Neuromuscular Plasticity in a Mouse Neurotoxic Model of Spinal Motoneuronal Loss

 ,
,  , ,
, ,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
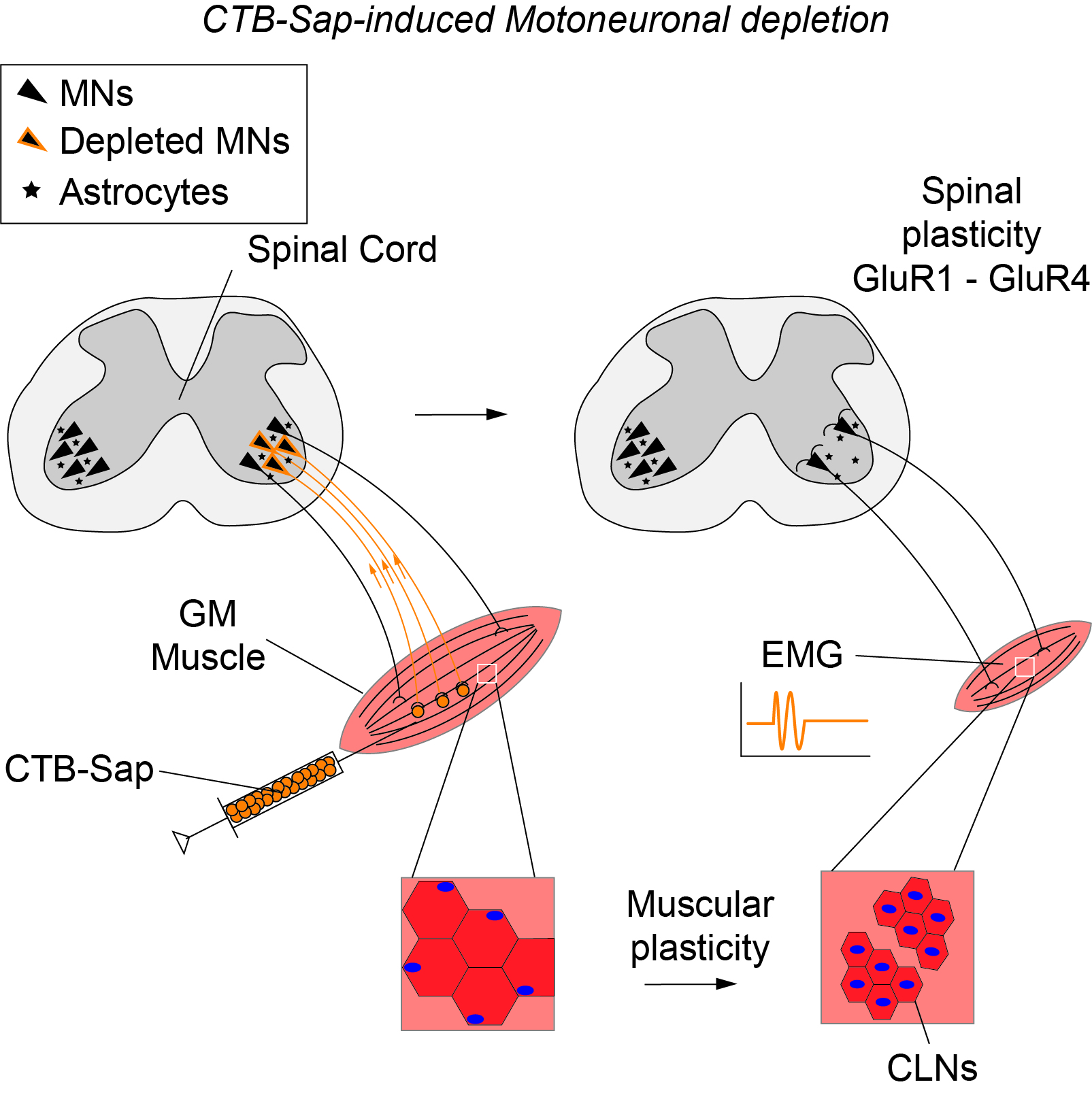
1. Introduction
2. Results
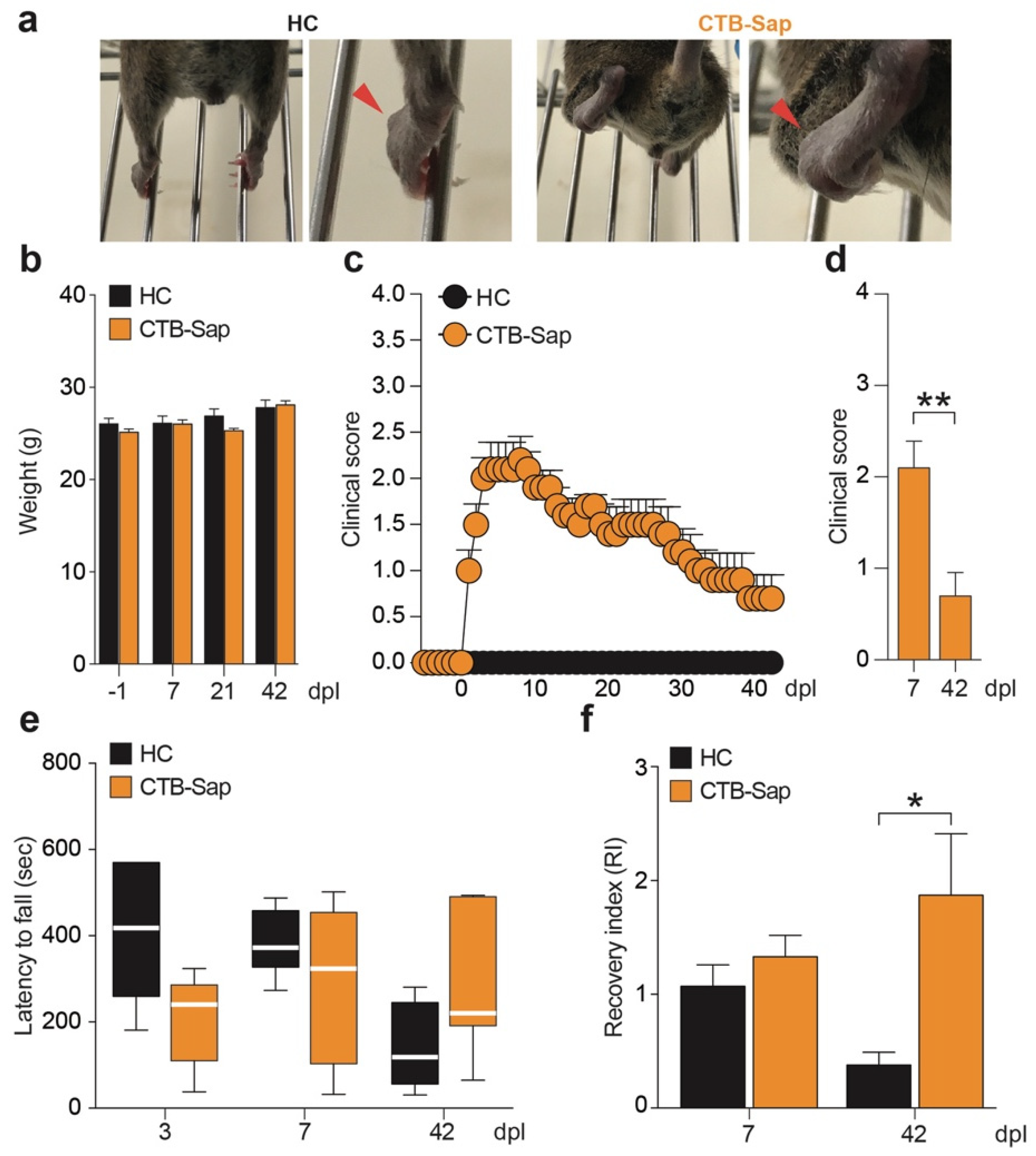
2.1. Behavioral Impairment Induced by CTB-Sap Lesion
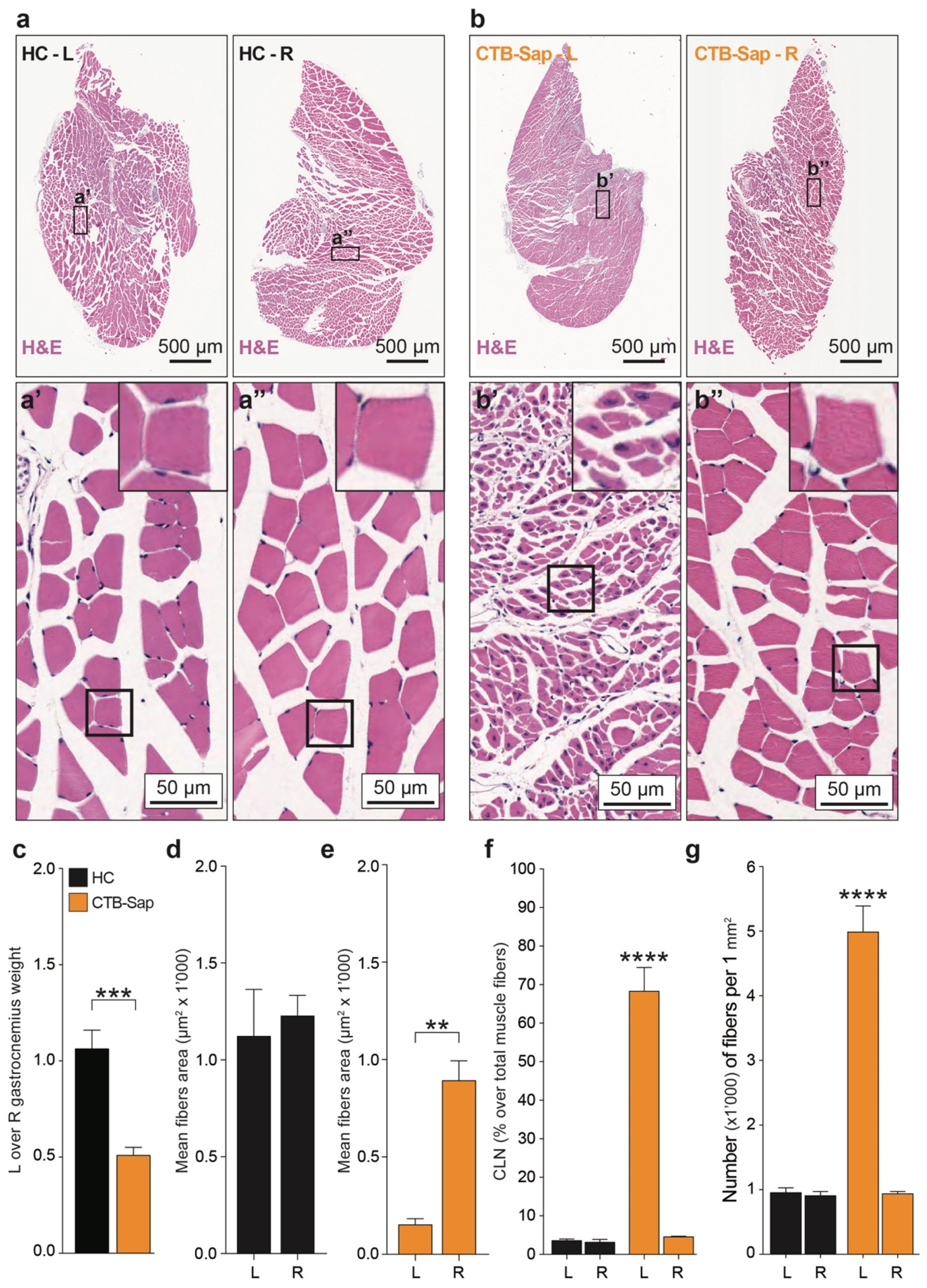
2.2. Anatomical Changes of Denervated Muscle
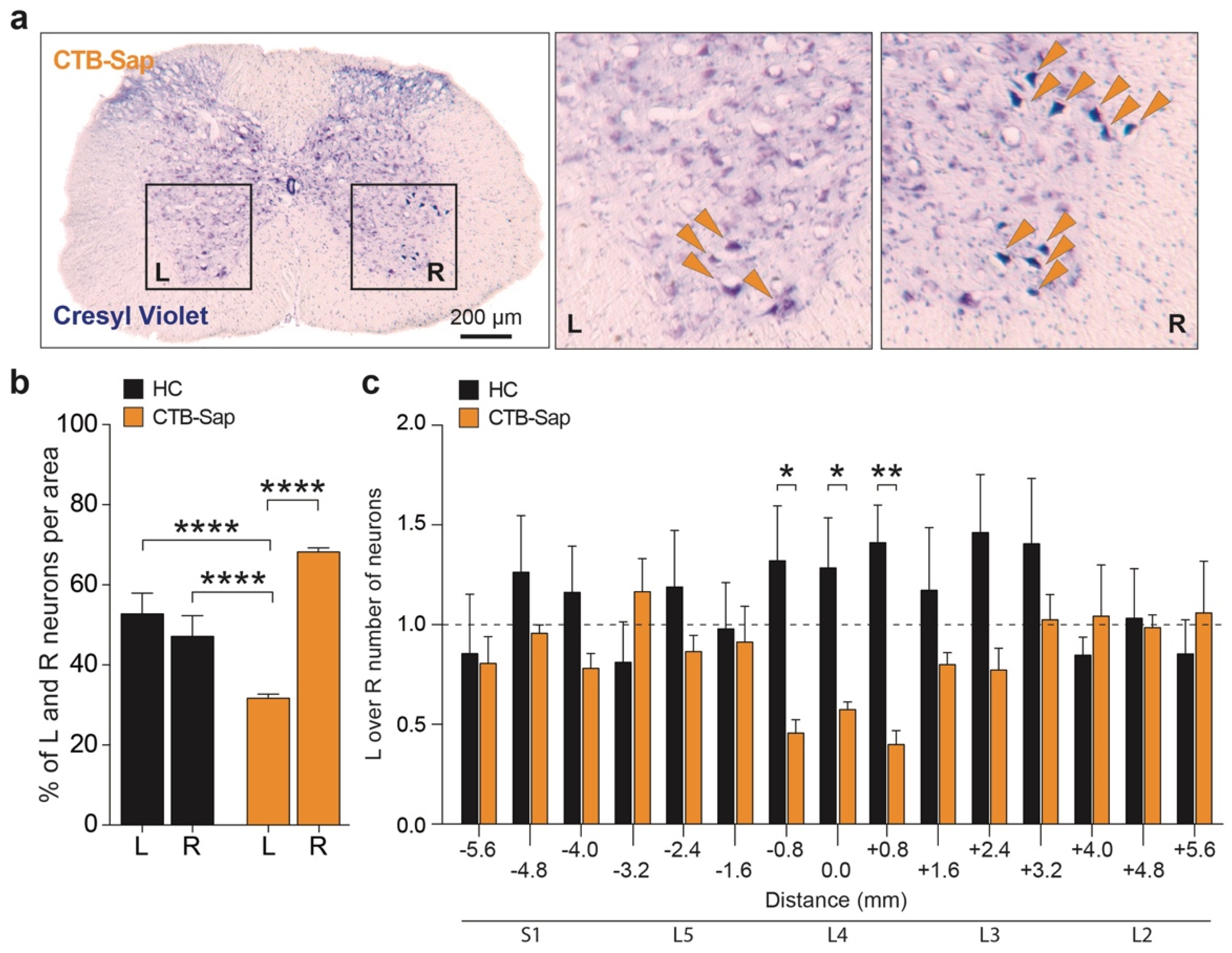
2.3. Plasticity Mechanisms in the SCs of CTB-Sap-Lesioned Mice
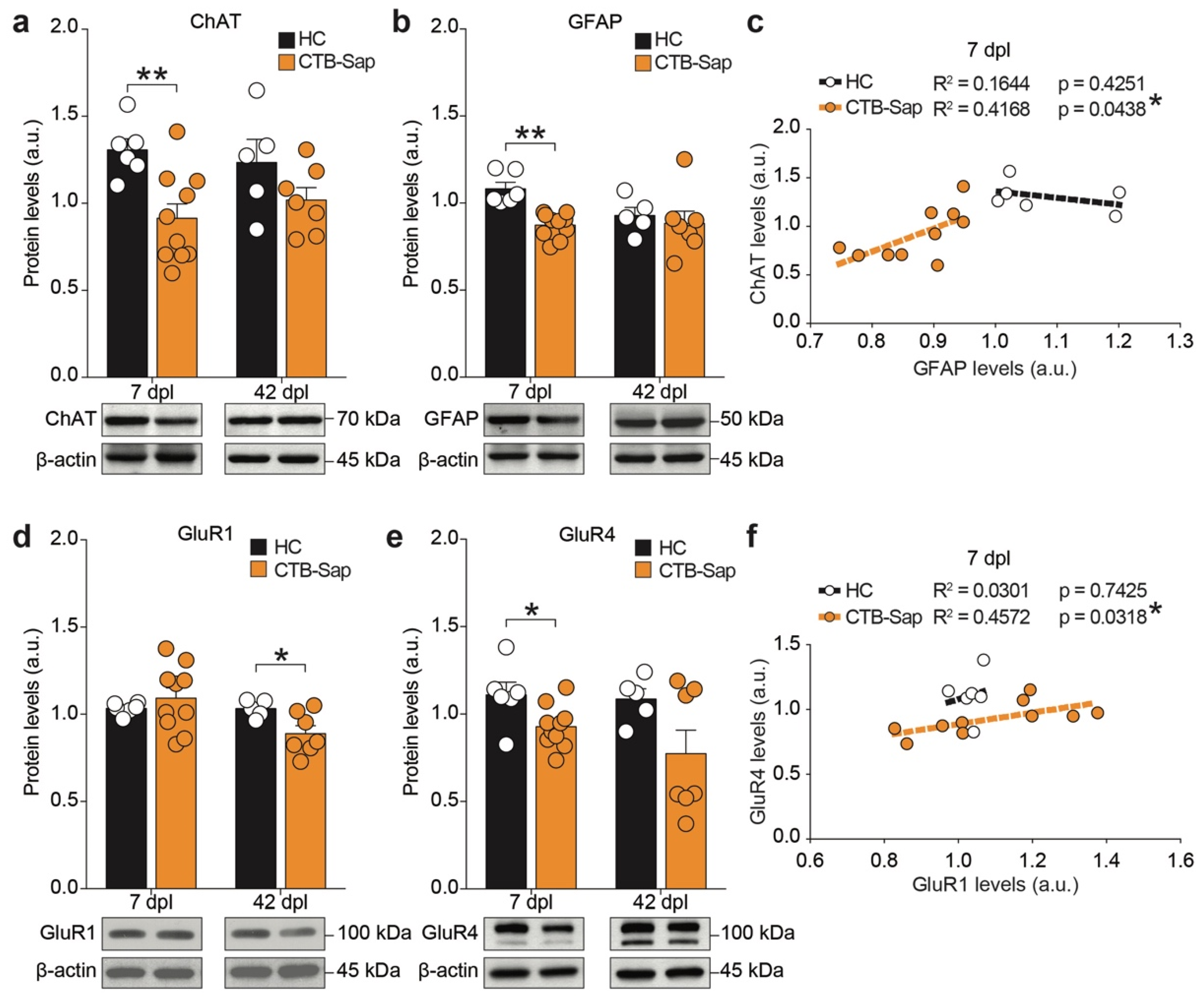
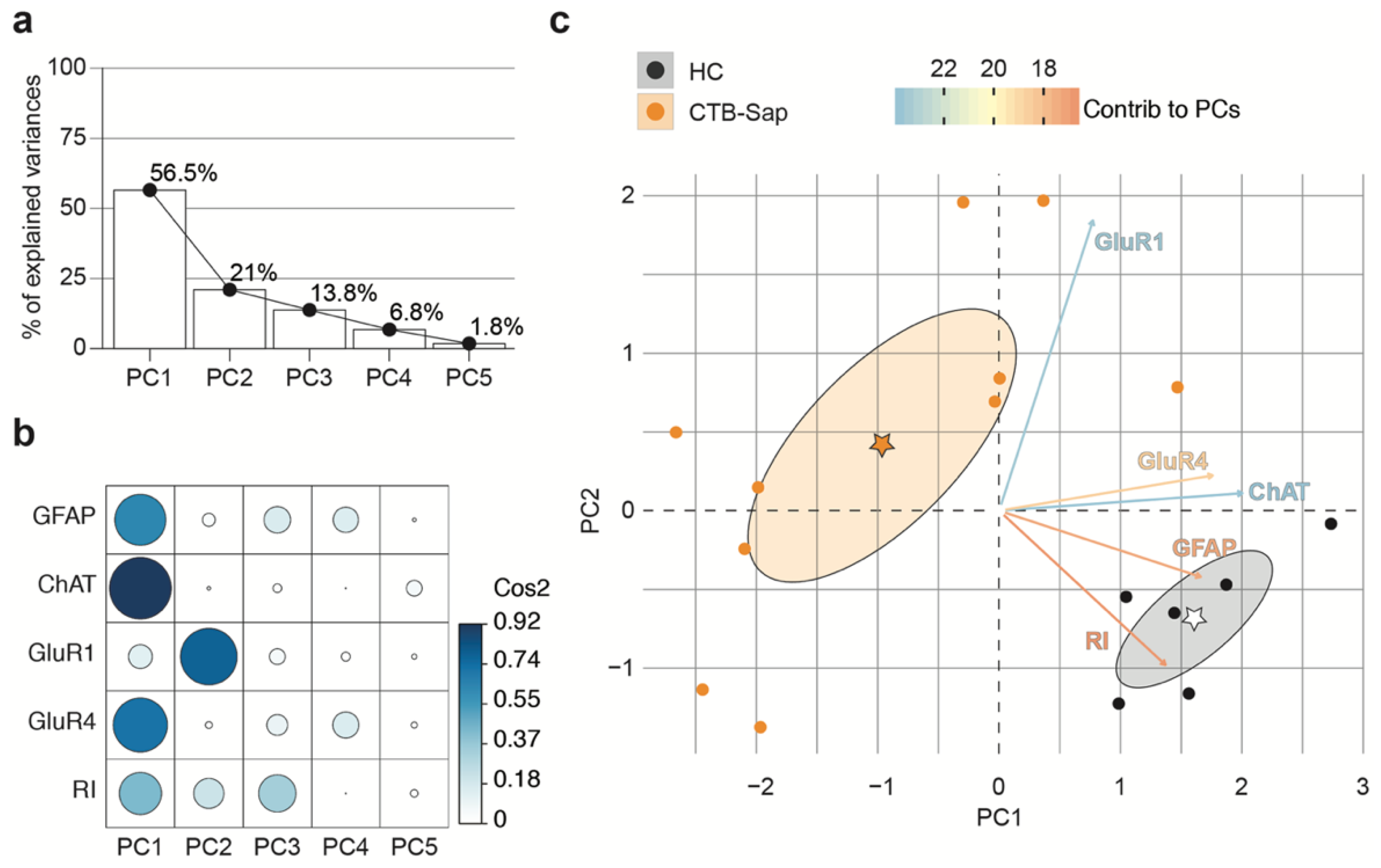
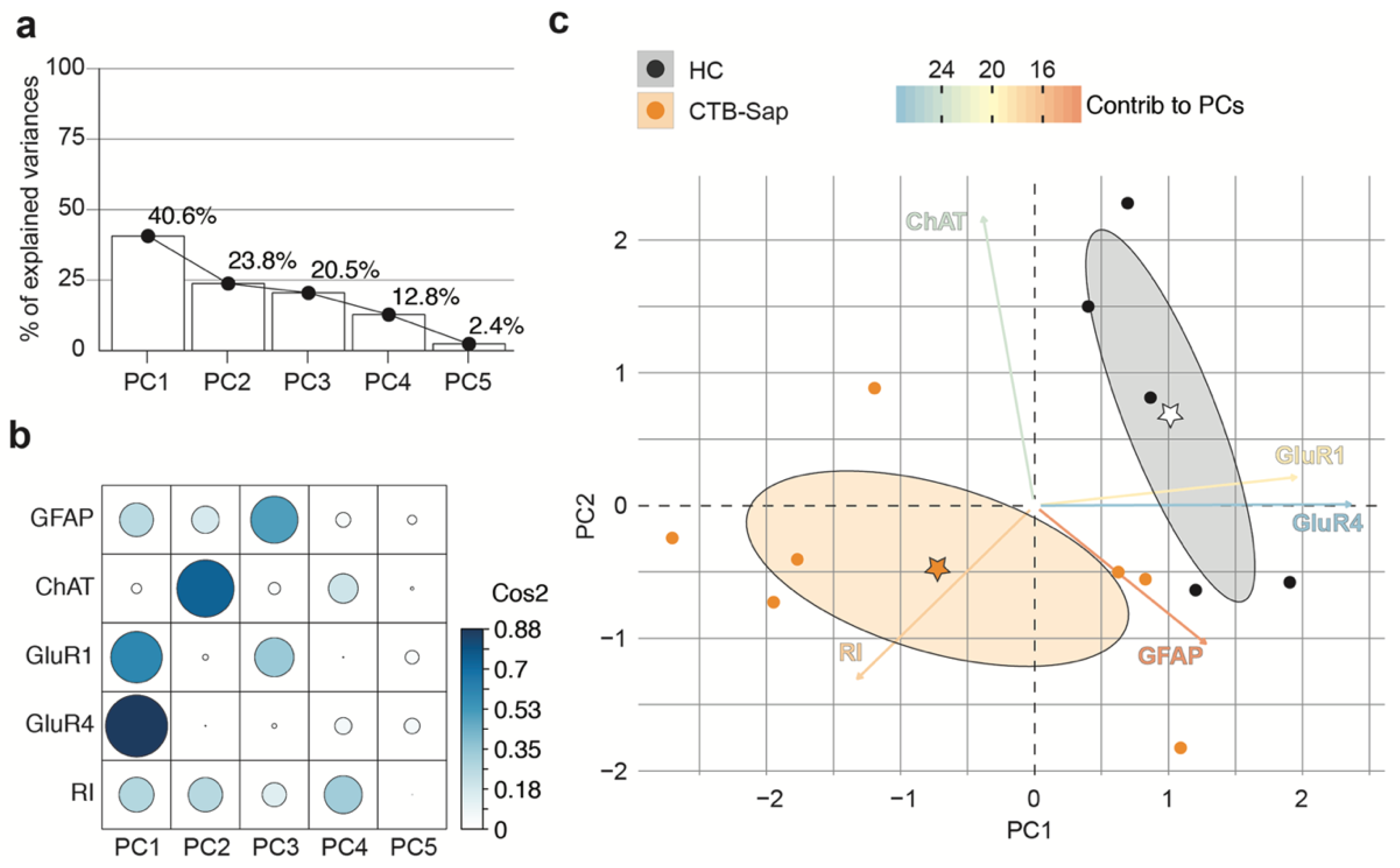
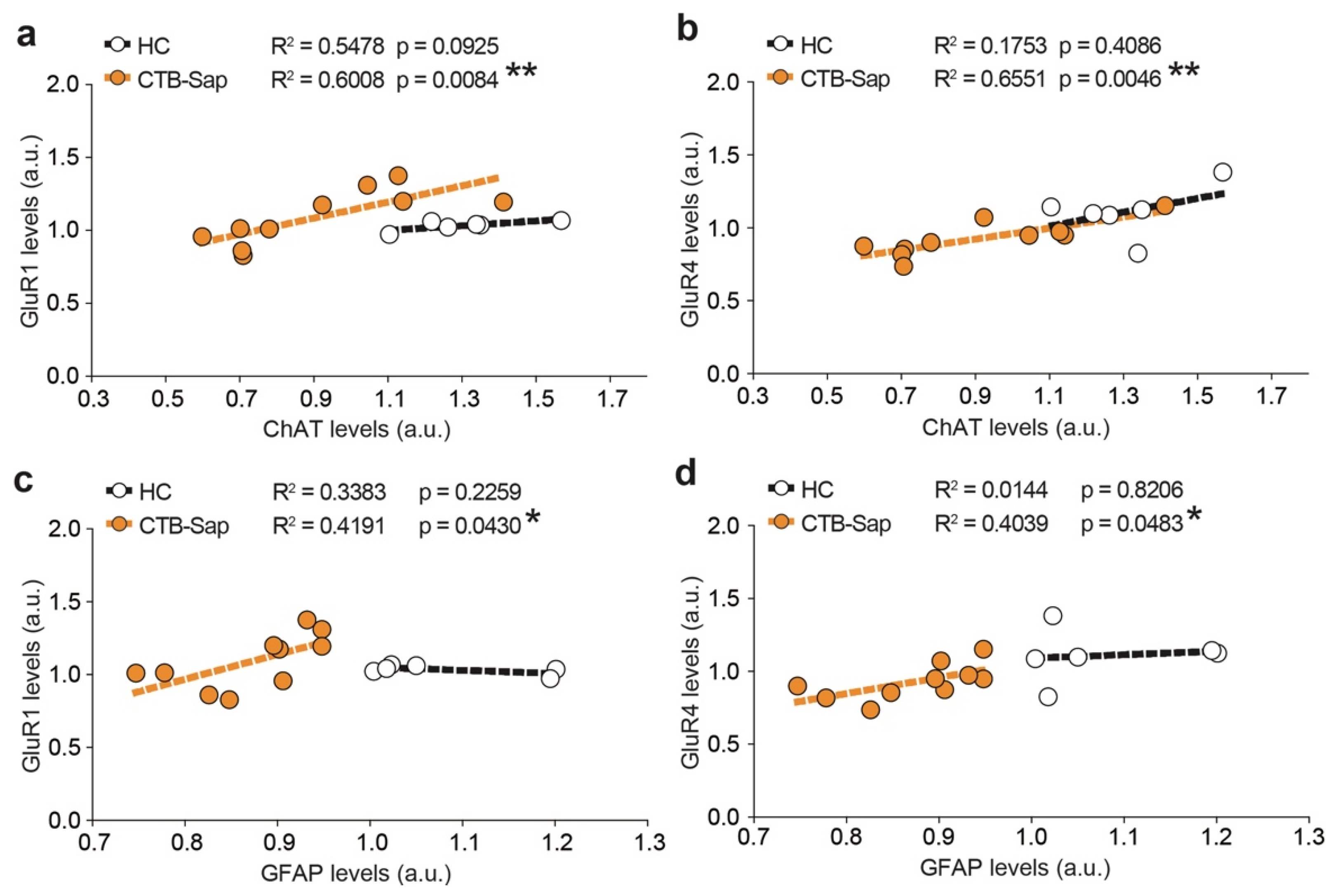
2.4. GluR1 and GluR4 Are Involved in Neuronal Plasticity upon Selective Spinal MN Depletion
3. Discussion
4. Materials and Methods
4.1. Subjects and Experimental Design
4.2. Neurotoxic Ablation of Spinal MNs
4.3. Behavioral Tests
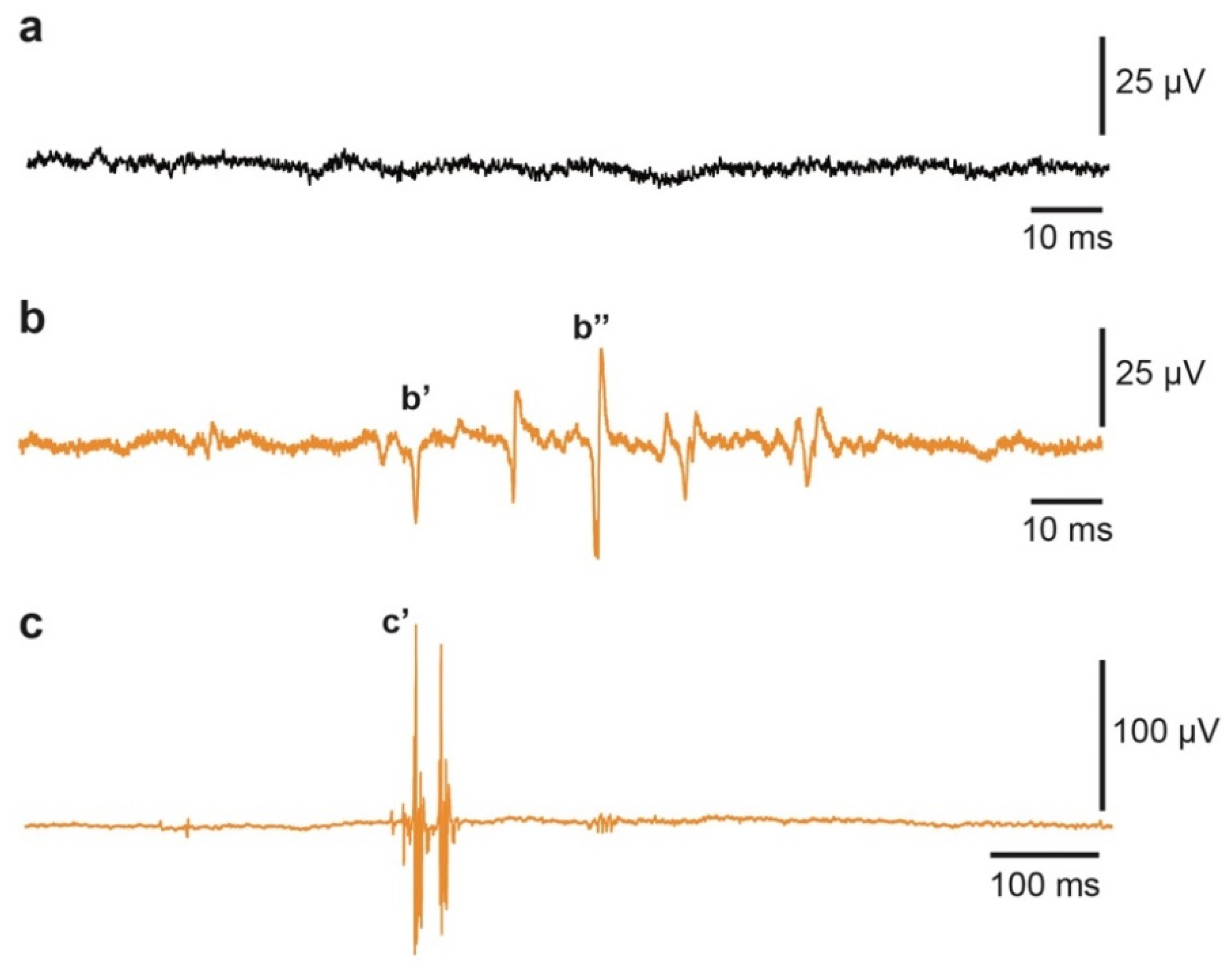
4.4. Electromyography
4.5. Ex Vivo Tissue Pathology (Muscles and SCs)
4.6. Immunoblotting
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| CLN | Centrally located nuclei |
| ChAT | Choline acetyltransferase |
| Cos2 | Square cosine |
| CTB-Sap | Cholera toxin-B saporin |
| Dpl | Day post lesion |
| EMG | Electromyography |
| FUS | Fused in sarcoma |
| GFAP | Glial fibrillary acidic protein |
| GM | Gastrocnemius muscle |
| HC | Healthy control |
| MN | Motoneurons |
| PCA | Principal component analysis |
| RI | Recovery index |
| ROI | Region of interest |
| SC | Spinal cord |
| SMA | Spinal muscular atrophy |
| SOD1 | Superoxide dismutase 1 |
| TDP-43 | TAR-DNA binding protein |
References
- Gordon, P.H. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals, C. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisen, A. Amyotrophic lateral sclerosis: A 40-year personal perspective. J. Clin. Neurosci. 2009, 16, 505–512. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Manzano, R.; Vaz, R.; Osta, R.; Brites, D. Synaptic Failure: Focus in an Integrative View of ALS. Brain Plast. 2016, 1, 159–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Bories, C.; Amendola, J.; Lamotte d’Incamps, B.; Durand, J. Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2007, 25, 451–459. [Google Scholar] [CrossRef]
