The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury


Abstract
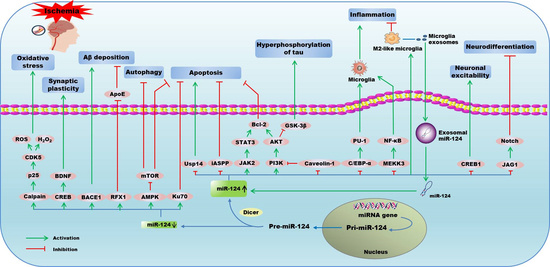
:
1. Introduction
2. MiR-124 in Ischemic Encephalopathy
2.1. MiR-124 and Ischemic Stroke
2.1.1. Ischemic Stroke
2.1.2. Mechanisms of miR-124 in Ischemic Stroke
2.1.3. Exosomal miR-124 in Ischemic Stroke
2.2. MiR-124 and AD
2.2.1. Alzheimer’s Disease
2.2.2. Mechanisms of miR-124 in Alzheimer’s Disease
2.3. MiR-124 and PD
2.3.1. Parkinson’s Disease
2.3.2. Mechanisms of miR-124 in Parkinson’s Disease
2.4. MiR-124 and Epilepsy
2.4.1. Epilepsy
2.4.2. Mechanisms of miR-124 in Epilepsy
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IE | Ischemic encephalopathy |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| MiR-124 | MicroRNA-124 |
| BBB | Blood-brain barrier |
| MiRNAs | MicroRNAs |
| CNS | Central nervous system |
| CIRI | Cerebral ischemia reperfusion injury |
| VSNL-1 | Visinin-like protein-1 |
| CDK4 | Cyclin-dependent kinase family 4 |
| Sox9 | SRY-box transcription factor |
| OGD | Oxygen and glucose deprivation |
| JAG1 | Jagged-1 |
| REST | RE1-silencing transcription factor |
| Usp14 | Ubiquitin specific peptidase 14 |
| PI3K | Phosphoinositide 3-kinase |
| mTOR | Mammalian target of rapamycin |
| PKB/AKT | Protein kinase B |
| GAP-43 | Growth associated protein-43 |
| TNF-α | Necrosis factor-α |
| IL-10 | Interleukin -10 |
| C/EBP-α | CCAAT/enhancer-binding protein alpha |
| Bcl-2 | B-cell Lymphoma-2 |
| iASPP | Inhibitory member of the apoptosis-stimulating proteins of p53 family |
| Bcl-xl | B-cell lymphoma-extra large |
| I/R | Ischemia/reperfusion |
| JAK2 | Janus tyrosine kinase 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| Ku70 | X-ray repair cross-complementing protein 6 |
| GLT1 | Glutamate transporter 1 |
| Aβ | β-amyloid |
| NFTs | Neurofibrillary tangles |
| BACE1 | Beta-site amyloid precursor protein cleaving enzyme1 |
| GSK3β | Glycogen synthase kinase 3β |
| RFX1 | Regulatory factor X1 |
| ApoE | Apolipoprotein E |
| CREB | cAMP-response element-binding protein |
| BDNF | Brain-derived neurotrophic factor |
| PTPN1 | Tyrosine-protein phosphatase non-receptor type 1 |
| SNpc | Substantia nigra pars compacta |
| MPTP | 1-methyl-4-pheny-1,2,3,6-tetrahydropyridine |
| MPP | Methyl phenyl pyridinium |
| AMPK | Adenosine 5’-monophosphate-activated protein kinase |
| ANAX5 | Annexin 5 |
| ERK | Extracellular regulated protein kinases |
| Casp-3 | Caspase-3 |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| MEKK3 | Mitogen-activated protein kinase kinase kinase 3 |
| p62 | Sequestosome 1 |
| p-p38 | Phosphor-p38 mitogen-activated protein kinase |
| CRE | cAMP-response element |
| NRSF | Neuron restrictive silencer factor |
References
- Chen, Z.X.; Xu, Q.Q.; Shan, C.S.; Shi, Y.H.; Wang, Y.; Chang, R.C.; Zheng, G.Q. Borneol for Regulating the Permeability of the Blood-Brain Barrier in Experimental Ischemic Stroke: Preclinical Evidence and Possible Mechanism. Oxid. Med. Cell Longev. 2019, 2019, 2936737. [Google Scholar] [CrossRef]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef]
- Graf, R.; Kataoka, K.; Rosner, G.; Heiss, W.D. Cortical deafferentation in cat focal ischemia: Disturbance and recovery of sensory functions in cortical areas with different degrees of cerebral blood flow reduction. J. Cereb. Blood Flow Metab. 1986, 6, 566–573. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Mao, X.; Xie, L.; Jin, K. Ablation of neurogenesis attenuates recovery of motor function after focal cerebral ischemia in middle-aged mice. PLoS ONE 2012, 7, e46326. [Google Scholar] [CrossRef]
- Escobar, I.; Xu, J.; Jackson, C.W.; Perez-Pinzon, M.A. Altered Neural Networks in the Papez Circuit: Implications for Cognitive Dysfunction after Cerebral Ischemia. J. Alzheimers Dis. 2019, 67, 425–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.H.; Liu, H. Life and death in the trash heap: The ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral Ischemia. Ageing Res. Rev. 2017, 34, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, J.C. Ischemic stroke: experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, M.; Xu, Z.Q.; Gao, C.Y.; Fang, C.Q.; Deng, J.; Yan, J.C.; Wang, Y.J.; Zhou, H.D. Vascular risk aggravates the progression of Alzheimer’s disease in a Chinese cohort. J Alzheimers Dis. 2010, 20, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Vemuganti, R. Mechanisms of Parkinson’s disease-related proteins in mediating secondary brain damage after cerebral ischemia. J Cereb. Blood Flow Metab. 