TASK-3 Gene Knockdown Dampens Invasion and Migration and Promotes Apoptosis in KATO III and MKN-45 Human Gastric Adenocarcinoma Cell Lines

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
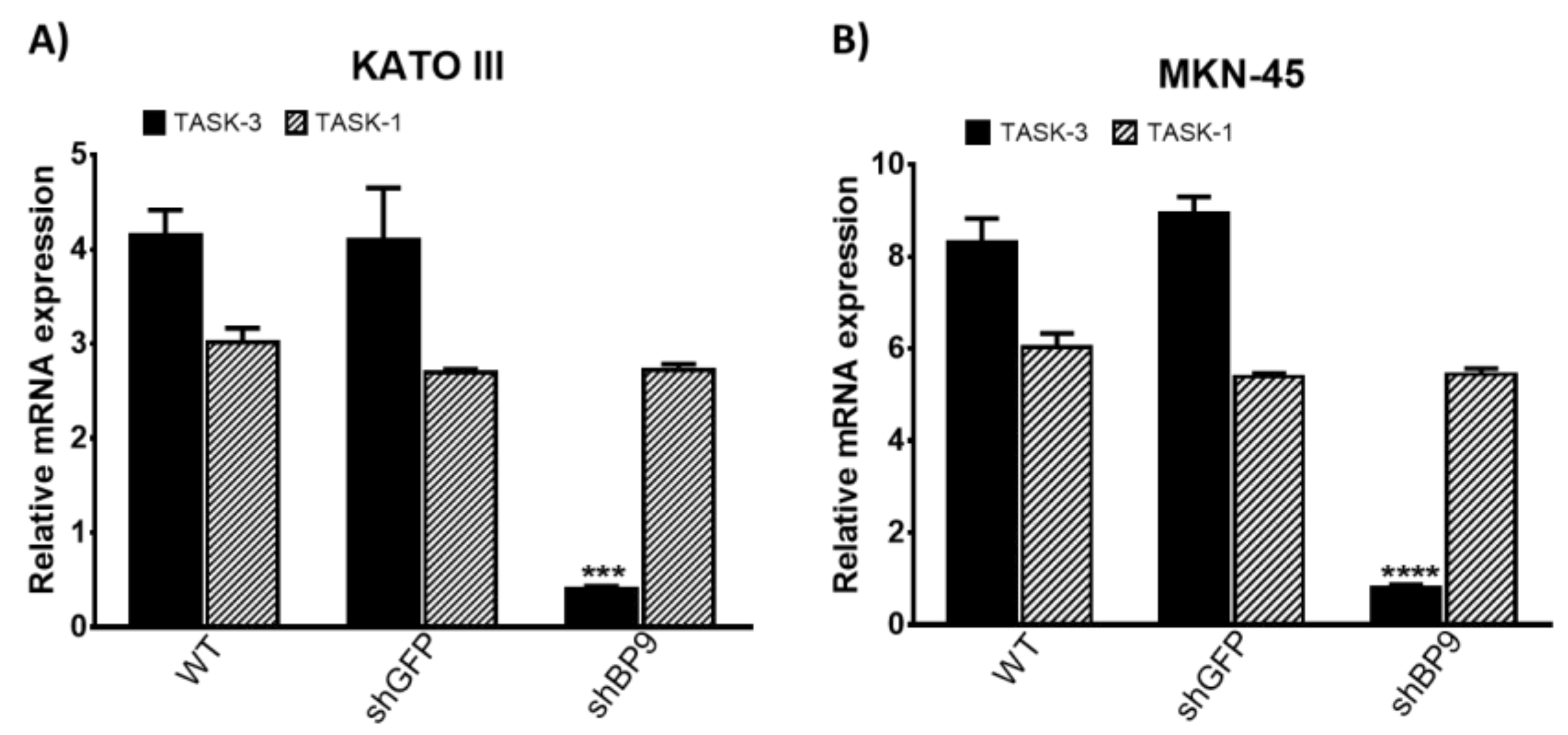
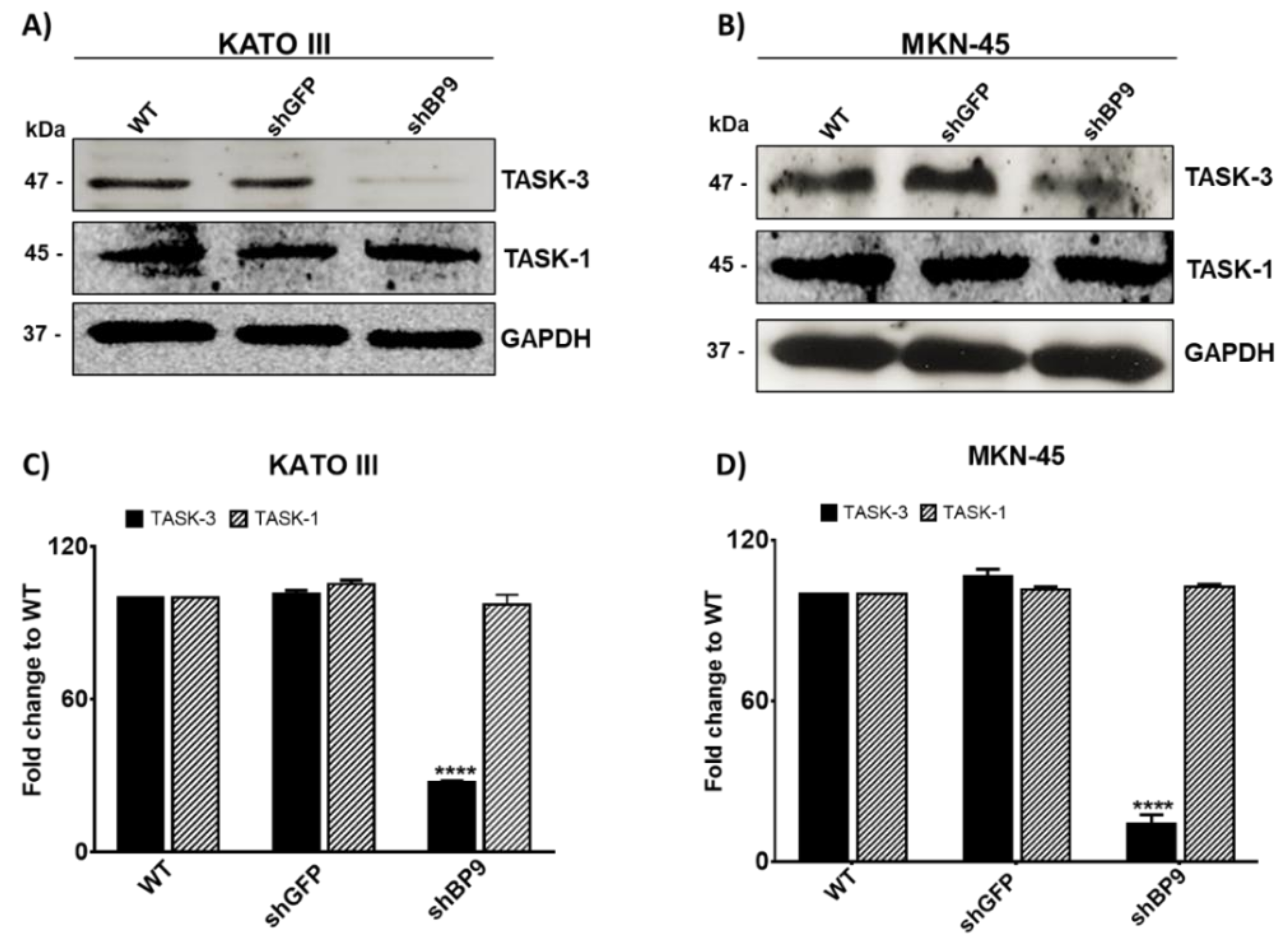
2.1. Expression and Knockdown of TASK-3 in KATO III and MKM-45 Cell Lines
2.2. TASK-3 Knockdown Inhibits Cell Proliferation and Viability in KATO III and MKN-45 Cells
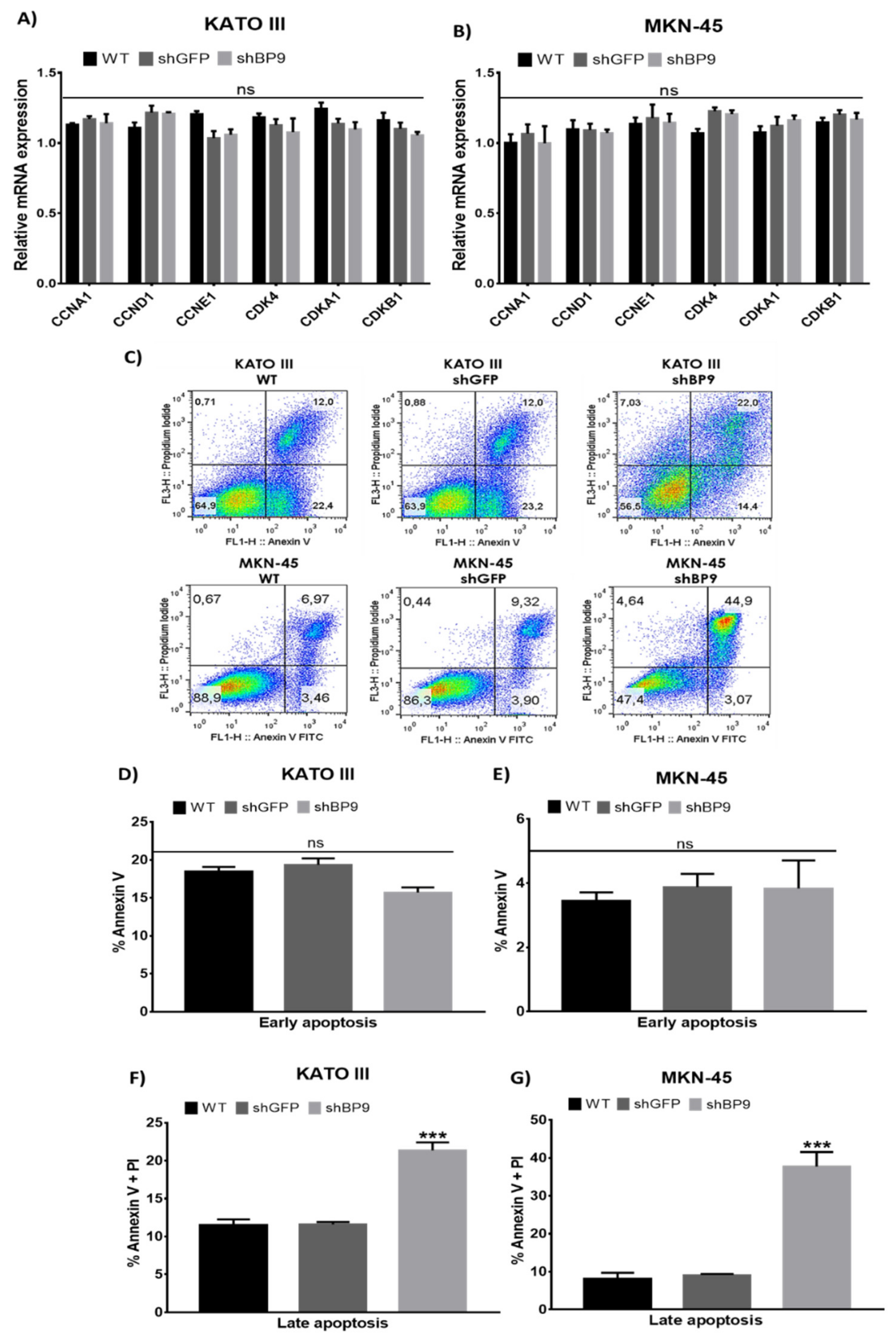
2.3. TASK-3 Knockdown in KATO III and MKN-45 Cells Triggers an Increase in Apoptosis without Altering the Cell Cycle
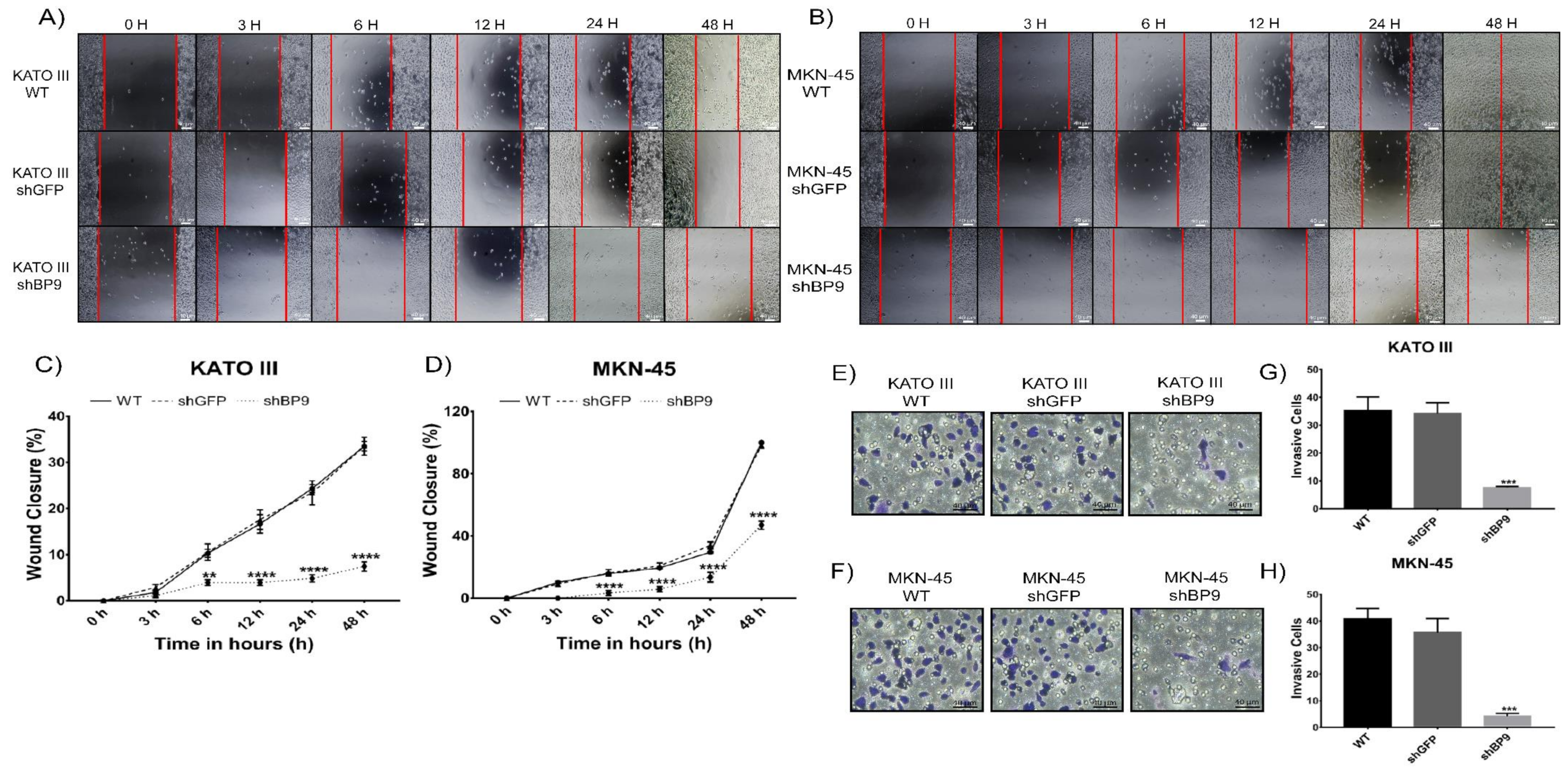
2.4. Inhibition of Cell Migration and Invasion Following TASK-3 Depletion
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Isolation and Quantitative RT-qPCR
4.3. Protein Extraction and Western Blotting
4.4. Generation of Retroviral Particles
4.5. Retroviral Infection
4.6. Proliferation and Cell Viability Assays
4.7. Apoptosis Assay
4.8. Wound Healing Assays
4.9. Cell Invasion Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taheri-Araghi, S.; Brown, S.D.; Sauls, J.T.; McIntosh, D.B.; Jun, S. Single-Cell Physiology. Annu. Rev. Biophys. 2015, 44, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.P.; Youle, R.J. Balancing cell growth and death. Curr. Opin. Cell Biol. 2012, 24, 802–803. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiol. Bethesda 2010, 25, 85–101. [Google Scholar] [CrossRef]
