Novel JAG1 Deletion Variant in Patient with Atypical Alagille Syndrome


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
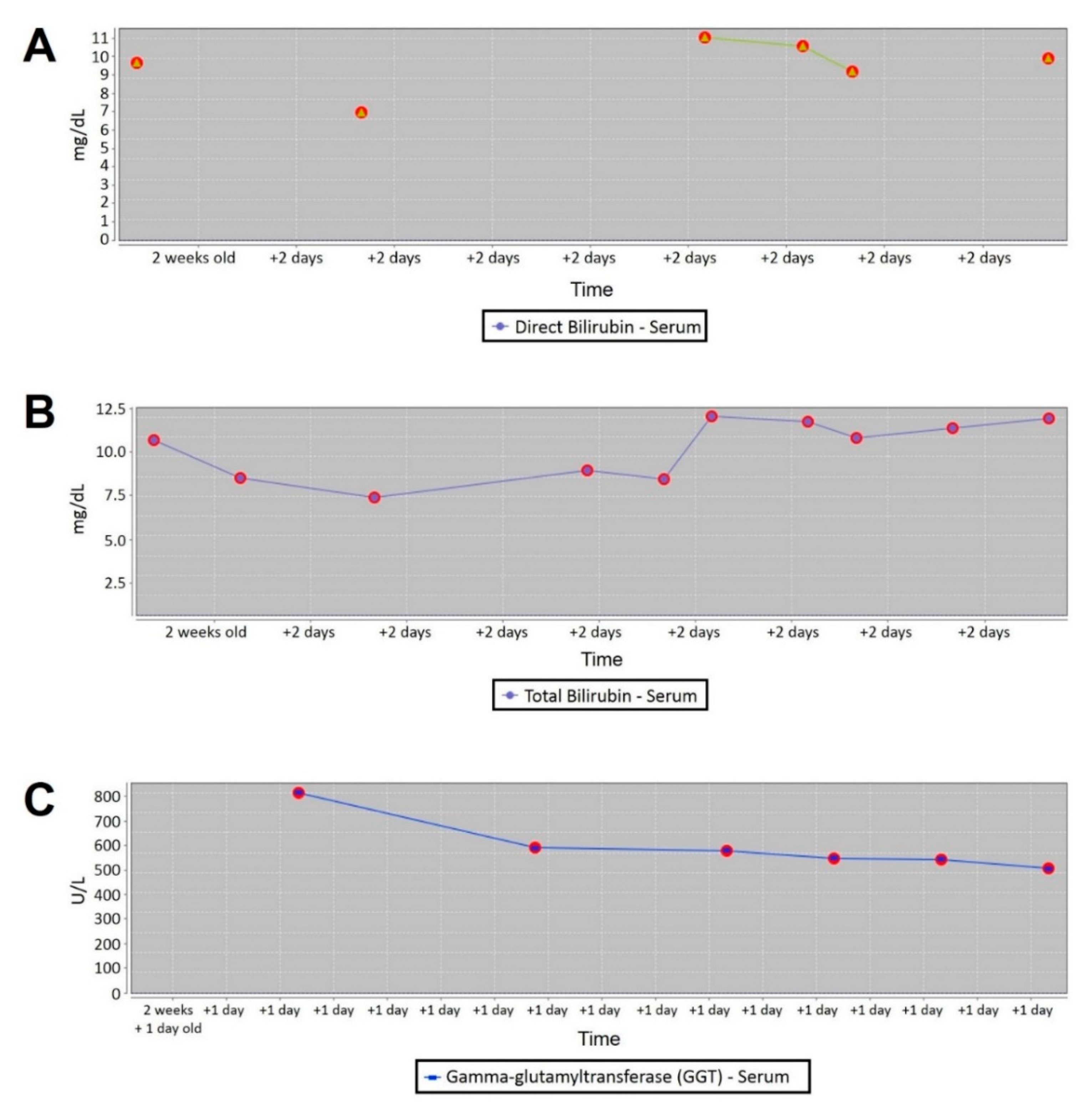
2. Case Presentation
3. Discussion
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, J.; Yang, B.; Paik, N.; Choe, Y.H.; Paik, Y.H. A case of Alagille syndrome presenting with chronic cholestasis in an adult. Clin. Mol. Hepatol. 2017, 23, 260–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurkiewicz, D.; Gliwicz, D.; Ciara, E.; Gerfen, J.; Pelc, M.; Piekutowska-Abramczuk, D.; Kugaudo, M.; Chrzanowska, K.; Spinner, N.B.; Krajewska-Walasek, M. Spectrum of JAG1 gene mutations in Polish patients with Alagille syndrome. J. Appl. Genet. 2014, 55, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.; Kamath, B.M.; Chitayat, D. Alagille syndrome: Clinical perspectives. Appl. Clin. Genet. 2016, 9, 75–82. [Google Scholar] [PubMed] [Green Version]
- Emerick, K.M.; Rand, E.B.; Goldmuntz, E.; Krantz, I.D.; Spinner, N.B.; Piccoli, D.A. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology 1999, 29, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Alagille, D.; Borde, J.; Habib, E.C.; Thomassin, N. Surgical attempts in atresia of the intrahepatic bile ducts with permeable extrahepatic bile duct. Study of 14 cases in children. Arch. Fr. Pediatr. 1969, 26, 51–71. [Google Scholar] [PubMed]
- Di Pinto, D.; Adragna, M. Renal manifestations in children with Alagille syndrome. Arch. Argent. Pediatr. 2018, 116, 149–153. [Google Scholar]
- Turnpenny, P.D.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.C.; Le Hoang, P.; Hutchinson, A.; Chao, G.; Gerfen, J.; Loomes, K.M.; Krantz, I.; Kamath, B.M.; Spinner, N.B. Alagille syndrome in a Vietnamese cohort: Mutation analysis and assessment of facial features. Am. J. Med. Genet. A 2012, 158, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Kamath, B.M.; Bason, L.; Piccoli, D.A.; Krantz, I.D.; Spinner, N.B. Consequences of JAG1 mutations. J. Med. Genet. 2003, 40, 891–895. [Google Scholar] [CrossRef] [Green Version]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Online Mendelian Inheritance in Man. Available online: https://www.omim.org/ (accessed on 21 November 2019).
- Jones, E.A.; Clement-Jones, M.; Wilson, D.I. JAGGED1 expression in human embryos: Correlation with the Alagille syndrome phenotype. J. Med. Genet. 2000, 37, 658–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Main, H.; Radenkovic, J.; Jin, S.B.; Lendahl, U.; Andersson, E.R. Notch signaling maintains neural rosette polarity. PLoS ONE 2013, 8, e62959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grochowski, C.M.; Loomes, K.M.; Spinner, N.B. Jagged1 (JAG1): Structure, expression, and disease associations. Gene 2016, 576, 381–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.Y.; Treece, A.L.; Russo, P.A.; Wen, J.W. An Atypical Presentation of Alagille Syndrome. Pediatr. Dev. Pathol. 