2017, 37, 1910–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaenisch, N.; Liebmann, L.; Guenther, M.; Hubner, C.A.; Frahm, C.; Witte, O.W. Reduced tonic inhibition after stroke promotes motor performance and epileptic seizures. Sci. Rep. 2016, 6, 26173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousad, S.A.; Isenovic, E.R. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2017, 15, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Hamzei, T.S.; Kho, W.; Aswendt, M.; Collmann, F.M.; Green, C.; Adamczak, J.; Tennstaedt, A.; Hoehn, M. Dynamic Modulation of Microglia/Macrophage Polarization by miR-124 after Focal Cerebral Ischemia. J. Neuroimmune Pharmacol. 2016, 11, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkul, A.; Akyol, A.; Yenisey, C.; Arpaci, E.; Kiylioglu, N.; Tataroglu, C. Oxidative stress in acute ischemic stroke. J. Clin. Neurosci. 2007, 14, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Qu, Y.; Zhu, J.; Zhang, L.; Huang, L.; Liu, H.; Li, S.; Mu, D. MiR-30d-5p Plays an Important Role in Autophagy and Apoptosis in Developing Rat Brains After Hypoxic-Ischemic Injury. J. Neuropathol. Exp. Neurol. 2017, 76, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Meng, A. MicroRNA-124 expression in the brains of rats during early cerebral ischemia and reperfusion injury is associated with cell apoptosis involving STAT3. Exp. Ther. Med. 2019, 17, 2870–2876. [Google Scholar] [CrossRef] [Green Version]
- Kanagaraj, N.; Beiping, H.; Dheen, S.T.; Tay, S.S. Downregulation of miR-124 in MPTP-treated mouse model of Parkinson’s disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience 2014, 272, 167–179. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Khoshnam, S.E.; Winlow, W.; Farbood, Y.; Moghaddam, H.F.; Farzaneh, M. Emerging Roles of microRNAs in Ischemic Stroke: As Possible Therapeutic Agents. J. Stroke 2017, 19, 166–187. [Google Scholar] [CrossRef]
- Deo, M.; Yu, J.Y.; Chung, K.H.; Tippens, M.; Turner, D.L. Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev. Dyn. 2006, 235, 2538–2548. [Google Scholar] [CrossRef] [Green Version]
- Akerblom, M.; Sachdeva, R.; Barde, I.; Verp, S.; Gentner, B.; Trono, D.; Jakobsson, J. MicroRNA-124 is a subventricular zone neuronal fate determinant. J. Neurosci. 2012, 32, 8879–8889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ye, Y.; Zhu, Z.; Mo, L.; Lin, C.; Wang, Q.; Wang, H.; Gong, X.; He, X.; Lu, G.; et al. MiR-124 Regulates Apoptosis and Autophagy Process in MPTP Model of Parkinson’s Disease by Targeting to Bim. Brain Pathol. 2016, 26, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.C.; Pastrana, E.; Tavazoie, M.; Doetsch, F. MiR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 2009, 12, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamzei, T.S.; Kho, W.; Riou, A.; Wiedermann, D.; Hoehn, M. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials 2016, 91, 151–165. [Google Scholar] [CrossRef]
- Rajasethupathy, P.; Fiumara, F.; Sheridan, R.; Betel, D.; Puthanveettil, S.V.; Russo, J.J.; Sander, C.; Tuschl, T.; Kandel, E. Characterization of small RNAs in Aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron 2009, 63, 803–817. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, V.A.; Firestein, B.L. The dendritic tree and brain disorders. Mol. Cell Neurosci. 2012, 50, 10–20. [Google Scholar] [CrossRef]
- Sonntag, K.C.; Woo, T.U.; Krichevsky, A.M. Converging miRNA functions in diverse brain disorders: A case for miR-124 and miR-126. Exp. Neurol. 2012, 235, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Soreq, H.; Wolf, Y. NeurimmiRs: MicroRNAs in the neuroimmune interface. Trends Mol. Med. 2011, 17, 548–555. [Google Scholar] [CrossRef]
- Volny, O.; Kasickova, L.; Coufalova, D.; Cimflova, P.; Novak, J. MicroRNAs in Cerebrovascular Disease. Adv. Exp. Med. Biol. 2015, 888, 155–195. [Google Scholar] [PubMed]
- Wang, W.; Wang, X.; Chen, L.; Zhang, Y.; Xu, Z.; Liu, J.; Jiang, G.; Li, J.; Zhang, X.; Wang, K.; et al. The microRNA miR-124 suppresses seizure activity and regulates CREB1 activity. Expert Rev. Mol. Med. 2016, 18, e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Chopp, M.; Zhang, R.L.; Tao, T.; Wang, X.L.; Kassis, H.; Hozeska-Solgot, A.; Zhang, L.; Chen, C.; Zhang, Z.G. MicroRNA profiling in subventricular zone after stroke: MiR-124a regulates proliferation of neural progenitor cells through Notch signaling pathway. PLoS ONE 2011, 6, e23461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.Z.; Yin, J.B.; Yang, J.J.; Cao, L. Regulatory factor X1 depresses ApoE-dependent Abeta uptake by miRNA-124 in microglial response to oxidative stress. Neuroscience 2017, 344, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Xiang, Y.; Li, D.; Liang, J.; Zhang, X.; Zhou, F.; Qiao, M.; Nie, Y.; He, Y.; Cheng, J.; et al. MiR-124-3p attenuates hyperphosphorylation of Tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3beta pathway in N2a/APP695swe cells. Oncotarget 2017, 8, 24314–24326. [Google Scholar]
