The miRNA-Mediated Post-Transcriptional Regulation of Maize in Response to High Temperature

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Phenotypic and Physiological Responses of Maize Leaves to HT Stress
2.2. Analysis of Small RNA-seq Data
2.3. Identification of Known and Novel Differentially Expressed miRNAs (DEMs)
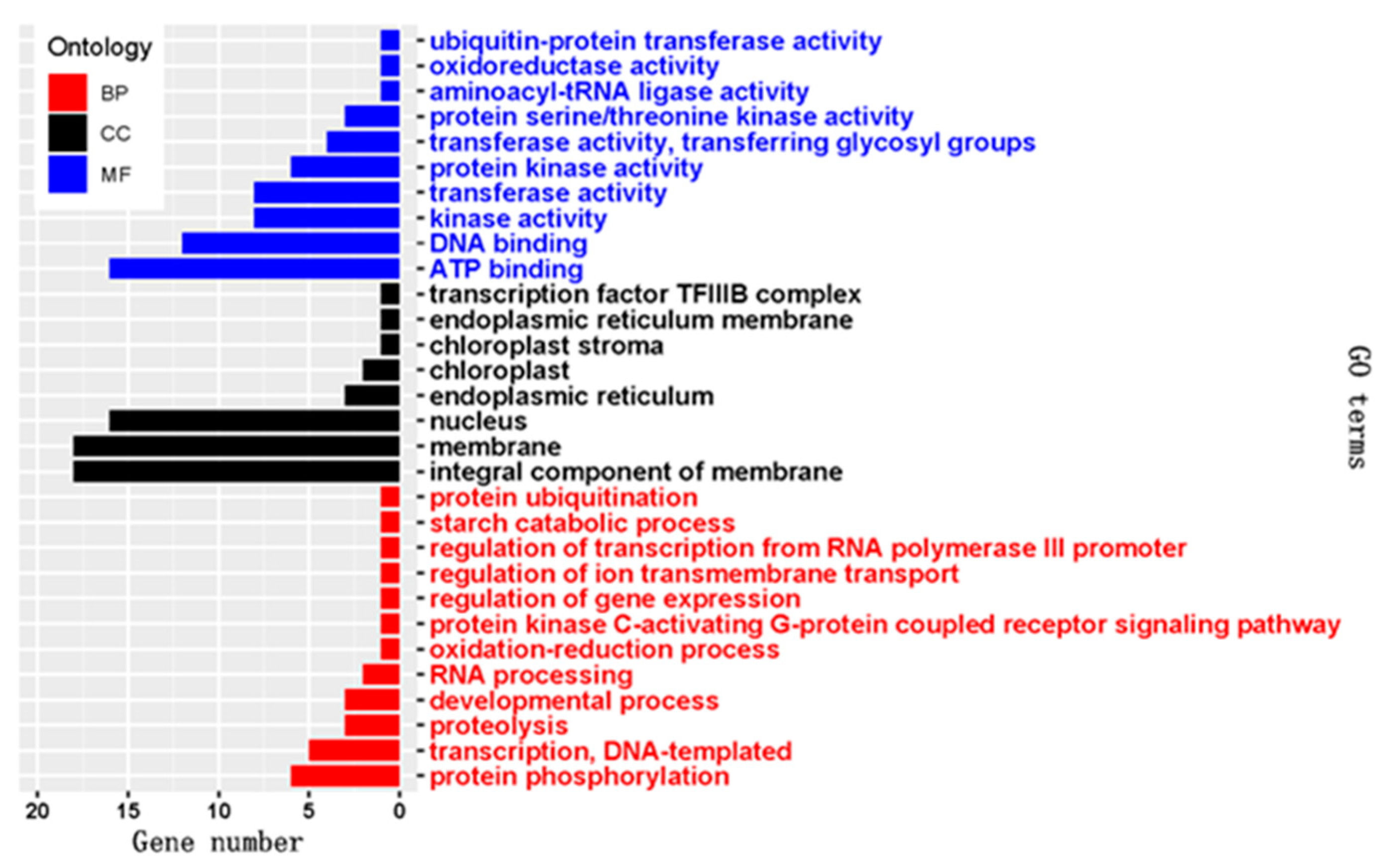
2.4. Target Prediction and Functional Annotation of the Known and Novel DEMs
2.5. HT-Responsive Transcriptome in Maize Leaves
2.6. Target mRNAs Negatively Regulated by DEMs
2.7. Quantitative Real-Time PCR Validation
3. Discussion
3.1. miRNAs of Maize in Response to High Temperature (HT)
3.2. Transcriptomics of HT-Stressed Leaves of Maize
3.3. Integrated Analyses of miRNA–mRNA Transcriptomics
4. Materials and Methods
4.1. Plant Material, Heat Treatment, and Sample Collection
4.2. Determination of Physiological Parameters of Maize Leaves
4.3. RNA Isolation, sRNA Library Preparation and Sequencing
4.4. Identification of Known and Novel miRNAs
4.5. Degradome Sequencing and Target Identification
4.6. mRNA Library Construction and Sequencing
4.7. Verification of RNA-seq Data by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HT | High Temperature |
| ROS | Reactive Oxidative Species |
| SOD | Superoxide Dismutase |
| CAT | Catalase |
| POD | Peroxidase |
| MDA | Malonaldehyde |
| TF | Transcription Factor |
| GRF | Growth Regulation Factor |
| qRT-PCR | Quantitative Real-Time PCR |
| AP2/EREBP | APETALA2/Ethylene-Responsive Element Binding Protein |
References
- Awasthi, R.; Bhandari, K.; Nayyar, H. Temperature stress and redox homeostasis in agricultural crops. Front. Environ. Sci. 2015, 3, 1–24. [Google Scholar] [CrossRef]
- Lobell, D.B.; Hammer, G.L.; Mclean, G. The critical role of extreme heat for maize production in the United States. Nat. Clim. Chang. 2013, 3, 497–501. [Google Scholar] [CrossRef]
- Hawkins, E.; Fricker, T.E.; Challinor, A.J.; Ferro, C.A.T.; Ho, C.K.; Osborne, T.M. Increasing influence of heat stress on French maize yields from the 1960s to the 2030s. Glob. Chang. Biol. 2013, 19, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Lobell, D.B.; Bänziger, M.; Magorokosho, C.; Vivek, B. Nonlinear heat effects on African maize as evidenced by historical yield trials. Nat. Clim. Chang. 2011, 1, 42–45. [Google Scholar] [CrossRef]
- Hu, X.; Li, Y.; Yang, H.; Liu, Q.; Li, C. Heat Shock Protein 70 May Improve the Ability of Antioxidant Defense Induced by the Combination of Drought and Heat in Maize Leaves. Acta Agron. Sin. 2010, 36, 636–644. [Google Scholar] [CrossRef]
- IPCC. Summary for Policymakers. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G., Tignor, M.M.B., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; pp. 1–33. [Google Scholar]
- De, Z.A.; Colcombet, J.; Hirt, H. The Role of MAPK Modules and ABA during Abiotic Stress Signaling. Trends Plant Sci. 2016, 21, 677–685. [Google Scholar] [CrossRef]
- Zhang, B. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef]
- Mcgettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, Q.; Chen, G.; Wang, L.; Jin, B. Regulation of Non-coding RNAs in Heat Stress Responses of Plants. Front. Plant Sci. 2016, 7, 1213. [Google Scholar] [CrossRef]
- Zhang, X.; Rerksiri, W.; Liu, A.; Zhou, X.; Xiong, H.; Xiang, J.; Chen, X.; Xiong, X. Transcriptome profile reveals heat response mechanism at molecular and metabolic levels in rice flag leaf. Gene 2013, 530, 185–192. [Google Scholar] [CrossRef]
- Gonzalez-Schain, N.; Dreni, L.; Lawas, L.M.F.; Galbiati, M.; Colombo, L.; Heuer, S.; Jagadish, K.S.V.; Kater, M.M. Genome-Wide Transcriptome Analysis During Anthesis Reveals New Insights into the Molecular Basis of Heat Stress Responses in Tolerant and Sensitive Rice Varieties. Plant Cell Physiol. 2016, 57, 57–68. [Google Scholar] [CrossRef]
- Mangelsen, E.; Kilian, J.; Harter, K.; Jansson, C.; Wanke, D.; Sundberg, E. Transcriptome Analysis of High-Temperature Stress in Developing Barley Caryopses: Early Stress Responses and Effects on Storage Compound Biosynthesis. Mol. Plant 2011, 4, 97–115. [Google Scholar] [CrossRef]
- Tao, L.; Xu, X.; Ying, L.; Wang, H.; Li, Z.; Li, Z. Comparative transcriptome analysis reveals differential transcription in heat-susceptible and heat-tolerant pepper (Capsicum annum L.) cultivars under heat stress. J. Plant Biol. 2015, 58, 411–424. [Google Scholar] [CrossRef]
- Shi, J.; Yan, B.; Lou, X.; Ma, H.; Ruan, S. Comparative transcriptome analysis reveals the transcriptional alterations in heat-resistant and heat-sensitive sweet maize (Zea mays L.) varieties under heat stress. BMC Plant Biol. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Frey, F.P.; Urbany, C.; Huettel, B.; Reinhardt, R.; Stich, B. Genome-wide expression profiling and phenotypic evaluation of European maize inbreds at seedling stage in response to heat stress. BMC Genom. 2015, 16, 123. [Google Scholar] [CrossRef]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.R.; Baurle, I. Arabidopsis miR156 Regulates Tolerance to Recurring Environmental Stress through SPL Transcription Factors. Plant Cell 2014, 26, 1792–1807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Z.; Gong, P.; Zhang, J.; Ziaf, K.; Li, H.; Xiao, F.; Ye, Z. Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol. Lett. 2011, 33, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ma, Y.; Liu, N.; Xu, J.; Hu, Q.; Li, Y.; Wu, Y.; Xie, S.; Zhu, L.; Min, L.; et al. microRNAs involved in auxin signalling modulate male sterility under high-temperature stress in cotton (Gossypium hirsutum). Plant J. 2017, 91, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Xin, C.; Wang, X.; Cai, J.; Zhou, Q.; Liu, F.; Dai, T.; Cao, W.; Jiang, D. Changes of transcriptome and proteome are associated with the enhanced post-anthesis high temperature tolerance induced by pre-anthesis heat priming in wheat. Plant Growth Regul. 2016, 79, 135–145. [Google Scholar] [CrossRef]
- Liao, J.; Zhou, H.; Peng, Q.; Zhong, P.; Zhang, H.; He, C.; Huang, Y. Transcriptome changes in rice (Oryza sativa L.) in response to high night temperature stress at the early milky stage. BMC Genom. 2015, 16, 18. [Google Scholar] [CrossRef]