- Quinlan, K.A.; Kajtaz, E.; Ciolino, J.D.; Imhoff-Manuel, R.D.; Tresch, M.C.; Heckman, C.J.; Tysseling, V.M. Chronic electromyograms in treadmill running SOD1 mice reveal early changes in muscle activation. J. Physiol. 2017, 595, 5387–5400. [Google Scholar] [CrossRef]
- Llewellyn-Smith, I.J.; Martin, C.L.; Arnolda, L.F.; Minson, J.B. Tracer-toxins: Cholera toxin B-saporin as a model. J. Neurosci. Methods 2000, 103, 83–90. [Google Scholar] [CrossRef]
- Wiley, R.G.; Kline, I.R. Neuronal lesioning with axonally transported toxins. J. Neurosci. Methods 2000, 103, 73–82. [Google Scholar] [CrossRef]
- Gulino, R.; Perciavalle, V.; Gulisano, M. Expression of cell fate determinants and plastic changes after neurotoxic lesion of adult mice spinal cord by cholera toxin-B saporin. Eur. J. Neurosci. 2010, 31, 1423–1434. [Google Scholar] [CrossRef]
- Gulino, R.; Litrico, L.; Leanza, G. Long-term survival and development of fetal ventral spinal grafts into the motoneuron-depleted rat spinal cord: Role of donor age. Brain Res. 2010, 1323, 41–47. [Google Scholar] [CrossRef]
- Tremblay, E.; Martineau, E.; Robitaille, R. Opposite Synaptic Alterations at the Neuromuscular Junction in an ALS Mouse Model: When Motor Units Matter. J. Neurosci. 2017, 37, 8901–8918. [Google Scholar] [CrossRef] [PubMed]
- Dukkipati, S.S.; Garrett, T.L.; Elbasiouny, S.M. The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2018, 596, 1723–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, J.; Amendola, J.; Bories, C.; Lamotte d’Incamps, B. Early abnormalities in transgenic mouse models of amyotrophic lateral sclerosis. J. Physiol. Paris 2006, 99, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 37968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, A.; Wen, J.; Rinaldi, D.; Houot, M.; Sayah, S.; Camuzat, A.; Fournier, C.; Fontanella, S.; Routier, A.; Couratier, P.; et al. Early Cognitive, Structural, and Microstructural Changes in Presymptomatic C9orf72 Carriers Younger Than 40 Years. JAMA Neurol. 2018, 75, 236–245. [Google Scholar] [CrossRef]
