Inducible Loss of the Aryl Hydrocarbon Receptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21

Abstract
:1. Introduction
2. Results
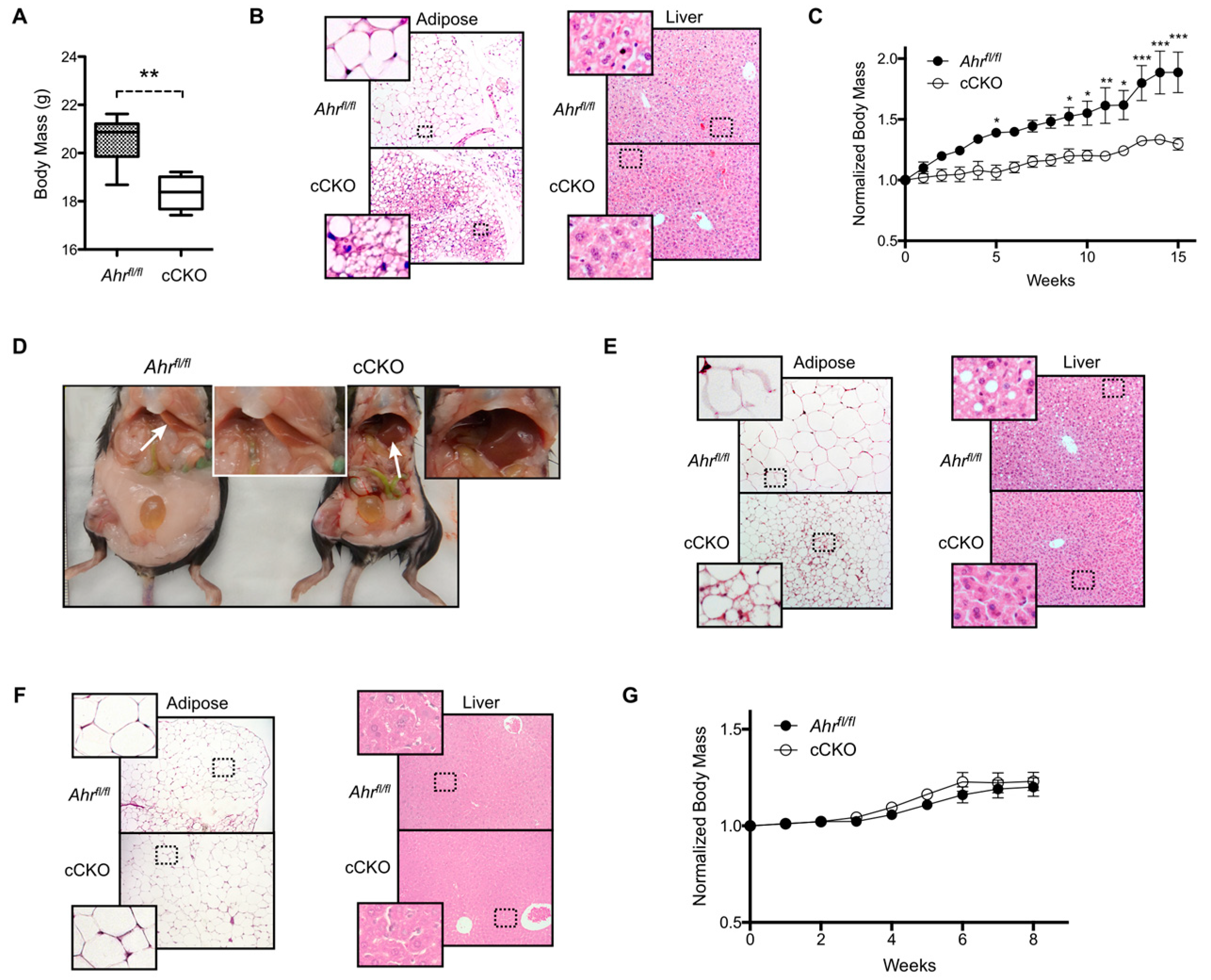
2.1. Constitutive Loss of Hepatocyte AHR Expression in Female Mice