- Liu, X.; Li, F.; Zhao, S.; Luo, Y.; Kang, J.; Zhao, H.; Yan, F.; Li, S.; Ji, X. MicroRNA-124-mediated regulation of inhibitory member of apoptosis-stimulating protein of p53 family in experimental stroke. Stroke 2013, 44, 1973–1980. [Google Scholar] [CrossRef]
- Doeppner, T.R.; Doehring, M.; Bretschneider, E.; Zechariah, A.; Kaltwasser, B.; Muller, B.; Koch, J.C.; Bahr, M.; Hermann, D.M.; Michel, U. MicroRNA-124 protects against focal cerebral ischemia via mechanisms involving Usp14-dependent REST degradation. Acta Neuropathol. 2013, 126, 251–265. [Google Scholar] [CrossRef]
- Song, Y.; Li, Z.; He, T.; Qu, M.; Jiang, L.; Li, W.; Shi, X.; Pa, J.; Zhang, L.; Wang, Y.; et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics 2019, 9, 2910–2923. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, J.; Shao, Y.; Wan, D. Catalpol may improve axonal growth via regulating miR-124 regulated PI3K/AKT/mTOR pathway in neurons after ischemia. Ann. Transl. Med. 2019, 7, 306. [Google Scholar] [CrossRef]
- Wang, C.; Wei, Z.; Jiang, G.; Liu, H. Neuroprotective mechanisms of miR-124 activating PI3K/Akt signaling pathway in ischemic stroke. Exp. Ther. Med. 2017, 13, 3315–3318. [Google Scholar] [CrossRef] [Green Version]
- Shu, K.; Zhang, Y. Protodioscin protects PC12 cells against oxygen and glucose deprivation-induced injury through miR-124/AKT/Nrf2 pathway. Cell Stress Chaperones 2019, 24, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Gui, H.; Li, Q.; Luo, Z.M.; Zheng, M.J.; Duan, J.L.; Liu, X. MicroRNA-124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci. Ther. 2013, 19, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Liu, J.L.; Li, J.P.; Xiao, F.; Zhang, Z.X.; Zhang, L. MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J. Mol. Neurosci. 2014, 52, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Doeppner, T.R.; Kaltwasser, B.; Sanchez-Mendoza, E.H.; Caglayan, A.B.; Bahr, M.; Hermann, D.M. Lithium-induced neuroprotection in stroke involves increased miR-124 expression, reduced RE1-silencing transcription factor abundance and decreased protein deubiquitination by GSK3beta inhibition-independent pathways. J. Cereb. Blood Flow Metab. 2017, 37, 914–926. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.N.; Li, W.F.; Li, F.; Zhang, Z.; Dai, Y.D.; Xu, A.L.; Qi, C.; Gao, J.M.; Gao, J. Resveratrol improves learning and memory in normally aged mice through microRNA-CREB pathway. Biochem. Biophys. Res. Commun. 2013, 435, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, D.; Huang, H.Z.; Wang, Z.H.; Hou, T.Y.; Yang, X.; Pang, P.; Wei, N.; Zhou, Y.F.; Dupras, M.J.; et al. A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol. Psychiatr. 2018, 83, 395–405. [Google Scholar] [CrossRef]
- An, F.; Gong, G.; Wang, Y.; Bian, M.; Yu, L.; Wei, C. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 2017, 8, 114065–114071. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Wang, H.; Ye, Y.; Shu, Y.; Deng, Y.; He, X.; Lu, G.; Zhang, S. MiR-124 regulates cell apoptosis and autophagy in dopaminergic neurons and protects them by regulating AMPK/mTOR pathway in Parkinson’s disease. Am. J. Transl. Res. 2016, 8, 2127–2137. [Google Scholar]
- Geng, L.; Liu, W.; Chen, Y. MiR-124-3p attenuates MPP (+)-induced neuronal injury by targeting STAT3 in SH-SY5Y cells. Exp. Biol. Med. 2017, 242, 1757–1764. [Google Scholar] [CrossRef]
- Dong, R.F.; Zhang, B.; Tai, L.W.; Liu, H.M.; Shi, F.K.; Liu, N.N. The Neuroprotective Role of MiR-124-3p in a 6-Hydroxydopamine-Induced Cell Model of Parkinson’s Disease via the Regulation of ANAX5. J. Cell Biochem. 2018, 119, 269–277. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Q.; Zhang, J.; Pan, W.; Zhao, J.; Xu, Y. Long non-coding RNA MALAT1 contributes to cell apoptosis by sponging miR-124 in Parkinson disease. Cell Biosci. 2017, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Ye, Y.; Mao, H.; Lu, F.; He, X.; Lu, G.; Zhang, S. MicroRNA-124 regulates the expression of MEKK3 in the inflammatory pathogenesis of Parkinson’s disease. J. Neuroinflamm. 2018, 15, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Zhu, Z.; Wu, J.; Zhang, Y.; Zhang, H.; Sun, X.; Qian, C.; Wang, B.; Xie, L.; Zhang, S.; et al. MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson’s disease. FASEB J. 2019, 33, 8648–8665. [Google Scholar] [CrossRef] [PubMed]
