR1) promoter in other to positively response against Aluminum stress in rice. | Anti-ASR5 | Arenhart et al. (2014) [145] |
| Developmental + immunity | ROS and defense responsive genes were repressed by HBI1 indicating defense function of HBI1 and also indirectly plays a role in repressing growth through activation of growth-inhibiting HLH genes. HBI1 was also learned to bind to positive activators brassinosteroids function. | Anti-YFP | Fan et al. (2014) [112] |
| Genetics | In genetic imprinting, some subsets of genes are expressed according to their parental origin. Paternally expressed genes (PEGs) were associated to maternal-specific H3K27me3. | Anti-H3K27me3 | Zhang et al. (2014) [146] |
| Abiotic + developmental | Abscisic, Stress, Ripening (ASR1) from tomato is upregulated in drought stress which acts primarily in the cell wall. | Anti-ASR1 | Ricardi et al. (2014) [134] |
| Developmental | SQUAMOSA Promoter Binding Protein-Like3 (SPL3) bind GTAC motif of phosphate starvation responsive gene promoters like PLDZ2, miR399f, and Pht1;5. | Anti-HA | Lei et al. (2016) [147] |
| Developmental | Combinatorial action affect MADS-box transcription factors FLC and SVP in flowering shows gibberellins’ processing genes. | Anti-GFP | Mateos et al. (2015) [137] |
| Genetics | In circadian clock of Maize hybrids, expression of morning-phased genes from binding with ZmCCA1 encourages growth vigor and photosynthesis. | Anti-CCA1 | Ko et al. (2016) [110] |
| Photosynthesis | Discovers E-box variant binding motif for Phytochrome interacting factor 4/5 (PIF4 and PIF5) in Cryptochromes (CRYs) during exposure to low blue light and CRY2 association with PIF4/5. | Anti-HA | Pedmale et al. (2016) [148] |
| Cellular | Shows chromatin domain organization at the nuclei periphery of Arabidopsis. The domain is a clear translation of a repressed environment that contains jumping genes, heterochromatic marks and silenced coding genes. | Anti-GFP | Bi et al. (2017) [149] |
| Immunity | Using both ChIP-seq and RNA-seq, 655 MYC2 binding were identified in response to Jasmonic acid genes. Also found MYC2 TFs that function in late defense stage. | Anti-GFP | Du et al. (2017) [40] |
| Immunity | After flagellin (flg22) treatment, HD2B targets chromatin were hyperacetylated responsible in plant immune defense and phosphorylation while hypoacetylated marks function in metabolic regulation, plastid organization, and chloroplast. | Anti-GFP | Latrasse et al. (2017) [133] |
| Biochemical | A zinc finger TF of rice ZFP36 inhibits ROS production by binding to ascorbate peroxidase known to have specificity to hydrogen peroxide. | Anti-ZFP36 | Huang et al. (2018) [150] |
| Developmental | Maize GIF in leaves and stems promotes meristematic determinacy and shoot architecture. ChIP-seq has found several GIF1 targets including mostly some transcriptional regulators like UB3, ZMPLATZ5, ZMARR7, bHLH and MYB family members. | Anti-GFP | Zhang et al. (2018) [114] |
| Developmental | FRUITFULL (FUL), a TF that directly repressed APETALA2 expression promotes meristem arrest and maintains the sequential expression of meristem maintenance factor WUSCHEL. | Anti-GFP | Balanzà et al. (2018) [151] |
| Abiotic factor | bZIP10 found to be active in Zinc regulation in Brachypodium which relate to oxidative stress and a motif homologous to Arabidopsis was found TGDCGACA. | Anti-GFP | Martin et al. (2018) [152] |
| Abiotic factor | Growth-Regulating Factor 4 (GRF4) TF co-interacts with growth inhibition regulator DELLA to regulate carbon, nitrogen metabolism and growth. | Anti-FLAG | Li et al. (2018) [153] |
| Abiotic factor | Rice OsTF1L mapped drought related stress and lignin biosynthesis genes. | Anti-MYC and anti-RNA Pol II | Bang et al. (2019) [154] |
| Epigenetics | Genome-wide ADCP1 is linked with chromosome enrichment site (pericentrome) and co-localization with H3K9me2. | Anti-GFP | Zhao et al. (2019) [155] |
| General | GmBZL3 is a brassinesteroids signaling molecule cross talking with many pathways like disease-related, immunity response pathways and hormone signaling. | GmBZL3 antibody | Song et al. (2019) [156] |
| Developmental | A Leucine zipper domain TF FD plays a crucial role in floral transition. | Anti-GFP | Collani et al. (2019) [157] |
| General | Found new Oryza VIP1 response element (OVRE) cis-element in abiotic and biotic responses. | Anti-FLAG | Liu et al. (2019) [39] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, I.I.; Kong, S.L.; Akmar Abdullah, S.N.; Munusamy, U. RNA-seq and ChIP-seq as Complementary Approaches for Comprehension of Plant Transcriptional Regulatory Mechanism. Int. J. Mol. Sci. 2020, 21, 167. https://doi.org/10.3390/ijms21010167
Muhammad II, Kong SL, Akmar Abdullah SN, Munusamy U. RNA-seq and ChIP-seq as Complementary Approaches for Comprehension of Plant Transcriptional Regulatory Mechanism. International Journal of Molecular Sciences. 2020; 21(1):167. https://doi.org/10.3390/ijms21010167
Chicago/Turabian StyleMuhammad, Isiaka Ibrahim, Sze Ling Kong, Siti Nor Akmar Abdullah, and Umaiyal Munusamy. 2020. "RNA-seq and ChIP-seq as Complementary Approaches for Comprehension of Plant Transcriptional Regulatory Mechanism" International Journal of Molecular Sciences 21, no. 1: 167. https://doi.org/10.3390/ijms21010167