- Junttila, M.R.; Evan, G.I. p53—A Jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Luebeck, E.G. Cancer: Genomic evolution of metastasis. Nature 2010, 467, 1053–1055. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Cancer Today. Available online: http://gco.iarc.fr/today/online-analysismultibars?v=2018&mode=cancer&mode_population=countries&population=900&populations=900&key=asr&sex=0&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=10&group_cancer=1&include_nmsc=1&include_nmsc_other=1&type_multiple=%257B%2522inc%2522%253Atrue%252C%2522mort%2522%253Afalse%252C%2522prev%2522%253Afalse%257D&orientation=horizontal&type_sort=0&type_nb_items=%257B%2522top%2522%253Atrue%252C%2522bottom%2522%253Afalse%257D&population_group_globocan_id=. (accessed on 5 June 2019).
- Padmanabhan, N.; Ushijima, T.; Tan, P. How to stomach an epigenetic insult: The gastric cancer epigenome. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.C.; Sohn, B.H.; Cheong, J.H.; Kim, S.B.; Lee, J.E.; Park, K.C.; Lee, S.H.; Park, J.L.; Park, Y.Y.; Lee, H.S.; et al. Clinical and genomic landscape of gastric cancer with a mesenchymal phenotype. Nat. Commun. 2018, 9, 1777. [Google Scholar] [CrossRef] [PubMed]
- Ilson, D.H. Advances in the treatment of gastric cancer. Curr. Opin. Gastroenterol. 2017, 33, 473–476. [Google Scholar] [CrossRef]