- Papma, J.M.; Jiskoot, L.C.; Panman, J.L.; Dopper, E.G.; den Heijer, T.; Donker Kaat, L.; Pijnenburg, Y.A.L.; Meeter, L.H.; van Minkelen, R.; Rombouts, S.; et al. Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology 2017, 89, 1256–1264. [Google Scholar] [CrossRef]
- Mills, K.R. The basics of electromyography. J. Neurol. Neurosurg. Psychiatry 2005, 76, ii32-35. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, A.; Gaetani, E.; Palladino, M.; Gatto, I.; Smith, R.C.; Neri, V.; Marcantoni, M.; Giarretta, I.; Silver, M.; Straino, S.; et al. Sonic hedgehog gene therapy increases the ability of the dystrophic skeletal muscle to regenerate after injury. Gene Ther. 2014, 21, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, A.; Weber, C.; Friedrich, O. A novel quantitative morphometry approach to assess regeneration in dystrophic skeletal muscle. Neuromuscul. Disord. 2014, 24, 596–603. [Google Scholar] [CrossRef]
- Folker, E.S.; Baylies, M.K. Nuclear positioning in muscle development and disease. Front Physiol. 2013, 4, 363. [Google Scholar] [CrossRef] [PubMed]
- Umansky, K.B.; Gruenbaum-Cohen, Y.; Tsoory, M.; Feldmesser, E.; Goldenberg, D.; Brenner, O.; Groner, Y. Runx1 Transcription Factor Is Required for Myoblasts Proliferation during Muscle Regeneration. PLoS Genet. 2015, 11, e1005457. [Google Scholar] [CrossRef] [PubMed]
- Krivickas, L.S.; Yang, J.I.; Kim, S.K.; Frontera, W.R. Skeletal muscle fiber function and rate of disease progression in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 636–643. [Google Scholar] [CrossRef]
- Morgan, B.R.; Coates, J.R.; Johnson, G.C.; Bujnak, A.C.; Katz, M.L. Characterization of intercostal muscle pathology in canine degenerative myelopathy: A disease model for amyotrophic lateral sclerosis. J. Neurosci. Res. 2013, 91, 1639–1650. [Google Scholar] [CrossRef]