Reduces Body Mass and Adiposity under Standard Feeding Conditions and High-Fat Diet Challenge
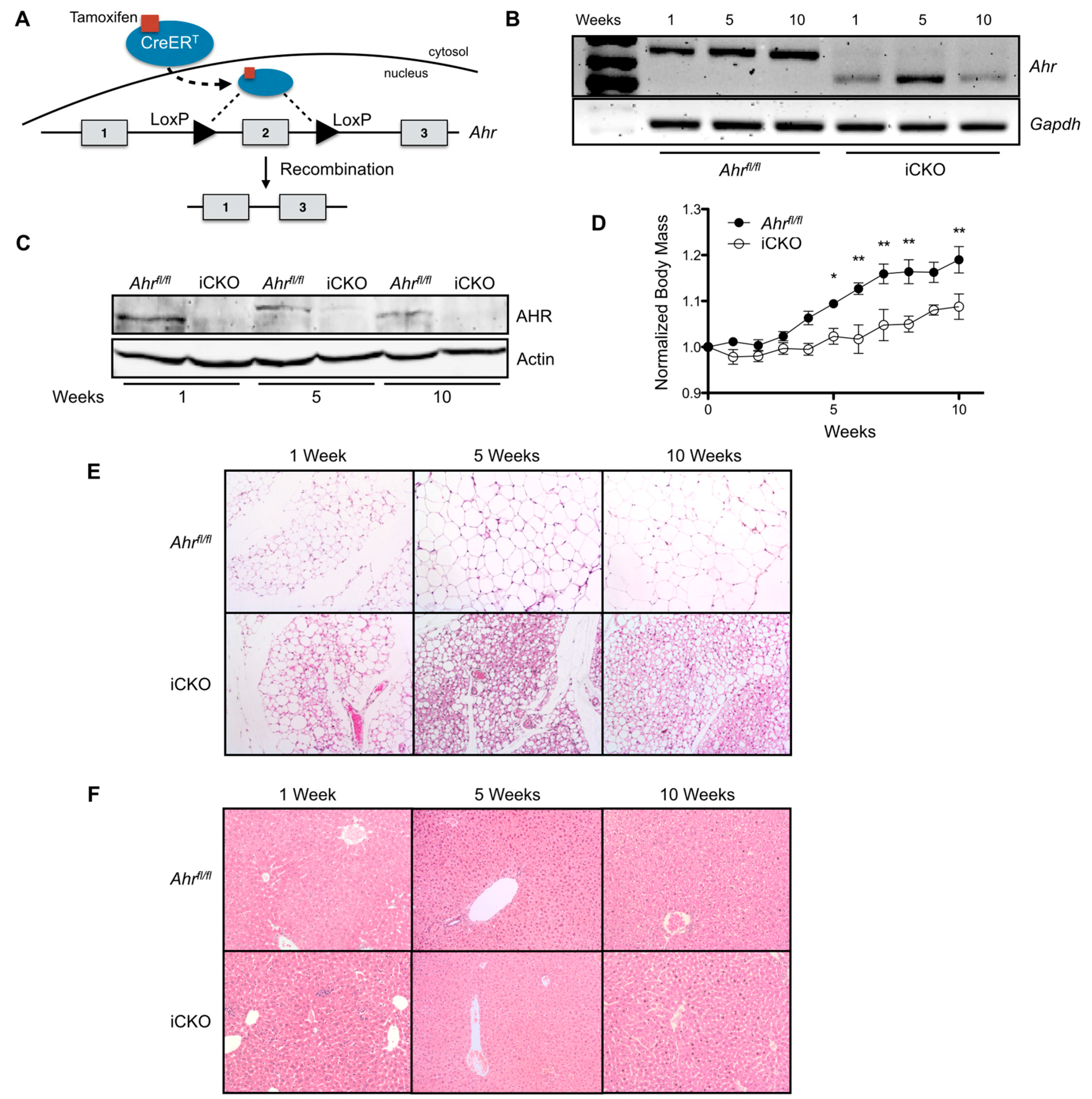
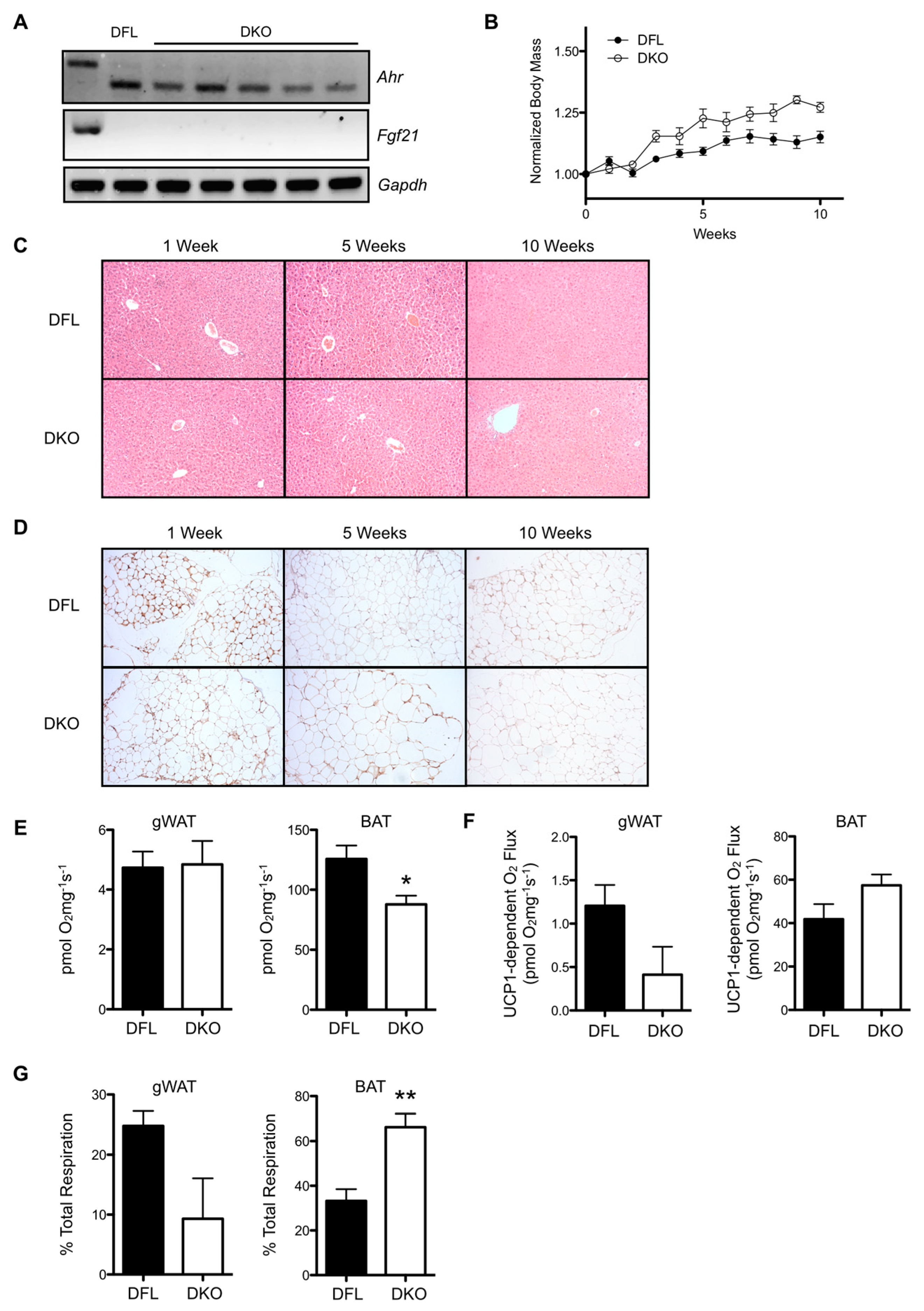
2.2. Induced Loss of Hepatocyte AHR Reduces Weight Gain and Adiposity, and