- Brennan, G.P.; Dey, D.; Chen, Y.; Patterson, K.P.; Magnetta, E.J.; Hall, A.M.; Dube, C.M.; Mei, Y.T.; Baram, T.Z. Dual and Opposing Roles of MicroRNA-124 in Epilepsy Are Mediated through Inflammatory and NRSF-Dependent Gene Networks. Cell Rep. 2016, 14, 2402–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schouten, M.; Fratantoni, S.A.; Hubens, C.J.; Piersma, S.R.; Pham, T.V.; Bielefeld, P.; Voskuyl, R.A.; Lucassen, P.J.; Jimenez, C.R.; Fitzsimons, C.P. MicroRNA-124 and -137 cooperativity controls caspase-3 activity through BCL2L13 in hippocampal neural stem cells. Sci. Rep. 2015, 5, 12448. [Google Scholar] [CrossRef] [Green Version]
- Feigin, V.L.; Roth, G.A.; Naghavi, M.; Parmar, P.; Krishnamurthi, R.; Chugh, S.; Mensah, G.A.; Norrving, B.; Shiue, I.; Ng, M.; et al. Global burden of stroke and risk factors in 188 countries, during 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016, 15, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Toni, D.; Fiorelli, M.; Gentile, M.; Bastianello, S.; Sacchetti, M.L.; Argentino, C.; Pozzilli, C.; Fieschi, C. Progressing neurological deficit secondary to acute ischemic stroke. A study on predictability, pathogenesis, and prognosis. Arch. Neurol. 1995, 52, 670–675. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhao, H.; Steinberg, G.K.; Yenari, M.A. Cellular and molecular events underlying ischemia-induced neuronal apoptosis. Drug News Perspect. 2003, 16, 497–503. [Google Scholar] [CrossRef]
- Felling, R.J.; Song, H. Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp. Neurol. 2015, 268, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Hou, X.; Ren, G.; Zhang, Y.; Cheng, H. Dynamic changes in miR-124 levels in patients with acute cerebral infarction. Int. J. Neurosci. 2019, 129, 649–653. [Google Scholar] [CrossRef]
- Chen, S.H.; Sun, H.; Zhang, Y.M.; Xu, H.; Yang, Y.; Wang, F.M. Effects of acupuncture at Baihui (GV 20) and Zusanli (ST 36) on peripheral serum expression of MicroRNA 124, laminin and integrin beta1 in rats with cerebral ischemia reperfusion injury. Chin. J. Integr. Med. 2016, 22, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Shen, C.; Hirokawa, G.; Ji, X.; Takahashi, R.; Shimada, K.; Kishimoto, C.; Iwai, N. Plasma miR-124 as a biomarker for cerebral infarction. Biomed. Res. 2011, 32, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Q.; Ji, Y.; Peng, J.; Zhou, X.; Chen, X.; Zhao, H.; Xu, T.; Chen, L.; Xu, Y. Increased Brain-Specific MiR-9 and MiR-124 in the Serum Exosomes of Acute Ischemic Stroke Patients. PLoS ONE 2016, 11, e0163645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainer, T.H.; Leung, L.Y.; Chan, C.; Leung, Y.K.; Abrigo, J.M.; Wang, D.; Graham, C.A. Plasma miR-124-3p and miR-16 concentrations as prognostic markers in acute stroke. Clin. Biochem. 2016, 49, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Jeyaseelan, K.; Lim, K.Y.; Armugam, A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke 2008, 39, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Chen, Y.; Gao, Y.; Zuo, Y.; Zhou, X. Effect of single-nucleotide polymorphism in pri-microRNA-124 on poststroke motor function recovery. J. Cell Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Talhada, D.; Rai, A.; Ferreira, R.; Ferreira, L.; Bernardino, L.; Ruscher, K. MicroRNA-124-loaded nanoparticles increase survival and neuronal differentiation of neural stem cells in vitro but do not contribute to stroke outcome in vivo. PLoS ONE 2018, 13, e0193609. [Google Scholar] [CrossRef]
- Yu, Y.L.; Chou, R.H.; Shyu, W.C.; Hsieh, S.C.; Wu, C.S.; Chiang, S.Y.; Chang, W.J.; Chen, J.N.; Tseng, Y.J.; Lin, Y.H.; et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol. Med. 2013, 5, 531–547. [Google Scholar] [CrossRef]
- Wang, L.; Chopp, M.; Zhang, R.L.; Zhang, L.; Letourneau, Y.; Feng, Y.F.; Jiang, A.; Morris, D.C.; Zhang, Z.G. The Notch pathway mediates expansion of a progenitor pool and neuronal differentiation in adult neural progenitor cells after stroke. Neuroscience 2009, 158, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, S.R.; Choi, S.S.; Yeo, H.G.; Chang, K.T.; Lee, H.J. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed. Res. Int. 2014, 2014, 297241. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, R.; Venkat, P.; Chopp, M.; Zacharek, A.; Yan, T.; Cui, X.; Seyfried, D.; Chen, J. D-4F increases microRNA-124a and reduces neuroinflammation in diabetic stroke rats. Oncotarget 2017, 8, 95481–95494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarev, E.D.; Veremeyko, T.; Weiner, H.L. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 2013, 61, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef] [PubMed]