- Wang, R.; Xu, L.; Zhu, X.; Zhai, L.; Wang, Y.; Yu, R.; Gong, Y.; Limera, C.; Liu, L. Transcriptome-Wide Characterization of Novel and Heat-Stress-Responsive microRNAs in Radish (Raphanus Sativus L.) Using Next-Generation Sequencing. Plant Mol. Biol. Rep. 2015, 33, 867–880. [Google Scholar] [CrossRef]
- Zhou, R.; Wang, Q.; Jiang, F.; Cao, X.; Sun, M.; Liu, M.; Wu, Z. Identification of miRNAs and their targets in wild tomato at moderately and acutely elevated temperatures by high-throughput sequencing and degradome analysis. Sci. Rep. 2016, 6, 33777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, M.; Wang, Y.; Yao, Y.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Mangrauthia, S.K.; Bhogireddy, S.; Agarwal, S.; Prasanth, V.V.; Voleti, S.R.; Neelamraju, S.; Subrahmanyam, D. Genome-wide changes in microRNA expression during short and prolonged heat stress and recovery in contrasting rice cultivars. J. Exp. Bot. 2017, 68, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, P.; Xia, J.; Zhou, X.; Gao, S.; Zhang, X.; Coutino, G.; Vazquez, F.; Zhang, W.; Jin, H. siRNAs from miRNA sites mediate DNA methylation of target genes. Nucleic Acids Res. 2010, 38, 6883–6894. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, L.Q.; Zheng, W.Y.; Wang, R.F.; Yang, L.X. Genome-Wide Identification of MicroRNAs Responsive to High Temperature in Rice (Oryza sativa) by High-Throughput Deep Sequencing. J. Agron. Crop Sci. 2015, 201, 379–388. [Google Scholar] [CrossRef]
- Allakhverdiev, S.I.; Kreslavski, V.D.; Klimov, V.V.; Los, D.A.; Carpentier, R.; Mohanty, P. Heat stress: An overview of molecular responses in photosynthesis. Photosynth. Res. 2008, 98, 541–550. [Google Scholar] [CrossRef]
- Song, Y.; Chen, Q.; Ci, D.; Shao, X.; Zhang, D. Effects of high temperature on photosynthesis and related gene expression in poplar. BMC Plant Biol. 2014, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Wu, H.; Peng, H.; Yao, Y.; Ni, Z.; Li, Z.; Zhou, C.; Sun, Q. Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using Wheat Genome Array. BMC Genom. 2008, 9, 432. [Google Scholar] [CrossRef] [PubMed]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Xu, S.; Li, J.; Zhang, X.; Wei, H.; Cui, L. Effects of heat acclimation pretreatment on changes of membrane lipid peroxidation, antioxidant metabolites, and ultrastructure of chloroplasts in two cool-season turfgrass species under heat stress. Environ. Exp. Bot. 2006, 56, 274–285. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Y.; Tian, J.; Huang, K.; Shi, T.; Dai, X.; Zhang, W. Transcriptional Profiling and Identification of Heat-Responsive Genes in Perennial Ryegrass by RNA-Sequencing. Front. Plant Sci. 2017, 8, 1032. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yu, L.; Xuan, J.; Lu, Y.; Lu, S.; Zhu, W. De novo transcriptome sequencing and gene expression profiling of spinach (Spinacia oleracea L.) leaves under heat stress. Sci. Rep.-UK 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, C.; Cai, X.; Wang, Q.; Dai, S. Heat-Responsive Photosynthetic and Signaling Pathways in Plants: Insight from Proteomics. Int. J. Mol. Sci. 2017, 18, 2191. [Google Scholar] [CrossRef]