- Persson, P.B.; Bondke Persson, A. Channels and channelopathies. Acta Physiol. 2016, 218, 149–151. [Google Scholar] [CrossRef]
- Albury, C.L.; Stuart, S.; Haupt, L.M.; Griffiths, L.R. Ion channelopathies and migraine pathogenesis. Mol. Genet. Genomics 2017, 292, 729–739. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J.; Wilde, A.A.M. Channelopathies as Causes of Sudden Cardiac Death. Card. Electrophysiol. Clin. 2017, 9, 537–549. [Google Scholar] [CrossRef]
- Beyder, A.; Farrugia, G. Ion channelopathies in functional GI disorders. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G581–G586. [Google Scholar] [CrossRef]
- Chakroborty, S.; Stutzmann, G.E. Calcium channelopathies and Alzheimer’s disease: Insight into therapeutic success and failures. Eur. J. Pharmacol. 2014, 739, 83–95. [Google Scholar] [CrossRef]
- Peruzzo, R.; Biasutto, L.; Szabò, I.; Leanza, L. Impact of intracellular ion channels on cancer development and progression. Eur. Biophys. J. 2016, 45, 685–707. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Renaudo, A.; L’Hoste, S.; Guizouarn, H.; Borgèse, F.; Soriani, O. Cancer cell cycle modulated by a functional coupling between sigma-1 receptors and Cl− channels. J. Biol. Chem. 2007, 282, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion channels in the