Increases Multilocular Lipid Droplet Formation in gWAT.
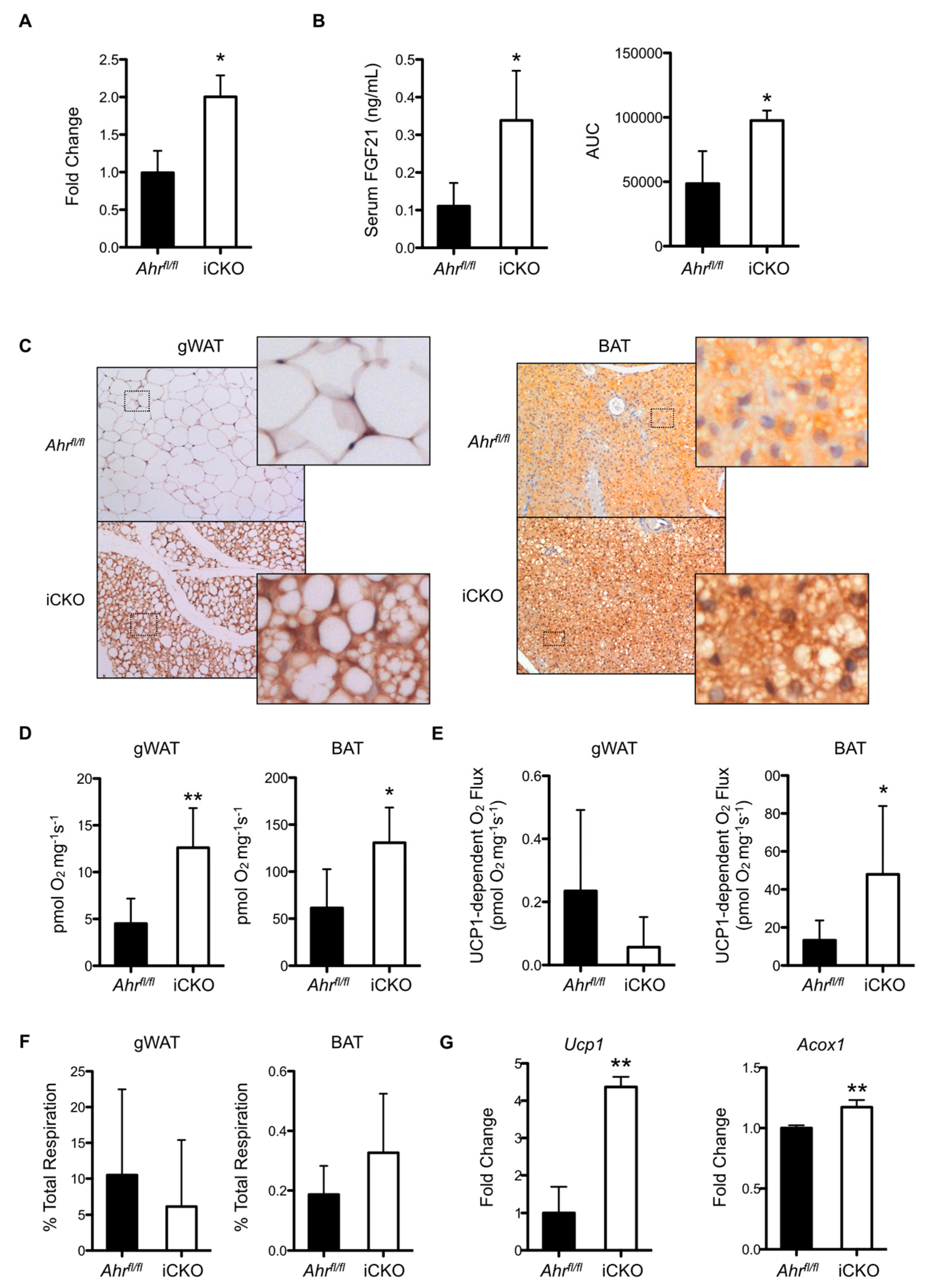
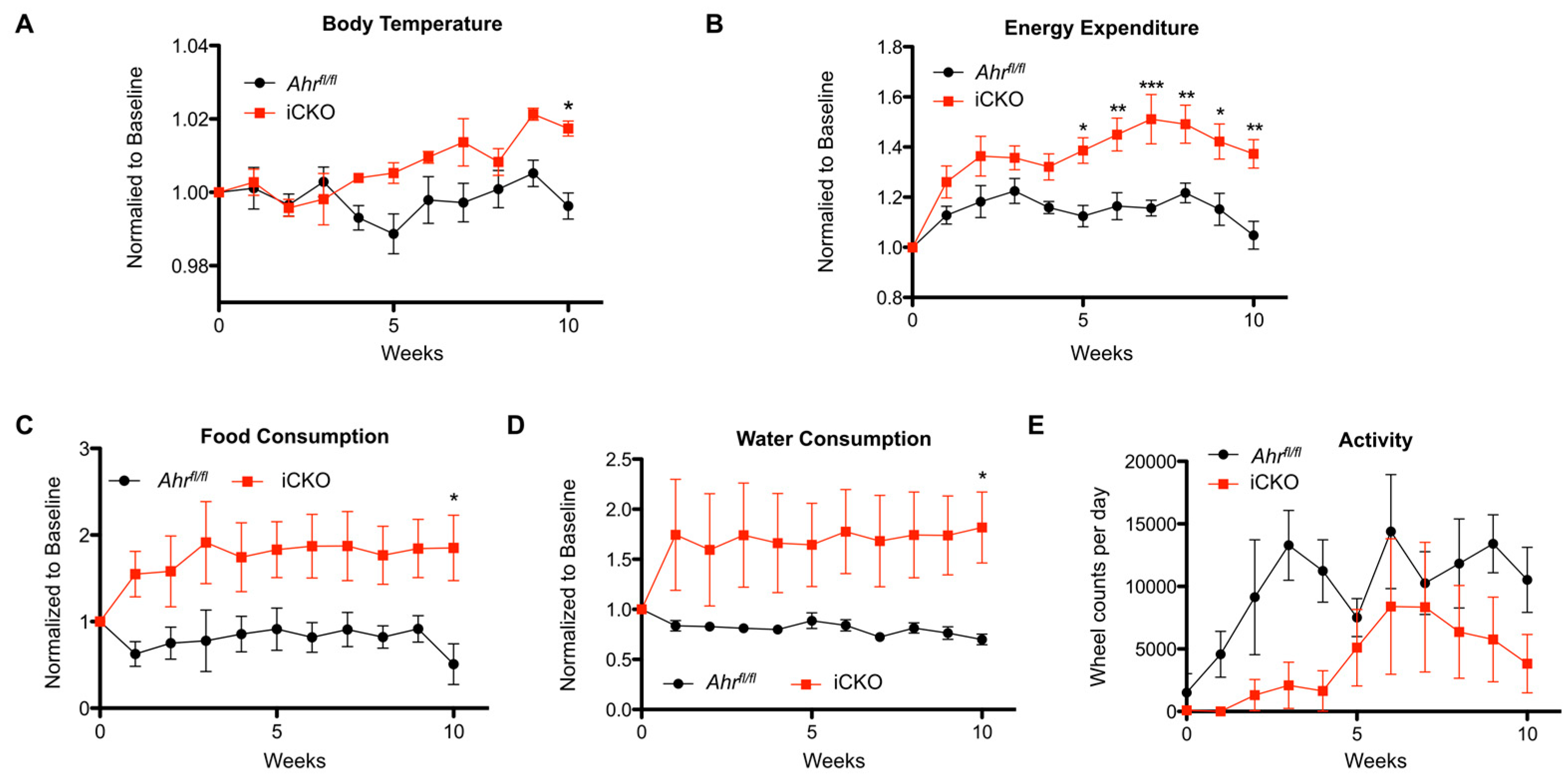
2.3. Induced AHR Loss Increases Hepatic FGF21 Production and BAT/gWAT Respiratory Capacity, and Is