- Pivonkova, H.; Anderova, M. Altered Homeostatic Functions in Reactive Astrocytes and Their Potential as a Therapeutic Target After Brain Ischemic Injury. Curr. Pharm. Des. 2017, 23, 5056–5074. [Google Scholar] [CrossRef]
- Hamill, C.E.; Goldshmidt, A.; Nicole, O.; McKeon, R.J.; Brat, D.J.; Traynelis, S.F. Special lecture: Glial reactivity after damage: Implications for scar formation and neuronal recovery. Clin. Neurosurg. 2005, 52, 29–44. [Google Scholar]
- Hu, G.Q.; Du, X.; Li, Y.J.; Gao, X.Q.; Chen, B.Q.; Yu, L. Inhibition of cerebral ischemia/reperfusion injury-induced apoptosis: Nicotiflorin and JAK2/STAT3 pathway. Neural Regen. Res. 2017, 12, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Sun, W.; Hayes, P.; Leskov, K.; Boothman, D.A.; Matsuyama, S. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat. Cell Biol. 2003, 5, 320–329. [Google Scholar] [CrossRef]
- Aryani, A.; Denecke, B. Exosomes as a Nanodelivery System: A Key to the Future of Neuromedicine? Mol. Neurobiol. 2016, 53, 818–834. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.G.; Chopp, M. Exosomes in stroke pathogenesis and therapy. J. Clin. Investig. 2016, 126, 1190–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.T.; Im, W.; Ban, J.J.; Lee, M.; Jung, K.H.; Lee, S.K.; Chu, K.; Kim, M. Exosome-Based Delivery of miR-124 in a Huntington’s Disease Model. J. Mov. Disord. 2017, 10, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol. Ther. Nucleic Acids 2017, 7, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesulam, M.M. Neuroplasticity failure in Alzheimer’s disease: Bridging the gap between plaques and tangles. Neuron 1999, 24, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332–384. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, Z.; Xiao, M.; Wang, C.; Tian, F. Chronic Cerebral Hypoperfusion Promotes Amyloid-Beta Pathogenesis via Activating beta/gamma-Secretases. Neurochem. Res. 2017, 42, 3446–3455. [Google Scholar] [CrossRef]
- Zhao, Y.; Gu, J.H.; Dai, C.L.; Liu, Q.; Iqbal, K.; Liu, F.; Gong, C.X. Chronic cerebral hypoperfusion causes decrease of O-GlcNAcylation, hyperphosphorylation of tau and behavioral deficits in mice. Front. Aging Neurosci. 2014, 6, 10. [Google Scholar] [CrossRef]
- Pluta, R.; Jablonski, M.; Ulamek-Koziol, M.; Kocki, J.; Brzozowska, J.; Januszewski, S.; Furmaga-Jablonska, W.; Bogucka-Kocka, A.; Maciejewski, R.; Czuczwar, S.J. Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol. Neurobiol. 2013, 48, 500–515. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of Amyloid Precursor Protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Shen, Y. Beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) as a biological candidate marker of Alzheimer’s disease. Scand. J. Clin. Lab. Invest. 2009, 69, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Wang, J.; Zhang, X.; Geng, Y.; Hu, Z.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Irie, K.; Murakami, K.; Masuda, Y.; Morimoto, A.; Ohigashi, H.; Ohashi, R.; Takegoshi, K.; Nagao, M.; Shimizu, T.; Shirasawa, T. Structure of beta-amyloid fibrils and its relevance to their neurotoxicity: Implications for the pathogenesis of Alzheimer’s disease. J. Biosci. Bioeng. 2005, 99, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowan, M.J.; Klyubin, I.; Cullen, W.K.; Anwyl, R. Synaptic plasticity in animal models of early Alzheimer’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Zuroff, L.; Daley, D.; Black, K.L.; Koronyo-Hamaoui, M. Clearance of cerebral Abeta in Alzheimer’s disease: Reassessing the role of microglia and monocytes. Cell Mol. Life Sci. 2017, 74, 2167–2201. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.Y.; Tse, W.; Smith, J.D.; Landreth, G.E. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 2012, 287, 2032–2044. [Google Scholar] [CrossRef] [Green Version]