regulation of apoptosis. Biochim. Biophys. Acta 2015, 1848, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.; Yang, M.; Dowle, A.A.; Thomas, J.R.; Brackenbury, W.J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer 2015, 14, 13. [Google Scholar] [CrossRef]
- Enyedi, P.; Czirjak, G. Molecular background of leak K+ currents: Two-pore domain potassium channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar] [CrossRef]
- Burgos, P.; Zúñiga, R.; Domínguez, P.; Delgado-López, F.; Plant, L.D.; Zúñiga, L. Differential expression of two-pore domain potassium channels in rat cerebellar granule neurons. Biochem. Biophys. Res. Commun. 2014, 453, 754–760. [Google Scholar] [CrossRef]
- Innamaa, A.; Jackson, L.; Asher, V.; van Shalkwyk, G.; Warren, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and prognostic significance of the oncogenic K2P potassium channel KCNK9 (TASK-3) in ovarian carcinoma. Anticancer Res. 2013, 33, 1401–1408. [Google Scholar]
- Mu, D.; Chen, L.; Zhang, X.; See, L.H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.Q.; Servoss, A.; et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 2003, 3, 297–302. [Google Scholar] [CrossRef]
- Kim, C.J.; Cho, Y.G.; Jeong, S.W.; Kim, Y.S.; Kim, S.Y.; Nam, S.W.; Lee, S.H.; Yoo, N.J.; Lee, Y.J.; Park, W.S. Altered expression of KCNK9 in colorectal cancers. Apmis 2004, 112, 588–594. [Google Scholar] [CrossRef]