Associated with Greater Energy Expenditure and Water Intake in the Absence of Increased Physical Activity
2.4. Combinatory Deletion of AHR and FGF21 from Hepatocytes Reverses the Effects of Hepatic AHR Deficiency
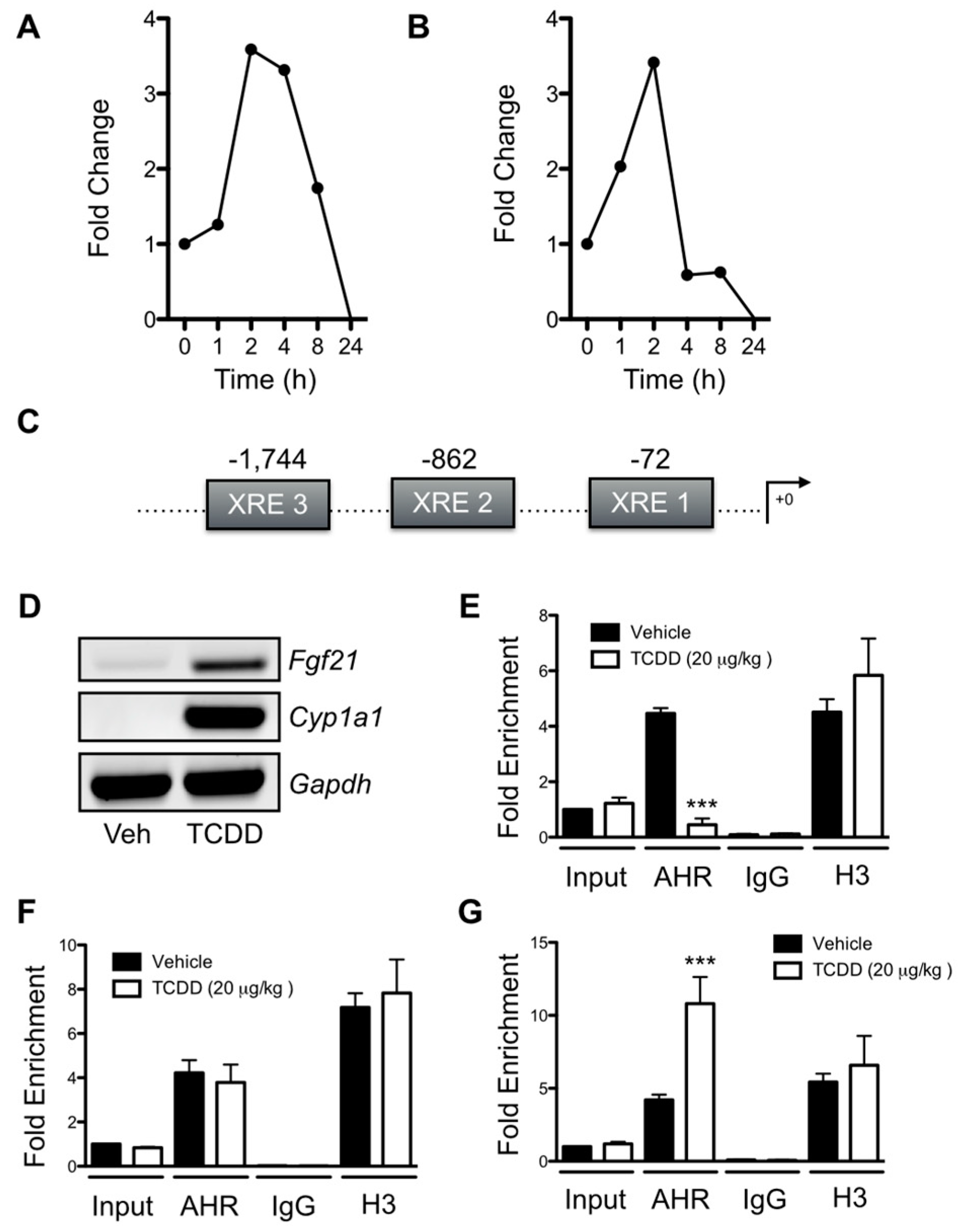
2.5. AHR Binding to a Novel XRE Site within the Fgf21 Promoter Is Associated with the Suppression of Fgf21 Transcription
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. RNA Extraction, Semi-Quantitative PCR, and Transcriptomic Analyses