- Pugazhenthi, S.; Wang, M.; Pham, S.; Sze, C.I.; Eckman, C.B. Downregulation of CREB expression in Alzheimer’s brain and in Abeta-treated rat hippocampal neurons. Mol. Neurodegener. 2011, 6, 60. [Google Scholar] [CrossRef]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Deisseroth, K.; Bito, H.; Tsien, R.W. Signaling from synapse to nucleus: Postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 1996, 16, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Finkbeiner, S.; Tavazoie, S.F.; Maloratsky, A.; Jacobs, K.M.; Harris, K.M.; Greenberg, M.E. CREB: A major mediator of neuronal neurotrophin responses. Neuron 1997, 19, 1031–1047. [Google Scholar] [CrossRef] [Green Version]
- Benito, E.; Barco, A. CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends Neurosci. 2010, 33, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, K.D.; Bak, M.S.; Kim, S.J.; Rhee, S.; Lee, Y.S. Restoring synaptic plasticity and memory in mouse models of Alzheimer’s disease by PKR inhibition. Mol. Brain 2017, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preethi, J.; Singh, H.K.; Charles, P.D.; Rajan, K.E. Participation of microRNA 124-CREB pathway: A parallel memory enhancing mechanism of standardised extract of Bacopa monniera (BESEB CDRI-08). Neurochem. Res. 2012, 37, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Arancibia, S.; Silhol, M.; Mouliere, F.; Meffre, J.; Hollinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kwon, H.J.; Lee, J.E.; Lee, Y.; Seoh, J.Y.; Han, P.L. Hyperoxygenation revitalizes Alzheimer’s disease pathology through the upregulation of neurotrophic factors. Aging Cell 2019, 18, e12888. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, V.; Dreyer, J.L. MicroRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol. Cell Neurosci. 2009, 42, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.Y.; Wang, G.Q.; Wang, N.N.; Yu, Q.Y.; Liu, R.L.; Shi, W.Q. The long-non-coding RNA NEAT1 is a novel target for Alzheimer’s disease progression via miR-124/BACE1 axis. Neurol. Res. 2019, 41, 489–497. [Google Scholar] [CrossRef]
- Hernandez, F.; Gomez, D.B.E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Huber, C.M.; Yee, C.; May, T.; Dhanala, A.; Mitchell, C.S. Cognitive Decline in Preclinical Alzheimer’s Disease: Amyloid-Beta versus Tauopathy. J. Alzheimers Dis. 2018, 61, 265–281. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Cheng, P.; Yu, K.; Han, Y.; Song, M.; Li, Y. Hyperforin attenuates aluminum-induced Abeta production and Tau phosphorylation via regulating Akt/GSK-3beta signaling pathway in PC12 cells. Biomed. Pharmacother. 2017, 96, 1–6. [Google Scholar] [CrossRef]
- ArunSundar, M.; Shanmugarajan, T.S.; Ravichandiran, V. 3,4-Dihydroxyphenylethanol Assuages Cognitive Impulsivity in Alzheimer’s Disease by Attuning HPA-Axis via Differential Crosstalk of alpha7 nAChR with MicroRNA-124 and HDAC6. ACS Chem. Neurosci. 2018, 9, 2904–2916. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.M. Parkinson’s disease: A review. Front. Biosci. 2014, 6, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Main, B.S.; Crack, P.J. Neuroinflammation and oxidative stress: Co-conspirators in the pathology of Parkinson’s disease. Neurochem. Int. 2013, 62, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [PubMed]
- Tang, H.; Gao, Y.; Zhang, Q.; Nie, K.; Zhu, R.; Gao, L.; Feng, S.; Wang, L.; Zhao, J.; Huang, Z.; et al. Chronic cerebral hypoperfusion independently exacerbates cognitive impairment within the pathopoiesis of Parkinson’s disease via microvascular pathologys. Behav. Brain Res. 2017, 333, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Grande, B.; Blackabey, V.; Gittens, B.; Pinteaux, E.; Denes, A. Loss of substance P and inflammation precede delayed neurodegeneration in the substantia nigra after cerebral ischemia. Brain Behav. Immun. 2013, 29, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Unal-Cevik, I.; Gursoy-Ozdemir, Y.; Yemisci, M.; Lule, S.; Gurer, G.; Can, A.; Muller, V.; Kahle, P.J.; Dalkara, T. Alpha-synuclein aggregation induced by brief ischemia negatively impacts neuronal survival in vivo: A study in [A30P]alpha-synuclein transgenic mouse. J. Cereb. Blood Flow Metab. 2011, 31, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Sudhof, T.C. Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Pan, X.; Zhang, J.; Ma, A.; Yang, S.; Ma, J.; Xie, A. Plasma levels of miR-137 and miR-124 are associated with Parkinson’s disease but not with Parkinson’s disease with depression. Neurol. Sci. 2017, 38, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Vila, M.; Jackson-Lewis, V.; Vukosavic, S.; Djaldetti, R.; Liberatore, G.; Offen, D.; Korsmeyer, S.J.; Przedborski, S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 2837–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, C.; Chen, S.; Ye, Y.; Guo, M.; Ren, Q.; Liu, L.; Zhang, H.; Xu, C.; Zhou, Q.; et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal. 2014, 26, 1680–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czapski, G.A.; Gassowska, M.; Wilkaniec, A.; Cieslik, M.; Adamczyk, A. Extracellular alpha-synuclein induces calpain-dependent overactivation of cyclin-dependent kinase 5 in vitro. FEBS Lett. 2013, 587, 3135–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Shao, X.Y.; Qi, G.J.; Chen, Q.; Bu, L.L.; Chen, L.J.; Shi, J.; Ming, J.; Tian, B. Cdk5-Dependent Activation of Neuronal Inflammasomes in Parkinson’s Disease. Mov. Disord. 2016, 31, 366–376. [Google Scholar] [CrossRef]