- Park, G.H.; Maeno-Hikichi, Y.; Awano, T.; Landmesser, L.T.; Monani, U.R. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J. Neurosci. 2010, 30, 12005–12019. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musaro, A. Metabolic Changes Associated With Muscle Expression of SOD1(G93A). Front. Physiol. 2018, 9, 831. [Google Scholar] [CrossRef]
- Lehmann, S.; Esch, E.; Hartmann, P.; Goswami, A.; Nikolin, S.; Weis, J.; Beyer, C.; Johann, S. Expression profile of pattern recognition receptors in skeletal muscle of SOD1((G93A)) amyotrophic lateral sclerosis (ALS) mice and sporadic ALS patients. Neuropathol. Appl. Neurobiol. 2018, 44, 606–627. [Google Scholar] [CrossRef]
- Pansarasa, O.; Rossi, D.; Berardinelli, A.; Cereda, C. Amyotrophic lateral sclerosis and skeletal muscle: An update. Mol. Neurobiol. 2014, 49, 984–990. [Google Scholar] [CrossRef]
- Gulino, R.; Cataudella, T.; Casamenti, F.; Pepeu, G.; Stanzani, S.; Leanza, G. Acetylcholine release from fetal tissue homotopically grafted to the motoneuron-depleted lumbar spinal cord. An in vivo microdialysis study in the awake rat. Exp. Neurol. 2007, 204, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.P.; Phelps, P.E.; Houser, C.R.; Crawford, G.D.; Salvaterra, P.M.; Vaughn, J.E. The morphology and distribution of neurons containing choline acetyltransferase in the adult rat spinal cord: An immunocytochemical study. J. Comp. Neurol. 1984, 229, 329–346. [Google Scholar] [CrossRef]
- Navaratnam, V.; Lewis, P.R. Cholinesterase-containing neurones in the spinal cord of the rat. Brain Res. 1970, 18, 411–425. [Google Scholar] [CrossRef]
- Phelps, C.J.; Sladek, J.R., Jr. Plasticity of catecholaminergic neurons in aged rat brain: Reinnervation and functional recovery after axotomy. Brain Res. Bull. 1984, 13, 727–736. [Google Scholar] [CrossRef]