4.3. Western Blot Analysis of AHR.
4.4. Serum FGF21 Analyses
4.5. Tryptic Digest of Serum Aliquots
4.6. Selective Reaction Monitoring (SRM)
4.7. Histological Analyses
4.8. High-Resolution Respirometry
4.9. Comprehensive Lab Animal Monitoring System
4.10. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACOX1 | Acyl-CoA Oxidase 1 |
| AHR | Aryl hydrocarbon receptor |
| AHSG | Alpha-2-HS-glycoprotein |
| ANTGL3 | Angiopoietin-like protein 3 |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| BAT | Brown adipose tissue |
| CYP1A1 | Cytochrome P450 1A1 |
| GWAT | Perigonadal white adipose tissue |
| IGFBP1/2 | Insulin-like growth factor binding protein 1/2 |
| FGF21 | Fibroblast growth factor 21 |
| SRM | Selective reaction monitoring |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| UCP1 | Uncoupling protein 1 |
| XRE | Xenobiotic response element |
References
- Beischlag, T.V.; Morales, J.L.; Hollingshead, B.D.; Perdew, G.H. The Aryl Hydrocarbon Receptor Complex and the Control of Gene Expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Vispute, S.G.; Liu, J.; Cheng, C.; Kharitonenkov, A.; Klaassen, C.D. Fibroblast growth factor (Fgf) 21 is a novel target gene of the aryl hydrocarbon receptor (AhR). Toxicol. Appl. Pharmacol. 2014, 278, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girer, N.G.; Murray, I.A.; Omiecinski, C.J.; Perdew, G.H. Hepatic Aryl Hydrocarbon Receptor Attenuates Fibroblast Growth Factor 21 Expression. J. Biol. Chem. 2016, 291, 15378–15387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Yan, J.; Liu, K.; Garbacz, W.G.; Wang, P.; Xu, M.; Ma, X.; Xie, W. Activation of aryl hydrocarbon receptor dissociates fatty liver from insulin resistance by inducing FGF21. Hepatology 2015, 61, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.M.; O’Dwyer, S.M.; Baker, R.K.; Covey, S.D.; Kieffer, T.J. FGF21-Mediated Improvements in Glucose Clearance Require Uncoupling Protein 1. Cell Rep. 2015, 13, 1521–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, K.A.; Sun, L. Turning WAT into BAT: A review on regulators controlling the browning of white adipocytes. Biosci. Rep. 2013, 33, e00065. [Google Scholar] [CrossRef]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 Is Liver Derived and Enhances Glucose Uptake During Refeeding and Overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.-S.; Lindberg, R.A.; et al. Fibroblast Growth Factor 21 Reverses Hepatic Steatosis, Increases Energy Expenditure, and Improves Insulin Sensitivity in Diet-Induced Obese Mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.D.; Li, C.Y.; Bina, H.A.; Lynes, S.E.; Michael, M.D.; Shanafelt, A.B.; Kharitonenkov, A.; Wasserman, D.H. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 2009, 150, 4084–4093. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.C.; Halstead, C.A.; Hansen, B.C.; Irizarry, A.R.; Martin, J.A.; Myers, S.R.; Reynolds, V.L.; Smith, H.W.; Wroblewski, V.J.; Kharitonenkov, A. LY2405319, an Engineered FGF21 Variant, Improves the Metabolic Status of Diabetic Monkeys. PLoS ONE 2013, 8, e65763. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Beals, J.M.; Micanovic, R.; Strifler, B.A.; Rathnachalam, R.; Wroblewski, V.J.; Li, S.; Koester, A.; Ford, A.M.; Coskun, T.; et al. Rational Design of a Fibroblast Growth Factor 21-Based Clinical Candidate, LY2405319. PLoS ONE 2013, 8, e58575. [Google Scholar] [CrossRef] [PubMed]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The Effects of LY2405319, an FGF21 Analog, in Obese Human Subjects with Type 2 Diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walisser, J.A.; Glover, E.; Pande, K.; Liss, A.L.; Bradfield, C.A. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. USA 2005, 102, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Kerley-Hamilton, J.S.; Trask, H.W.; Ridley, C.J.A.; DuFour, E.; Ringelberg, C.S.; Nurinova, N.; Wong, D.; Moodie, K.L.; Shipman, S.L.; Moore, J.H.; et al. Obesity Is Mediated by Differential Aryl Hydrocarbon Receptor Signaling in Mice Fed a Western Diet. Environ. Health Perspect. 2012, 120, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.-X.; Wang, C.; Zhang, Z.-M.; Jaeger, C.D.; Krager, S.L.; Bottum, K.M.; Liu, J.; Liao, D.-F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int. J. Obes. 2015, 39, 1300–1309. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.; Pineau, T.; Hilbert, D.M.; McPhail, T.; Lee, S.S.; Kimura, S.; Nebert, D.W.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 1995, 268, 722–726. [Google Scholar] [CrossRef]