- Kanagaraj, N. MicroRNA Expressions in the MPTP-Induced Parkinson’s Disease Model with Special Reference to miR-124 (Doctoral dissertation). National University of Singapore: Singapore, 2013; Retrieved from https://core.ac.uk/download/pdf/48682603.pdf. [Google Scholar]
- Schirinzi, T.; Madeo, G.; Martella, G.; Maltese, M.; Picconi, B.; Calabresi, P.; Pisani, A. Early synaptic dysfunction in Parkinson’s disease: Insights from animal models. Mov. Disord. 2016, 31, 802–813. [Google Scholar] [CrossRef]
- Gutierrez-Vargas, J.A.; Munera, A.; Cardona-Gomez, G.P. CDK5 knockdown prevents hippocampal degeneration and cognitive dysfunction produced by cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1937–1949. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, C.; Paiva, J.; Santos, T.; Ferreira, L.; Bernardino, L. MicroRNA-124 loaded nanoparticles enhance brain repair in Parkinson’s disease. J. Control Release 2016, 235, 291–305. [Google Scholar] [CrossRef]
- Azam, F.; Prasad, M.V.; Thangavel, N. Targeting oxidative stress component in the therapeutics of epilepsy. Curr. Top. Med. Chem. 2012, 12, 994–1007. [Google Scholar] [CrossRef]
- Fox, C.K.; Mackay, M.T.; Dowling, M.M.; Pergami, P.; Titomanlio, L.; Deveber, G. Prolonged or recurrent acute seizures after pediatric arterial ischemic stroke are associated with increasing epilepsy risk. Dev. Med. Child Neurol. 2017, 59, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, G.R.; Ding, S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 2016, 85, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, E.L.; Annegers, J.F.; Hauser, W.A.; O’Brien, P.C.; Whisnant, J.P. Population-based study of seizure disorders after cerebral infarction. Neurology 1996, 46, 350–355. [Google Scholar] [CrossRef]
- Bryndziar, T.; Sedova, P.; Kramer, N.M.; Mandrekar, J.; Mikulik, R.; Brown, R.J.; Klaas, J.P. Seizures Following Ischemic Stroke: Frequency of Occurrence and Impact on Outcome in a Long-Term Population-Based Study. J. Stroke Cerebrovasc. Dis. 2016, 25, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Bladin, C.F.; Alexandrov, A.V.; Bellavance, A.; Bornstein, N.; Chambers, B.; Cote, R.; Lebrun, L.; Pirisi, A.; Norris, J.W. Seizures after stroke: A prospective multicenter study. Arch. Neurol. 2000, 57, 1617–1622. [Google Scholar] [CrossRef] [Green Version]
- Waldbaum, S.; Patel, M. Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy? J. Bioenerg. Biomembr. 2010, 42, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Iori, V.; Frigerio, F.; Vezzani, A. Modulation of neuronal excitability by immune mediators in epilepsy. Curr. Opin. Pharmacol. 2016, 26, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Cavazos, J.E.; Cross, D.J. The role of synaptic reorganization in mesial temporal lobe epilepsy. Epilepsy Behav. 2006, 8, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Risbud, R.M.; Porter, B.E. Changes in microRNA expression in the whole hippocampus and hippocampal synaptoneurosome fraction following pilocarpine induced status epilepticus. PLoS ONE 2013, 8, e53464. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Han, X.; Blendy, J.A.; Porter, B.E. Decreased CREB levels suppress epilepsy. Neurobiol. Dis. 2012, 45, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.T.; Cai, R.R.; Zhang, P.F.; Lin, Y.X. Apoptosis and expression of Caspase 3 and Caspase 4 in neurocytes of refractory human temporal lobe epilepsy. Zhonghua Yi Xue Za Zhi 2016, 96, 522–525. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | Targets | Abbreviations | Function | Effects | Refs |
|---|---|---|---|---|---|
| Ischemic stroke | SRY-box transcription factor | Sox9 | Neuronal differentiation | Repressing the expression of Sox9 | [24] |
| Jagged-1 | JAG1 | Neuronal differentiation | Repressing JAG1/Notch signaling pathway | [33] | |