- Laborde, E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010, 17, 1373–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkasov, V.; Hofmann, S.; Druffel-Augustin, S.; Mogk, A.; Tyedmers, J.; Stoecklin, G.; Bukau, B. Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 2013, 23, 2452–2462. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gan, S. AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J. 2006, 46, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Mitsuda, N.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. The NAC transcription factors NST1 and NST2 of Arabidopsis regulate secondary wall thickenings and are required for anther dehiscence. Plant Cell 2005, 17, 2993–3006. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Fujita, Y.; Maruyama, K.; Seki, M.; Hiratsu, K.; Ohme-Takagi, M.; Tran, L.S.; Yamaguchi-Shinozaki, K.; Shinozaki, K. A dehydration-induced NAC protein, RD26, is involved in a novel ABA-dependent stress-signaling pathway. Plant J. 2004, 39, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Liao, K.; Du, H.; Xu, Y.; Song, H.; Li, X.; Xiong, L. A stress-responsive NAC transcription factor SNAC3 confers heat and drought tolerance through modulation of reactive oxygen species in rice. J. Exp. Bot. 2015, 66, 6803–6817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.W.; Schwab, R.; Czech, B.; Mica, E.; Weigel, D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell 2008, 20, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Jia, H.; Yan, Q.; Wang, X. Overexpression of a SBP-Box Gene (VpSBP16) from Chinese Wild Vitis Species in Arabidopsis Improves Salinity and Drought Stress Tolerance. Int. J. Mol. Sci. 2018, 19, 940. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, S.; Jin, J.; Fu, D.; Yang, X.; Weng, X.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Coordinated regulation of vegetative and reproductive branching in rice. Proc. Natl. Acad. Sci. USA 2015, 112, 15504–15509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef]
- Chen, L.; Han, J.; Deng, X.; Tan, S.; Li, L.; Li, L.; Zhou, J.; Peng, H.; Yang, G.; He, G.; Zhang, W. Expansion and stress responses of AP2/EREBP superfamily in Brachypodium distachyon. Sci. Rep. 2016, 6, 21623. [Google Scholar] [CrossRef] [PubMed]
- Nova-Franco, B.; Iniguez, L.P.; Valdes-Lopez, O.; Alvarado-Affantranger, X.; Leija, A.; Fuentes, S.I.; Ramirez, M.; Paul, S.; Reyes, J.L.; Girard, L.; et al. The micro-RNA172c-APETALA2-1 node as a key regulator of the common bean-Rhizobium etli nitrogen fixation symbiosis. Plant Physiol. 2015, 168, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Hossain, M.S.; Wang, J.; Valdes-Lopez, O.; Liang, Y.; Libault, M.; Qiu, L.; Stacey, G. miR172 regulates soybean nodulation. Mol. Plant Microbe Interact. 2013, 26, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.L.; Chua, N.H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, F.; Cao, H.; Peng, H.; Ni, Z.; Sun, Q.; Yao, Y. TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS ONE 2012, 7, e48445. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of plant microRNA targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef]