- Mendell, L.M.; Munson, J.B.; Arvanian, V.L. Neurotrophins and synaptic plasticity in the mammalian spinal cord. J. Physiol. 2001, 533, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulino, R.; Dimartino, M.; Casabona, A.; Lombardo, S.A.; Perciavalle, V. Synaptic plasticity modulates the spontaneous recovery of locomotion after spinal cord hemisection. Neurosci. Res. 2007, 57, 148–156. [Google Scholar] [CrossRef]
- Edgerton, V.R.; Tillakaratne, N.J.; Bigbee, A.J.; de Leon, R.D.; Roy, R.R. Plasticity of the spinal neural circuitry after injury. Annu. Rev. Neurosci. 2004, 27, 145–167. [Google Scholar] [CrossRef]
- Gulino, R.; Forte, S.; Parenti, R.; Gulisano, M. TDP-43 as a Modulator of Synaptic Plasticity in a Mouse Model of Spinal Motoneuron Degeneration. CNS Neurol. Disord. Drug Targets 2015, 14, 55–60. [Google Scholar] [CrossRef]
- Gulino, R.; Gulisano, M. Involvement of brain-derived neurotrophic factor and sonic hedgehog in the spinal cord plasticity after neurotoxic partial removal of lumbar motoneurons. Neurosci. Res. 2012, 73, 238–247. [Google Scholar] [CrossRef]
- Gulino, R.; Gulisano, M. Noggin and Sonic hedgehog are involved in compensatory changes within the motoneuron-depleted mouse spinal cord. J. Neurol. Sci. 2013, 332, 102–109. [Google Scholar] [CrossRef]
- Serapide, M.F.; Zappala, A.; Parenti, R.; Panto, M.R.; Cicirata, F. Laterality of the pontocerebellar projections in the rat. Eur. J. Neurosci. 2002, 15, 1551–1556. [Google Scholar] [CrossRef]
- Parenti, R.; Zappala, A.; Serapide, M.F.; Panto, M.R.; Cicirata, F. Projections of the basilar pontine nuclei and nucleus reticularis tegmenti pontis to the cerebellar nuclei of the rat. J. Comp. Neurol. 2002, 452, 115–127. [Google Scholar] [CrossRef]
- Cicirata, F.; Zappala, A.; Serapide, M.F.; Parenti, R.; Panto, M.R.; Paz, C. Different pontine projections to the two sides of the cerebellum. Brain Res. Brain Res. Rev. 2005, 49, 280–294. [Google Scholar] [CrossRef]