- Pocsai, K.; Kosztka, L.; Bakondi, G.; Gönczi, M.; Fodor, J.; Dienes, B.; Szentesi, P.; Kovács, I.; Feniger-Barish, R.; Kopf, E.; et al. Melanoma cells exhibit strong intracellular TASK-3-specific immunopositivity in both tissue sections and cell culture. Cell Mol. Life Sci. 2006, 63, 2364–2376. [Google Scholar] [CrossRef]
- Ko, J.H.; Gu, W.; Lim, I.; Bang, H.; Ko, E.A.; Zhou, T. Ion Channel Gene Expression in Lung Adenocarcinoma: Potential Role in Prognosis and Diagnosis. PLoS ONE 2014, 9, e86569. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Lazdunski, M. The 2P-domain K+ channels: Role in apoptosis and tumorigenesis. Pflugers Arch. 2004, 448, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Rouzaire-Dubois, B.; Dubois, J.M. K+ channel block-induced mammalian neuroblastoma cell swelling: A possible mechanism to influence proliferation. J. Physiol. 1998, 510, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Rouzaire-Dubois, B.; Milandri, J.B.; Bostel, S.; Dubois, J.M. Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers Arch. 2000, 440, 881–888. [Google Scholar] [PubMed]
- Zúñiga, R.; Valenzuela, C.; Concha, G.; Brown, N.; Zúñiga, L. TASK-3 Downregulation Triggers Cellular Senescence and Growth Inhibition in Breast Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, L.; Wiser, O.; Slavin, A.; Mu, D.; Powers, S.; Jan, L.Y.; Hoey, T. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc. Natl. Acad. Sci. USA 2003, 100, 7803–7807. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Luo, L.; Lal, B.; Ma, X.; Chen, L.; Hann, C.L.; Fulton, A.M.; Leahy, D.J.; Laterra, J.; Li, M. A monoclonal antibody against KCNK9 K+ channel extracellular domain inhibits tumour growth and metastasis. Nat. Commun. 2016, 7, 10339. [Google Scholar] [CrossRef] [Green Version]
- Walworth, N.C. Cell-cycle checkpoint kinases: Checking in on the cell cycle. Curr. Opin. Cell Biol. 2000, 12, 697–704. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, C.; Liu, D.L.; Kantheti, H.S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Bagal, S.K.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion channels as therapeutic targets: A drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [Google Scholar] [CrossRef]
- Huang, X.; Jan, L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, I.; Pocsai, K.; Czifra, G.; Sarkadi, L.; Szucs, G.; Nemes, Z.; Rusznák, Z. TASK-3 immunoreactivity shows differential distribution in the human gastrointestinal tract. Virchows Arch. 2005, 446, 402–410. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Expression of KCNK9 in Cancer. Available online: https://www.proteinatlas.org/ENSG00000169427-KCNK9/pathology (accessed on 1 October 2019).