- Nagasaki, H.; Itoh, J.; Hayashi, K.; Hibara, K.; Satoh-Nagasawa, N.; Nosaka, M.; Mukouhata, M.; Ashikari, M.; Kitano, H.; Matsuoka, M.; et al. The small interfering RNA production pathway is required for shoot meristem initiation in rice. Proc. Natl. Acad. Sci. USA 2007, 104, 14867–14871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbert, P.B.; Adler, H.T.; Parks, D.W.; Comai, L. The REVOLUTA gene is necessary for apical meristem development and for limiting cell divisions in the leaves and stems of Arabidopsis thaliana. Development 1995, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, D.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003, 36, 94–104. [Google Scholar] [CrossRef]
- Omidbakhshfard, M.A.; Proost, S.; Fujikura, U.; Mueller-Roeber, B. Growth-Regulating Factors (GRFs): A Small Transcription Factor Family with Important Functions in Plant Biology. Mol. Plant 2015, 8, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, F.; Messa, M.; Nández, R.; Czapla, H.; Zou, Y.; Strittmatter, S.M.; De Camilli, P. Sac2/INPP5F is an inositol 4-phosphatase that functions in the endocytic pathway. J. Cell Biol. 2015, 209, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mary Elizabeth, W.; Javad, T.; Evan, C.; Katherine, P.; Elizabeth, J.D.; James, E.T.; Michelle, H.; Daryll, B.D. Mutations in the Arabidopsis phosphoinositide phosphatase gene SAC9 lead to overaccumulation of PtdIns (4,5) P2 and constitutive expression of the stress-response pathway. Plant Physiol. 2005, 138, 686–700. [Google Scholar] [CrossRef]
- Liu, F.; Hu, W.; Vierstra, R.D. The Vacuolar Protein Sorting-38 Subunit of the Arabidopsis Phosphatidylinositol-3-Kinase Complex Plays Critical Roles in Autophagy, Endosome Sorting, and Gravitropism. Front. Plant Sci. 2018, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, H.K. Chlorophylls and carotenoids: Pigments of photosynthetic biomembranes. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1987; pp. 350–382. [Google Scholar]
- Elstner, E.F.; Heupel, A. Inhibition of nitrite formation from hydroxylammoniumchloride: A simple assay for superoxide dismutase. Anal. Biochem. 1976, 70, 616–620. [Google Scholar] [CrossRef]
- Brennan, T.; Frenkel, C. Involvement of Hydrogen Peroxide in the Regulation of Senescence in Pear. Plant Physiol. 1977, 59, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Heath, R.L.; Packer, L. Photoperoxidation in isolated chloroplasts: I. Kinetics and stoichiometry of fatty acid peroxidation. Arch. Biochem. Biophys. 1968, 125, 189–198. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, J. Effect of abscisic acid on active oxygen species, antioxidative defence system and oxidative damage in leaves of maize seedlings. Plant Cell Physiol. 2001, 42, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR_family | Targets Annotation | TF Family |