- Baba, T.; Mimura, J.; Nakamura, N.; Harada, N.; Yamamoto, M.; Morohashi, K.-I.; Fujii-Kuriyama, Y. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol. Cell. Biol. 2005, 25, 10040–10051. [Google Scholar] [CrossRef]
- Conforto, T.L.; Waxman, D.J. Sex-specific mouse liver gene expression: Genome-wide analysis of developmental changes from pre-pubertal period to young adulthood. Biol. Sex Differ. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Risch, N.; Ghosh, S.; Todd, J.A. Statistical evaluation of multiple-locus linkage data in experimental species and its relevance to human studies: Application to nonobese diabetic (NOD) mouse and human insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 1993, 53, 702–714. [Google Scholar] [PubMed]
- Weiss, L.A.; Abney, M.; Cook, E.H.; Ober, C. Sex-Specific Genetic Architecture of Whole Blood Serotonin Levels. Am. J. Hum. Genet. 2005, 76, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, J.; Gustafsson, J.-Å. Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl. Recept. Signal. 2006, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macotela, Y.; Boucher, J.; Tran, T.T.; Kahn, C.R. Sex and Depot Differences in Adipocyte Insulin Sensitivity and Glucose Metabolism. Diabetes 2009, 58, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Hazlett, H.F.; Nemani, K.V.; Trask, H.W.; West, R.J.; Lupien, L.E.; Collins, A.J.; Ringelberg, C.S.; et al. Inhibition of the aryl hydrocarbon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicol. Appl. Pharmacol. 2016, 300, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.G.; Jornayvaz, F.R.; Petersen, M.C.; Pesta, D.; Guigni, B.A.; Serr, J.; Zhang, D.; Kahn, M.; Samuel, V.T.; Jurczak, M.J.; et al. Cellular Mechanisms by Which FGF21 Improves Insulin Sensitivity in Male Mice. Endocrinology 2013, 154, 3099–3109. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Yu, X.-C.; Liu, Z.; Hu, Y.; Sturgis, L.T.; Miranda, M.L.; Liu, Q. The Angiopoietin-like Proteins ANGPTL3 and ANGPTL4 Inhibit Lipoprotein Lipase Activity through Distinct Mechanisms. J. Biol. Chem. 2009, 284, 1419–1424. [Google Scholar] [CrossRef]
- Wang, Y.; McNutt, M.C.; Banfi, S.; Levin, M.G.; Holland, W.L.; Gusarova, V.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 11630–11635. [Google Scholar] [CrossRef]
- Mathews, S.T.; Singh, G.P.; Ranalletta, M.; Cintron, V.J.; Qiang, X.; Goustin, A.S.; Jen, K.-L.C.; Charron, M.J.; Jahnen-Dechent, W.; Grunberger, G. Improved Insulin Sensitivity and Resistance to Weight Gain in Mice Null for the Ahsg Gene. Diabetes 2002, 51, 2450–2458. [Google Scholar] [CrossRef] [Green Version]
- Rajwani, A.; Ezzat, V.; Smith, J.; Yuldasheva, N.Y.; Duncan, E.R.; Gage, M.; Cubbon, R.M.; Kahn, M.B.; Imrie, H.; Abbas, A.; et al. Increasing Circulating IGFBP1 Levels Improves Insulin Sensitivity, Promotes Nitric Oxide Production, Lowers Blood Pressure, and Protects Against Atherosclerosis. Diabetes 2012, 61, 915–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wei, W.; Krzeszinski, J.Y.; Wang, Y.; Wan, Y. A Liver-Bone Endocrine Relay by IGFBP1 Promotes Osteoclastogenesis and Mediates FGF21-Induced Bone Resorption. Cell Metab. 2015, 22, 811–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, S.; Li, Z.; Lemieux, I.; Alméras, N.; Tremblay, A.; Bergeron, J.; Poirier, P.; Deshaies, Y.; Després, J.-P.; Picard, F. Circulating IGFBP-2 levels are incrementally linked to correlates of the metabolic syndrome and independently associated with VLDL triglycerides. Atherosclerosis 2014, 237, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Porter, C. Quantification of UCP1 function in human brown adipose tissue. Adipocyte 2017, 6, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Saraf, M.K.; Herndon, D.N.; Porter, C.; Toliver-Kinsky, T.; Radhakrishnan, R.; Chao, T.; Chondronikola, M.; Sidossis, L.S. Morphological changes in subcutaneous white adipose tissue after severe burn injury. J. Burn Care Res. Off. Publ. Am. Burn Assoc. 2016, 37, e96–e103. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.D.L.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef]