2018, 21, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Penton, A.L.; Leonard, L.D.; Spinner, N.B. Notch signaling in human development and disease. Semin. Cell Dev. Biol. 2012, 23, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Orssaud, C.; Robert, M.P.; Roche, O. Relevance of Identifying JAG1 Mutations in Patients with Isolated Posterior Embryotoxon. J. Glaucoma 2016, 25, 923–925. [Google Scholar] [CrossRef]
- Krantz, I.D.; Colliton, R.P.; Genin, A.; Rand, E.B.; Li, L.; Piccoli, D.A.; Spinner, N.B. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am. J. Hum. Genet. 1998, 62, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Grochowski, C.M.; Rajagopalan, R.; Falsey, A.M.; Loomes, K.M.; Piccoli, D.A.; Krantz, I.D.; Devoto, M.; Spinner, N.B. Exome sequencing reveals compound heterozygous mutations in ATP8B1 in a JAG1/NOTCH2 mutation-negative patient with clinically diagnosed Alagille syndrome. Am. J. Med. Genet. A 2015, 167, 891–893. [Google Scholar] [CrossRef]
- Masek, J.; Andersson, E.R. The developmental biology of genetic Notch disorders. Development 2017, 144, 1743–1763. [Google Scholar] [CrossRef] [Green Version]
- Luca, V.C.; Kim, B.C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger, R.S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [CrossRef] [Green Version]
- Canalis, E.; Zanotti, S. Hajdu-Cheney Syndrome, a Disease Associated with NOTCH2 Mutations. Curr. Osteoporos. Rep. 2016, 14, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, Y.; Kadokawa, Y.; Okabe, M.; Ikawa, M.; Coleman, J.R.; Tsujimoto, Y. Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development 1999, 126, 3415–3424. [Google Scholar] [PubMed]
- Youngstrom, D.W.; Dishowitz, M.I.; Bales, C.B.; Carr, E.; Mutyaba, P.L.; Kozloff, K.M.; Shitaye, H.; Hankenson, K.D.; Loomes, K.M. Jagged1 expression by osteoblast-lineage cells regulates trabecular bone mass and periosteal expansion in mice. Bone 2016, 91, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kola, S.; Koneti, N.R.; Golla, J.P.; Akka, J.; Gundimeda, S.D.; Mundluru, H.P. Mutational analysis of JAG1 gene in non-syndromic tetralogy of Fallot children. Clin. Chim. Acta 2011, 412, 2232–2236. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mead, T.J.; Yutzey, K.E. Notch pathway regulation of neural crest cell development in vivo. Dev. Dyn. 2012, 241, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.M.; Bird, L.; Barnes, P.; Barth, R.; Hudgins, L. Lateral meningocele syndrome: Vertical transmission and expansion of the phenotype. Am. J. Med. Genet. A 2005, 133, 115–121. [Google Scholar] [CrossRef]