- Cicirata, F.; Serapide, M.F.; Parenti, R.; Panto, M.R.; Zappala, A.; Nicotra, A.; Cicero, D. The basilar pontine nuclei and the nucleus reticularis tegmenti pontis subserve distinct cerebrocerebellar pathways. Prog. Brain Res. 2005, 148, 259–282. [Google Scholar] [CrossRef]
- Lule, D.; Diekmann, V.; Kassubek, J.; Kurt, A.; Birbaumer, N.; Ludolph, A.C.; Kraft, E. Cortical plasticity in amyotrophic lateral sclerosis: Motor imagery and function. Neurorehabil. Neural. Repair. 2007, 21, 518–526. [Google Scholar] [CrossRef]
- Perry, S.; Han, Y.; Das, A.; Dickman, D. Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum. Mol. Genet. 2017, 26, 4153–4167. [Google Scholar] [CrossRef]
- Konrad, C.; Jansen, A.; Henningsen, H.; Sommer, J.; Turski, P.A.; Brooks, B.R.; Knecht, S. Subcortical reorganization in amyotrophic lateral sclerosis. Exp. Brain Res. 2006, 172, 361–369. [Google Scholar] [CrossRef]
- Pullen, A.H.; Athanasiou, D. Increase in presynaptic territory of C-terminals on lumbar motoneurons of G93A SOD1 mice during disease progression. Eur. J. Neurosci. 2009, 29, 551–561. [Google Scholar] [CrossRef]
- Poujois, A.; Schneider, F.C.; Faillenot, I.; Camdessanche, J.P.; Vandenberghe, N.; Thomas-Anterion, C.; Antoine, J.C. Brain plasticity in the motor network is correlated with disease progression in amyotrophic lateral sclerosis. Hum. Brain Mapp. 2013, 34, 2391–2401. [Google Scholar] [CrossRef]
- Sica, R.E. Is amyotrophic lateral sclerosis a primary astrocytic disease? Med. Hypotheses 2012, 79, 819–822. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, D.; Le Verche, V.; Jacquier, A.; Ikiz, B.; Przedborski, S.; Re, D.B. Inflammation in ALS and SMA: Sorting out the good from the evil. Neurobiol. Dis. 2010, 37, 493–502. [Google Scholar] [CrossRef]
- Vicario, N.; Zappala, A.; Calabrese, G.; Gulino, R.; Parenti, C.; Gulisano, M.; Parenti, R. Connexins in the Central Nervous System: Physiological Traits and Neuroprotective Targets. Front. Physiol. 2017, 8, 1060. [Google Scholar] [CrossRef]