- Nagy, D.; Gönczi, M.; Dienes, B.; Szöőr, Á.; Fodor, J.; Nagy, Z.; Tóth, A.; Fodor, T.; Bai, P.; Szücs, G.; et al. Silencing the KCNK9 potassium channel (TASK-3) gene disturbs mitochondrial function, causes mitochondrial depolarization, and induces apoptosis of human melanoma cells. Arch. Dermatol. Res. 2014, 306, 885–902. [Google Scholar] [CrossRef] [PubMed]
- Gierten, J.; Ficker, E.; Bloehs, R.; Schlömer, K.; Kathöfer, S.; Scholz, E.; Zitron, E.; Kiesecker, C.; Bauer, A.; Becker, R.; et al. Regulation of two-pore-domain (K2P) potassium leak channels by the tyrosine kinase inhibitor genistein. Br. J. Pharmacol. 2008, 154, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Bista, P.; Pawlowski, M.; Cerina, M.; Ehling, P.; Leist, M.; Meuth, P.; Aissaoui, A.; Borsotto, M.; Heurteaux, C.; Decher, N.; et al. Differential phospholipase C-dependent modulation of TASK and TREK two-pore domain K+ channels in rat thalamocortical relay neurons. J. Physiol. 2015, 593, 127–144. [Google Scholar] [CrossRef] [Green Version]
- Leithner, K.; Hirschmugl, B.; Li, Y.; Tang, B.; Papp, R.; Nagaraj, C.; Stacher, E.; Stiegler, P.; Lindenmann, J.; Olschewski, A.; et al. TASK-1 Regulates Apoptosis and Proliferation in a Subset of Non-Small Cell Lung Cancers. PLoS ONE 2016, 11, e0157453. [Google Scholar] [CrossRef]
- Rusznák, Z.; Bakondi, G.; Kosztka, L.; Pocsai, K.; Dienes, B.; Fodor, J.; Telek, A.; Gönczi, M.; Szűcs, G.; Csernoch, L. Mitochondrial expression of the two-pore domain TASK-3 channels in malignantly transformed and non-malignant human cells. Virchows Arch. 2008, 452, 415–426. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, e1002459. [Google Scholar] [CrossRef] [Green Version]
- Trepat, X.; Chen, Z.; Jacobson, K. Cell migration. Compr. Physiol. 2012, 2, 2369–2392. [Google Scholar]
- Schwab, A.; Stock, C. Ion channels and transporters in tumour cell migration and invasion. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130102. [Google Scholar] [CrossRef] [Green Version]
- Luby-Phelps, K. Cytoarchitecture and physical properties of cytoplasm: Volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol. 2000, 192, 189–221. [Google Scholar] [PubMed]
- Lee, G.W.; Park, H.S.; Kim, E.J.; Cho, Y.W.; Kim, G.T.; Mun, Y.J.; Choi, E.J.; Lee, J.S.; Han, J.; Kang, D. Reduction of breast cancer cell migration via up-regulation of TASK-3 two-pore domain K+ channel. Acta Physiol. 2012, 204, 513–524. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cikutović-Molina, R.; Herrada, A.A.; González, W.; Brown, N.; Zúñiga, L. TASK-3 Gene Knockdown Dampens Invasion and Migration and Promotes Apoptosis in KATO III and MKN-45 Human Gastric Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 6077. https://doi.org/10.3390/ijms20236077
Cikutović-Molina R, Herrada AA, González W, Brown N, Zúñiga L. TASK-3 Gene Knockdown Dampens Invasion and Migration and Promotes Apoptosis in KATO III and MKN-45 Human Gastric Adenocarcinoma Cell Lines. International Journal of Molecular Sciences. 2019; 20(23):6077. https://doi.org/10.3390/ijms20236077
Chicago/Turabian StyleCikutović-Molina, Rocio, Andres A. Herrada, Wendy González, Nelson Brown, and Leandro Zúñiga. 2019. "TASK-3 Gene Knockdown Dampens Invasion and Migration and Promotes Apoptosis in KATO III and MKN-45 Human Gastric Adenocarcinoma Cell Lines" International Journal of Molecular Sciences 20, no. 23: 6077. https://doi.org/10.3390/ijms20236077