|---|---|---|
| miR1430 | Nuclear transcription factor Y subunit A-10 | NF-YA |
| miR156 | neighbor of tga1 | SBP/SPL |
| SBP-domain containing protein | ||
| SBP-domain protein 5 | ||
| Squamosa promoter-binding-like protein 11 | ||
| Squamosa promoter-binding-like protein 15 | ||
| Squamosa promoter-binding-like protein 18 | ||
| Squamosa promoter-binding-like protein 2 | ||
| Squamosa promoter-binding-like protein 5 | ||
| Squamosa promoter-binding-like protein 6 | ||
| tassel sheath4 | ||
| teosinte glume architecture1 | ||
| miR160 | Auxin response factor 16 | ARF |
| miR164 | NAC domain-containing protein 21/22 | NAC |
| NAC domain-containing protein 79 | ||
| miR166 | Homeobox-leucine zipper protein ATHB-14 | HD-ZIP |
| Homeobox-leucine zipper protein ATHB-14 | ||
| Homeobox-leucine zipper protein REVOLUTA | ||
| rolled leaf2 | ||
| miR169 | CCAAT-HAP2-transcription factor 26 | NF-YA |
| nuclear transcription factor y subunit a1 | ||
| Nuclear transcription factor Y subunit A-10 | ||
| Nuclear transcription factor Y subunit A-3 | ||
| Nuclear transcription factor Y subunit A-8 | ||
| Nuclear transcription factor Y subunit A-9 | ||
| SBP-transcription factor16 | SBP | |
| miR172 | Floral homeotic protein APETALA 2 | AP2 |
| Putative AP2/EREBP transcription factor superfamily protein | ||
| sister of indeterminate spikelet1 | ||
| tasselseed6 | ||
| miR396 | Growth-regulating factor | GRF |
| Growth-regulating factor 2 | ||
| Growth-regulating factor 6 | ||
| Putative growth-regulating factor 6 |
| GO ID | Ontology | Description | Gene Ratio |
|---|---|---|---|
| GO:0006351 | BP | transcription, DNA-templated | 54/323 |
| GO:0032502 | BP | developmental process | 30/323 |
| GO:0006816 | BP | calcium ion transport | 18/323 |
| GO:0070588 | BP | calcium ion transmembrane transport | 18/323 |
| GO:0007275 | BP | multicellular organism development | 14/323 |
| GO:0009725 | BP | response to hormone | 12/323 |
| GO:0009734 | BP | auxin-activated signaling pathway | 10/323 |
| GO:0055114 | BP | oxidation-reduction process | 5/323 |
| GO:0006508 | BP | proteolysis | 4/323 |
| GO:0005634 | CC | nucleus | 141/323 |
| GO:0016020 | CC | membrane | 40/323 |
| GO:0005737 | CC | cytoplasm | 31/323 |
| GO:0005783 | CC | endoplasmic reticulum (ER) | 1/323 |
| GO:0003677 | MF | DNA binding | 142/323 |
| GO:0008289 | MF | lipid binding | 51/323 |
| GO:0003700 | MF | transcription factor activity | 47/323 |
| GO:0042578 | MF | phosphoric ester hydrolase activity | 21/323 |
| GO:0005388 | MF | calcium-transporting ATPase activity | 18/323 |
| GO:0003824 | MF | catalytic activity | 9/323 |
| GO:0016491 | MF | oxidoreductase activity | 5/323 |
| GO ID | GO Ontology | Description | Gene Ratio |
|---|---|---|---|
| GO:0006629 | BP | lipid metabolic process | 5/19 |
| GO:0006468 | BP | protein phosphorylation | 5/19 |
| GO:0016310 | BP | phosphorylation | 5/19 |
| GO:0006139 | BP | nucleobase-containing compound metabolic process | 1/19 |