| Ubiquitin specific peptidase 14 | Usp14 | Neuronal differentiation Neurovascular remodeling | Decreasing the expression of Usp14, which mediates REST degradation | [37] | |
| Anti-apoptosis | Exosomal miR-124 derived from M2 microglia attenuates neuronal apoptosis via downregulating Usp14 | [38] | |||
| Phosphoinositide 3-kinase | PI3K | Anti-axonal growth | Inhibition of miR-124 activating PI3K/AKT/mTOR/GAP-43 pathway | [39] | |
| Anti-apoptosis | Activating PI3K/AKT/ Bcl-2 signaling pathway | [40] | |||
| Activating PI3K/AKT/Nrf2 signaling pathway | [41] | ||||
| Anti-oxidative stress | Activating PI3K/AKT/Nrf2 signaling pathway | [41] | |||
| Inhibitory member of the apoptosis-stimulating proteins of p53 family | iASPP | Pro-apoptosis | Downregulating the expression of iASPP | [36] | |
| B-cell lymphoma-2 B-cell lymphoma-extra large | Bcl-2/Bcl-xl | Anti-apoptosis | Upregulating the expression of Bcl-2 and Bcl-xl respectively | [42] | |
| Janus kinase 2 | JAK2 | Anti-apoptosis | Activating JAK2/STAT3/Bcl-2 signaling pathway | [16] | |
| X-ray repair cross-complementing protein 6 | Ku70 | Pro-apoptosis | Knockdown of miR-124 attenuates I/R-induced apoptosis via negatively regulating Ku 70 | [43] | |
| CCAAT/enhancer-binding protein alpha | C/EBP-α | Anti-inflammation | Inhibiting C/EBP-α/PU.1 signaling pathway | [22] | |
| RE1-silencing transcription factor | REST | Neuroplasticity Angiogenesis | Facilitating REST degradation | [44] | |
| Alzheimer’s disease | Regulatory factor X1 | RFX1 | Anti-Aβ deposition | Decreased miR-124 represses ApoE-dependent Aβ uptake by targeting and upregulating RFX1 | [34] |
| cAMP-response element binding protein | CREB | Suppressing synaptic plasticity | Decreased miR-124 increases the expression of CREB and subsequent BDNF synthesis | [45] | |
| Tyrosine-protein phosphatase non-receptor type 1 | PTPN1 | Suppressing synaptic plasticity | Downregulating the expression of PTPN1 | [46] | |
| Beta-site Amyloid precursor protein Cleaving Enzyme 1 | BACE1 | Anti- Aβ deposition | Decreased miR-124 upregulates BACE1 level | [47] | |
| Caveolin-1 | Cav-1 | Inhibiting hyperphosphorylation of tau | Repressing the expression of Cav-1, which further activates PI3K/AKT/GSK3β signaling pathway | [35] | |
| Parkinson’s disease | Bim | Bim | Anti-apoptosis Anti-autophagy | Inhibiting the expression of Bim and reducing Bax translocation to mitochondria and lysosomal membrane | [23] |
| Adenosine 5’-monophosphate-activated protein kinase | AMPK | Anti-apoptosis Anti-autophagy | Decreased miR-124 induces apoptosis and autophagy-associated proteins via activating AMPK/mTOR signaling pathway | [48] | |
| Signal transducer and activator of transcription-3 | STAT3 | Anti-apoptosis | Suppressing STAT3 expression | [49] | |
| AnnexinA5 | ANXA5 | Anti-apoptosis | Downregulating ANXA5/ERK signaling pathway | [50] | |
| Caspase-3 | Casp-3 | Anti-apoptosis | Restraining the increase of Casp-3 | [51] | |
| Mitogen-activated protein kinase kinase kinase 3 | MEKK3 | Anti-neuroinflammation | Suppressing MEKK3/NF-κB signaling pathway | [52] | |
| Sequestosome 1 Phospho-p38 mitogen-activated protein kinase | p62 p-p38 | Anti-inflammation | Suppressing the expression of p62 and p-p38 | [53] | |
| Calpain 1 | Calpain 1 | Anti-oxidative stress | Decreased miR-124 upregulates the expression of calpain1/p25/CDK5 pathway proteins | [17] | |
| Epilepsy | cAMP-response element-binding protein 1 | CREB1 | Anti-neuronal excitability | Repressing the expression of CREB1 | [32] |
| CCAAT/enhancer-binding protein alpha | C/EBPα | Anti-epileptogenic | Downregulating C/EBPα level, which then inhibits the expression and activity of NRSF | [54] | |
| Bcl-2-like 13 | Bcl2L13 | Anti-apoptosis | Diminishing the expression of Bcl2L13, which further represses the activation of caspase-3 | [55] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Feng, Z.; Du, L.; Huang, Y.; Ge, J.; Deng, Y.; Mei, Z. The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury. Int. J. Mol. Sci. 2020, 21, 120. https://doi.org/10.3390/ijms21010120
Liu X, Feng Z, Du L, Huang Y, Ge J, Deng Y, Mei Z. The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury. International Journal of Molecular Sciences. 2020; 21(1):120. https://doi.org/10.3390/ijms21010120
Chicago/Turabian StyleLiu, Xiaolu, Zhitao Feng, Lipeng Du, Yaguang Huang, Jinwen Ge, Yihui Deng, and Zhigang Mei. 2020. "The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury" International Journal of Molecular Sciences 21, no. 1: 120. https://doi.org/10.3390/ijms21010120