Autophagy Deficiency in Renal Proximal Tubular Cells Leads to an Increase in Cellular Injury and Apoptosis under Normal Fed Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
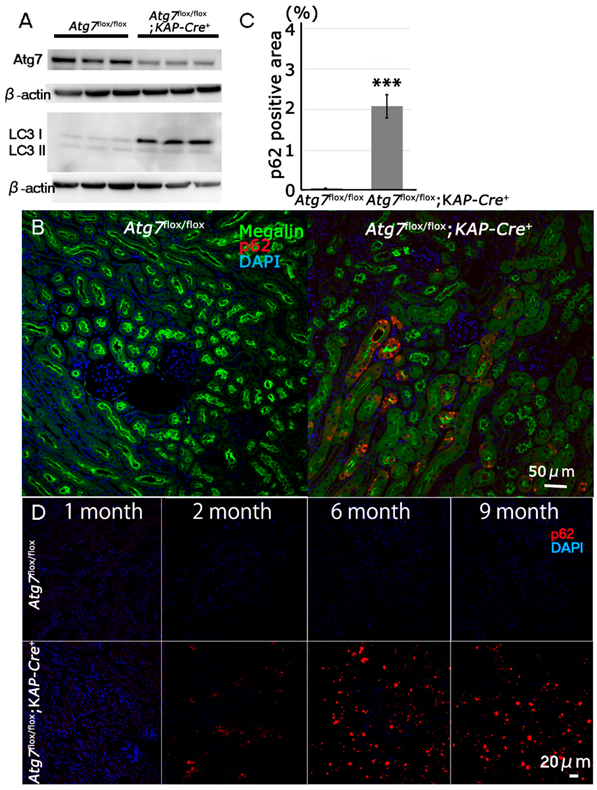
2.1. Newly Generated Atg7flox/flox;KAP-Cre+ Mice Exhibit Molecular Changes in Proximal Tubular Cells Including an Age-Dependent Increase in p62/SQSTM1 under Normal Fed Conditions
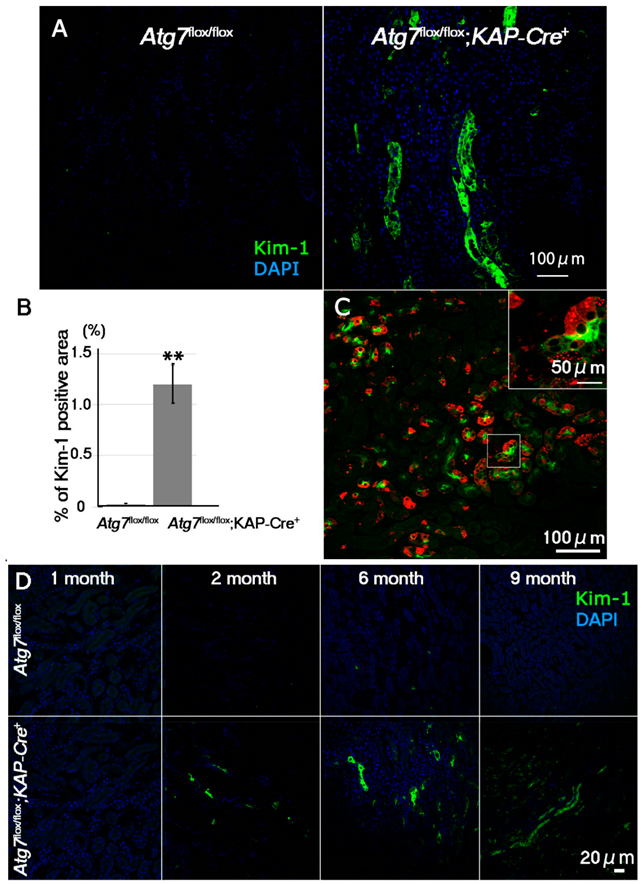
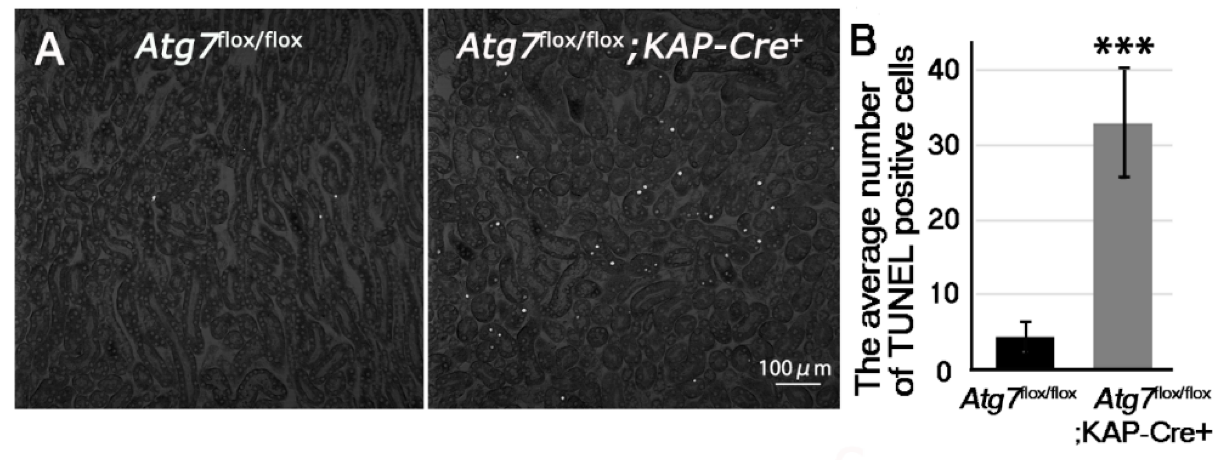
2.2. Increases in Positive Immunosignals of Kim-1 and TUNEL in Normal Fed Atg7flox/flox;KAP-Cre+ Mice
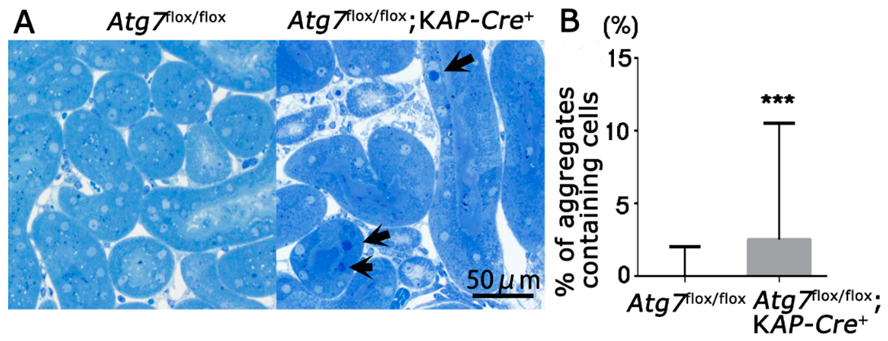
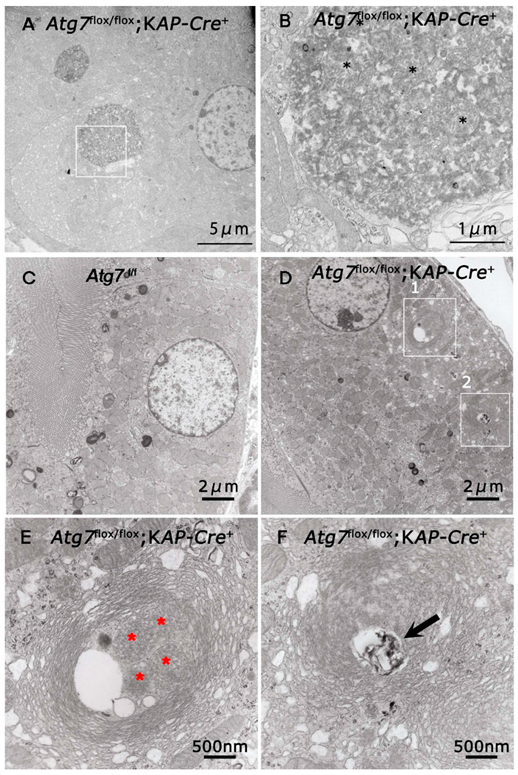
2.3. Accumulation of Abnormal Structures in the Renal Proximal Epithelial Tubular Cells of Atg7flox/flox;KAP-Cre+ Mice
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Urinary Examinations
4.3. Blood Examinations
4.4. Antibodies
4.5. Fixation and Embedding for Light and Electron Microscopy
4.6. Immunohistochemistry
4.7. TUNEL Staining
4.8. Immunoblot Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Christensen, E.I.; Nielsen, S. Structural and functional features of protein handling in the kidney proximal tubule. Semin. Nephrol. 1991, 11, 414–439. [Google Scholar] [PubMed]
- Maack, T.; Johnson, V.; Kau, S.T.; Figueiredo, J.; Sigulem, D. Renal filtration, transport, and metabolism of low-molecular-weight proteins: A review. Kidney Int. 1979, 16, 251–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, C.; Tanida, I.; Ohmuraya, M.; Oliva Trejo, J.A.; Kakuta, S.; Sunabori, T.; Uchiyama, Y. Lack of Cathepsin D in the Renal Proximal Tubular Cells Resulted in Increased Sensitivity against Renal Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2019, 20, 1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlton, J.R.; Portilla, D.; Okusa, M.D. A basic science view of acute kidney injury biomarkers. Nephrol. Dial. Transplant. 