- Vicario, N.; Calabrese, G.; Zappala, A.; Parenti, C.; Forte, S.; Graziano, A.C.E.; Vanella, L.; Pellitteri, R.; Cardile, V.; Parenti, R. Inhibition of Cx43 mediates protective effects on hypoxic/reoxygenated human neuroblastoma cells. J. Cell Mol. Med. 2017, 21, 2563–2572. [Google Scholar] [CrossRef] [Green Version]
- Kwak, S.; Hideyama, T.; Yamashita, T.; Aizawa, H. AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology 2010, 30, 182–188. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef]
- Kuner, R.; Groom, A.J.; Bresink, I.; Kornau, H.C.; Stefovska, V.; Muller, G.; Hartmann, B.; Tschauner, K.; Waibel, S.; Ludolph, A.C.; et al. Late-onset motoneuron disease caused by a functionally modified AMPA receptor subunit. Proc. Natl. Acad. Sci. USA 2005, 102, 5826–5831. [Google Scholar] [CrossRef] [Green Version]
- Nichols, N.L.; Vinit, S.; Bauernschmidt, L.; Mitchell, G.S. Respiratory function after selective respiratory motor neuron death from intrapleural CTB-saporin injections. Exp. Neurol. 2015, 267, 18–29. [Google Scholar] [CrossRef]
- Lind, L.A.; Murphy, E.R.; Lever, T.E.; Nichols, N.L. Hypoglossal Motor Neuron Death Via Intralingual CTB-saporin (CTB-SAP) Injections Mimic Aspects of Amyotrophic Lateral Sclerosis (ALS) Related to Dysphagia. Neuroscience 2018, 390, 303–316. [Google Scholar] [CrossRef]
- Nichols, N.L.; Craig, T.A.; Tanner, M.A. Phrenic long-term facilitation following intrapleural CTB-SAP-induced respiratory motor neuron death. Respir. Physiol. Neurobiol. 2018, 256, 43–49. [Google Scholar] [CrossRef]
- Gulino, R.; Parenti, R.; Gulisano, M. Novel Mechanisms of Spinal Cord Plasticity in a Mouse Model of Motoneuron Disease. Biomed. Res. Int. 2015, 2015, 654637. [Google Scholar] [CrossRef]
- Vercelli, A.; Mereuta, O.M.; Garbossa, D.; Muraca, G.; Mareschi, K.; Rustichelli, D.; Ferrero, I.; Mazzini, L.; Madon, E.; Fagioli, F. Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 31, 395–405. [Google Scholar] [CrossRef]
- Hatzipetros, T.; Kidd, J.D.; Moreno, A.J.; Thompson, K.; Gill, A.; Vieira, F.G. A Quick Phenotypic Neurological Scoring System for Evaluating Disease Progression in the SOD1-G93A Mouse Model of ALS. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [Green Version]