- Gordon, W.R.; Arnett, K.L.; Blacklow, S.C. The molecular logic of Notch signaling—A structural and biochemical perspective. J. Cell Sci. 2008, 121, 3109–3119. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, N.; Martinez-Trillos, A.; Montraveta, A.; Lopez-Guerra, M.; Rosich, L.; Nadeu, F.; Valero, J.G.; Aymerich, M.; Magnano, L.; Rozman, M.; et al. Mutations in RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica 2019, 104, 576–586. [Google Scholar] [CrossRef]
- Yuan, B.; Neira, J.; Pehlivan, D.; Santiago-Sim, T.; Song, X.; Rosenfeld, J.; Posey, J.E.; Patel, V.; Jin, W.; Adam, M.P.; et al. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet. Med. 2019, 21, 663. [Google Scholar] [CrossRef]
- Pachow, D.; Wick, W.; Gutmann, D.H.; Mawrin, C. The mTOR signaling pathway as a treatment target for intracranial neoplasms. Neuro Oncol. 2015, 17, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puvabanditsin, S.; Gueye-Ndiaye, S.; Puthenpura, V.; Gengel, N.; Tam, V.; Mehta, R. Microduplication of 17p[Dup(17)(12p11.2)]: Report of a Neonate With a Spina Bifida and Cardiac Anomalies and a Literature Review. Genet. Couns 2016, 27, 503–507. [Google Scholar] [PubMed]
- Kudinov, A.E.; Deneka, A.; Nikonova, A.S.; Beck, T.N.; Ahn, Y.H.; Liu, X.; Martinez, C.F.; Schultz, F.A.; Reynolds, S.; Yang, D.H.; et al. Musashi-2 (MSI2) supports TGF-beta signaling and inhibits claudins to promote non-small cell lung cancer (NSCLC) metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 6955–6960. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micaglio, E.; Andronache, A.A.; Carrera, P.; Monasky, M.M.; Locati, E.T.; Pirola, B.; Presi, S.; Carminati, M.; Ferrari, M.; Giamberti, A.; et al. Novel JAG1 Deletion Variant in Patient with Atypical Alagille Syndrome. Int. J. Mol. Sci. 2019, 20, 6247. https://doi.org/10.3390/ijms20246247
Micaglio E, Andronache AA, Carrera P, Monasky MM, Locati ET, Pirola B, Presi S, Carminati M, Ferrari M, Giamberti A, et al. Novel JAG1 Deletion Variant in Patient with Atypical Alagille Syndrome. International Journal of Molecular Sciences. 2019; 20(24):6247. https://doi.org/10.3390/ijms20246247
Chicago/Turabian StyleMicaglio, Emanuele, Andreea Alina Andronache, Paola Carrera, Michelle M. Monasky, Emanuela T. Locati, Barbara Pirola, Silvia Presi, Mario Carminati, Maurizio Ferrari, Alessandro Giamberti, and et al. 2019. "Novel JAG1 Deletion Variant in Patient with Atypical Alagille Syndrome" International Journal of Molecular Sciences 20, no. 24: 6247. https://doi.org/10.3390/ijms20246247