2014, 29, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy-Thandavan, S.; Jiang, M.; Schoenlein, P.; Dong, Z. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Renal Physiol. 2009, 297, F244–F256. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Eskelinen, E.L. New insights into the mechanisms of macroautophagy in mammalian cells. Int. Rev. Cell Mol. Biol. 2008, 266, 207–247. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Tanida-Miyake, E.; Ueno, T.; Kominami, E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem. 2001, 276, 1701–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I.; Tanida-Miyake, E.; Komatsu, M.; Ueno, T.; Kominami, E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J. Biol. Chem. 2002, 277, 13739–13744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sou, Y.S.; Tanida, I.; Komatsu, M.; Ueno, T.; Kominami, E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J. Biol. Chem. 2006, 281, 3017–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.; Livingston, M.; Hao, J.; Li, L.; Mei, C.; Dong, Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci. Rep. 2016, 6, 22171. [Google Scholar] [CrossRef]
- Ding, Y.; Davisson, R.L.; Hardy, D.O.; Zhu, L.J.; Merrill, D.C.; Catterall, J.F.; Sigmund, C.D. The kidney androgen-regulated protein promoter confers renal proximal tubule cell-specific and highly androgen-responsive expression on the human angiotensinogen gene in transgenic mice. J. Biol. Chem. 1997, 272, 28142–28148. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Havasi, A.; Borkan, S.C. Apoptosis and acute kidney injury. Kidney Int. 2011, 80, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Koike, M.; Nakanishi, H.; Saftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S.; et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Weinberg, J.M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Hu, W.; Feng, C.; Mao, X.; Jin, K.; Ye, Y. Increasing Proliferation of Intrinsic Tubular Cells after Renal Ischemia-reperfusion Injury in Adult Rat. Aging Dis. 2015, 6, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusaka, T.; Niimura, F.; Shimizu, A.; Pastan, I.; Saito, A.; Kobori, H.; Nishiyama, A.; Ichikawa, I. Liver angiotensinogen is the primary source of renal angiotensin II. J. Am. Soc. Nephrol. 2012, 23, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Koike, M.; Shibata, M.; Waguri, S.; Yoshimura, K.; Tanida, I.; Kominami, E.; Gotow, T.; Peters, C.; von Figura, K.; Mizushima, N.; et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am. J. Pathol. 2005, 167, 1713–1728. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, C.; Tanida, I.; Oliva Trejo, J.A.; Kakuta, S.; Uchiyama, Y. Autophagy Deficiency in Renal Proximal Tubular Cells Leads to an Increase in Cellular Injury and Apoptosis under Normal Fed Conditions. Int. J. Mol. Sci. 2020, 21, 155. https://doi.org/10.3390/ijms21010155
Suzuki C, Tanida I, Oliva Trejo JA, Kakuta S, Uchiyama Y. Autophagy Deficiency in Renal Proximal Tubular Cells Leads to an Increase in Cellular Injury and Apoptosis under Normal Fed Conditions. International Journal of Molecular Sciences. 2020; 21(1):155. https://doi.org/10.3390/ijms21010155
Chicago/Turabian StyleSuzuki, Chigure, Isei Tanida, Juan Alejandro Oliva Trejo, Soichiro Kakuta, and Yasuo Uchiyama. 2020. "Autophagy Deficiency in Renal Proximal Tubular Cells Leads to an Increase in Cellular Injury and Apoptosis under Normal Fed Conditions" International Journal of Molecular Sciences 21, no. 1: 155. https://doi.org/10.3390/ijms21010155