- Parenti, R.; Puzzo, L.; Vecchio, G.M.; Gravina, L.; Salvatorelli, L.; Musumeci, G.; Vasquez, E.; Magro, G. Immunolocalization of Wilms’ Tumor protein (WT1) in developing human peripheral sympathetic and gastroenteric nervous system. Acta Histochem. 2014, 116, 48–54. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Vecchio, G.M.; Musumeci, G.; Rita, A.; Parenti, R. Cytoplasmic expression of Wilms tumor transcription factor-1 (WT1): A useful immunomarker for young-type fibromatoses and infantile fibrosarcoma. Acta Histochem. 2014, 116, 1134–1140. [Google Scholar] [CrossRef]
- Vicario, N.; Parenti, R.; Arico, G.; Turnaturi, R.; Scoto, G.M.; Chiechio, S.; Parenti, C. Repeated activation of delta opiod receptors counteracts nerve injury-induced TNF-alpha up-regulation in the sciatic nerve of rats with neuropathic pain: A possible correlation with delta opiod receptors-mediated antiallodinic effect. Mol. Pain 2016, 12. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulino, R.; Vicario, N.; Giunta, M.A.S.; Spoto, G.; Calabrese, G.; Vecchio, M.; Gulisano, M.; Leanza, G.; Parenti, R. Neuromuscular Plasticity in a Mouse Neurotoxic Model of Spinal Motoneuronal Loss. Int. J. Mol. Sci. 2019, 20, 1500. https://doi.org/10.3390/ijms20061500
Gulino R, Vicario N, Giunta MAS, Spoto G, Calabrese G, Vecchio M, Gulisano M, Leanza G, Parenti R. Neuromuscular Plasticity in a Mouse Neurotoxic Model of Spinal Motoneuronal Loss. International Journal of Molecular Sciences. 2019; 20(6):1500. https://doi.org/10.3390/ijms20061500
Chicago/Turabian StyleGulino, Rosario, Nunzio Vicario, Maria A.S. Giunta, Graziana Spoto, Giovanna Calabrese, Michele Vecchio, Massimo Gulisano, Giampiero Leanza, and Rosalba Parenti. 2019. "Neuromuscular Plasticity in a Mouse Neurotoxic Model of Spinal Motoneuronal Loss" International Journal of Molecular Sciences 20, no. 6: 1500. https://doi.org/10.3390/ijms20061500