- Guerra, C.; Navarro, P.; Valverde, A.M.; Arribas, M.; Brüning, J.; Kozak, L.P.; Kahn, C.R.; Benito, M. Brown adipose tissue–specific insulin receptor knockout shows diabetic phenotype without insulin resistance. J. Clin. Investig. 2001, 108, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.E.; Rabelo, R. Regulation of the uncoupling protein gene expression. Eur. J. Endocrinol. 1997, 136, 251–264. [Google Scholar] [CrossRef]
- Keipert, S.; Kutschke, M.; Ost, M.; Schwarzmayr, T.; van Schothorst, E.M.; Lamp, D.; Brachthäuser, L.; Hamp, I.; Mazibuko, S.E.; Hartwig, S.; et al. Long-Term Cold Adaptation Does Not Require FGF21 or UCP1. Cell Metab. 2017, 26, 437–446.e5. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.-M.; Sanyal, S.; Angelin, B.; Alexson, S.E.H.; Rudling, M. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009, 583, 2882–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wong, K.; Walsh, K.; Gao, B.; Zang, M. Retinoic Acid Receptor β Stimulates Hepatic Induction of Fibroblast Growth Factor 21 to Promote Fatty Acid Oxidation and Control Whole-body Energy Homeostasis in Mice. J. Biol. Chem. 2013, 288, 10490–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bookout, A.L.; de Groot, M.H.M.; Owen, B.M.; Lee, S.; Gautron, L.; Lawrence, H.L.; Ding, X.; Elmquist, J.K.; Takahashi, J.S.; Mangelsdorf, D.J.; et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat. Med. 2013, 19, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine-Fridman, A.; Chen, L.; Elferink, C.J. Cytochrome P4501A1 Promotes G1 Phase Cell Cycle Progression by Controlling Aryl Hydrocarbon Receptor Activity. Mol. Pharmacol. 2004, 65, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T. Research Perspectives on the Regulation and Physiological Functions of FGF21 and its Association with NAFLD. Front. Endocrinol. 2015, 6, 147. [Google Scholar] [CrossRef]
- Feil, R.; Brocard, J.; Mascrez, B.; LeMeur, M.; Metzger, D.; Chambon, P. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10887–10890. [Google Scholar] [CrossRef]
- Harper, T.A.; Joshi, A.D.; Elferink, C.J. Identification of Stanniocalcin 2 as a Novel Aryl Hydrocarbon Receptor Target Gene. J. Pharmacol. Exp. Ther. 2013, 344, 579–588. [Google Scholar] [CrossRef]
- Mustafa, M.G.; Petersen, J.R.; Ju, H.; Cicalese, L.; Snyder, N.; Haidacher, S.J.; Denner, L.; Elferink, C. Biomarker discovery for early detection of hepatocellular carcinoma in hepatitis C-infected patients. Mol. Cell. Proteomics 2013, 12, 3640–3652. [Google Scholar] [CrossRef]
- Mustafa, G.M.; Larry, D.; Petersen, J.R.; Elferink, C.J. Targeted proteomics for biomarker discovery and validation of hepatocellular carcinoma in hepatitis C infected patients. World J. Hepatol. 2015, 7, 1312–1324. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Ahrfl/fl | iCKO | Fold Change | p Value | q Value |
|---|---|---|---|---|---|
| Angptl3 | 417.2 | 98.30 | −4.243 | 0.00005 | 0.0004340 |
| Ahsg | 6757 | 4283 | −1.578 | 0.00505 | 0.0241456 |
| Fgf21 | 0.788 | 1.520 | 1.929 | 0.00265 | 0.0140155 |
| Igfbp1 | 59.58 | 219.7 | 3.687 | 0.00005 | 0.0004340 |
| Igfbp2 | 258.2 | 514.8 | 1.994 | 0.00005 | 0.0004340 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girer, N.G.; Carter, D.; Bhattarai, N.; Mustafa, M.; Denner, L.; Porter, C.; Elferink, C.J. Inducible Loss of the Aryl Hydrocarbon Receptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21. Int. J. Mol. Sci. 2019, 20, 950. https://doi.org/10.3390/ijms20040950
Girer NG, Carter D, Bhattarai N, Mustafa M, Denner L, Porter C, Elferink CJ. Inducible Loss of the Aryl Hydrocarbon Receptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21. International Journal of Molecular Sciences. 2019; 20(4):950. https://doi.org/10.3390/ijms20040950
Chicago/Turabian StyleGirer, Nathaniel G., Dwayne Carter, Nisha Bhattarai, Mehnaz Mustafa, Larry Denner, Craig Porter, and Cornelis J. Elferink. 2019. "Inducible Loss of the Aryl Hydrocarbon Receptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21" International Journal of Molecular Sciences 20, no. 4: 950. https://doi.org/10.3390/ijms20040950