| GO:0090305 | BP | nucleic acid phosphodiester bond hydrolysis | 1/19 |
| GO:0006412 | BP | translation | 1/19 |
| GO:0055085 | BP | transmembrane transport | 1/19 |
| GO:0016021 | CC | integral component of membrane | 9/19 |
| GO:0016020 | CC | membrane | 9/19 |
| GO:0005840 | CC | ribosome | 1/19 |
| GO:0005622 | CC | intracellular | 1/19 |
| GO:0008889 | MF | glycerophosphodiester phosphodiesterase activity | 5/19 |
| GO:0008081 | MF | phosphoric diester hydrolase activity | 5/19 |
| GO:0004672 | MF | protein kinase activity | 5/19 |
| GO:0016301 | MF | kinase activity | 5/19 |
| GO:0005524 | MF | ATP binding | 5/19 |
| GO:0016740 | MF | transferase activity | 2/19 |
| GO:0008408 | MF | 3′-5′ exonuclease activity | 1/19 |
| GO:0019706 | MF | protein-cysteine S-palmitoyltransferase activity | 1/19 |
| GO:0022857 | MF | transmembrane transporter activity | 1/19 |
| miRNA ID | log2(FC) | pval | Transcript ID | log2(FC) | pval | Pathway |
|---|---|---|---|---|---|---|
| osa-MIR5079a-p3_2ss2CT21TA | 6.3 | 0.01 | Zm00001d027612_T003 | −1.9 | 0.03 | Porphyrin and chlorophyll metabolism |
| Aminoacyl-tRNA biosynthesis | ||||||
| miRn218 | 3.4 | 0.01 | Zm00001d016463_T005 | −3.7 | 0.02 | Steroid biosynthesis |
| Zm00001d022429_T030 | −12.6 | 0.00 | RNA degradation | |||
| Zm00001d022429_T036 | −12.6 | 0.00 | ||||
| miRn242 | −0.9 | 0.00 | Zm00001d051080_T001 | 4.6 | 0.00 | Purine metabolism |
| Zm00001d051080_T006 | 4.3 | 0.00 | ||||
| Zm00001d051080_T009 | 4.3 | 0.00 | ||||
| Zm00001d002141_T002 | 2.2 | 0.03 | Spliceosome | |||
| zma-miR164f-5p | −1.0 | 0.04 | Zm00001d043152_T001 | 4.7 | 0.02 | Pyrimidine metabolism |
| beta-Alanine metabolism | ||||||
| Pantothenate and CoA biosynthesis | ||||||
| zma-miR159a-5p | −1.1 | 0.01 | Zm00001d029550_T013 | 9.3 | 0.00 | Glycerolipid metabolism |
| Glycerophospholipid metabolism | ||||||
| Phosphatidylinositol signaling system | ||||||
| miRn248 | −1.4 | 0.01 | Zm00001d043998_T004 | 3.1 | 0.00 | Protein processing in ER |
| Ubiquitin mediated proteolysis | ||||||
| miRn194 | −1.6 | 0.04 | Zm00001d002141_T002 | 2.2 | 0.03 | Spliceosome |
| miRn202 | −2.3 | 0.01 | Zm00001d027314_T002 | 1.6 | 0.02 | Endocytosis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; An, P.; Li, H.; Wang, X.; Zhou, J.; Dong, P.; Zhao, Y.; Wang, Q.; Li, C. The miRNA-Mediated Post-Transcriptional Regulation of Maize in Response to High Temperature. Int. J. Mol. Sci. 2019, 20, 1754. https://doi.org/10.3390/ijms20071754
Zhang M, An P, Li H, Wang X, Zhou J, Dong P, Zhao Y, Wang Q, Li C. The miRNA-Mediated Post-Transcriptional Regulation of Maize in Response to High Temperature. International Journal of Molecular Sciences. 2019; 20(7):1754. https://doi.org/10.3390/ijms20071754
Chicago/Turabian StyleZhang, Moubiao, Panpan An, Hongping Li, Xiuling Wang, Jinlong Zhou, Pengfei Dong, Yali Zhao, Qun Wang, and Chaohai Li. 2019. "The miRNA-Mediated Post-Transcriptional Regulation of Maize in Response to High Temperature" International Journal of Molecular Sciences 20, no. 7: 1754. https://doi.org/10.3390/ijms20071754