Virtual Screening Using Pharmacophore Models Retrieved from Molecular Dynamic Simulations

 , and
, and
Abstract
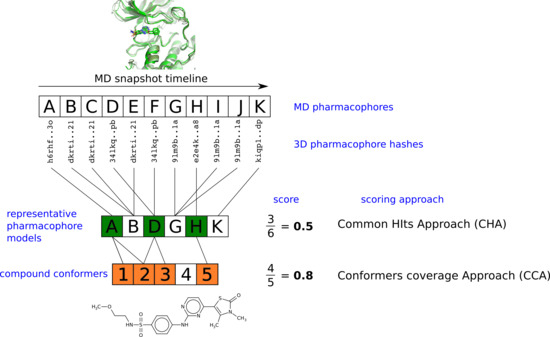
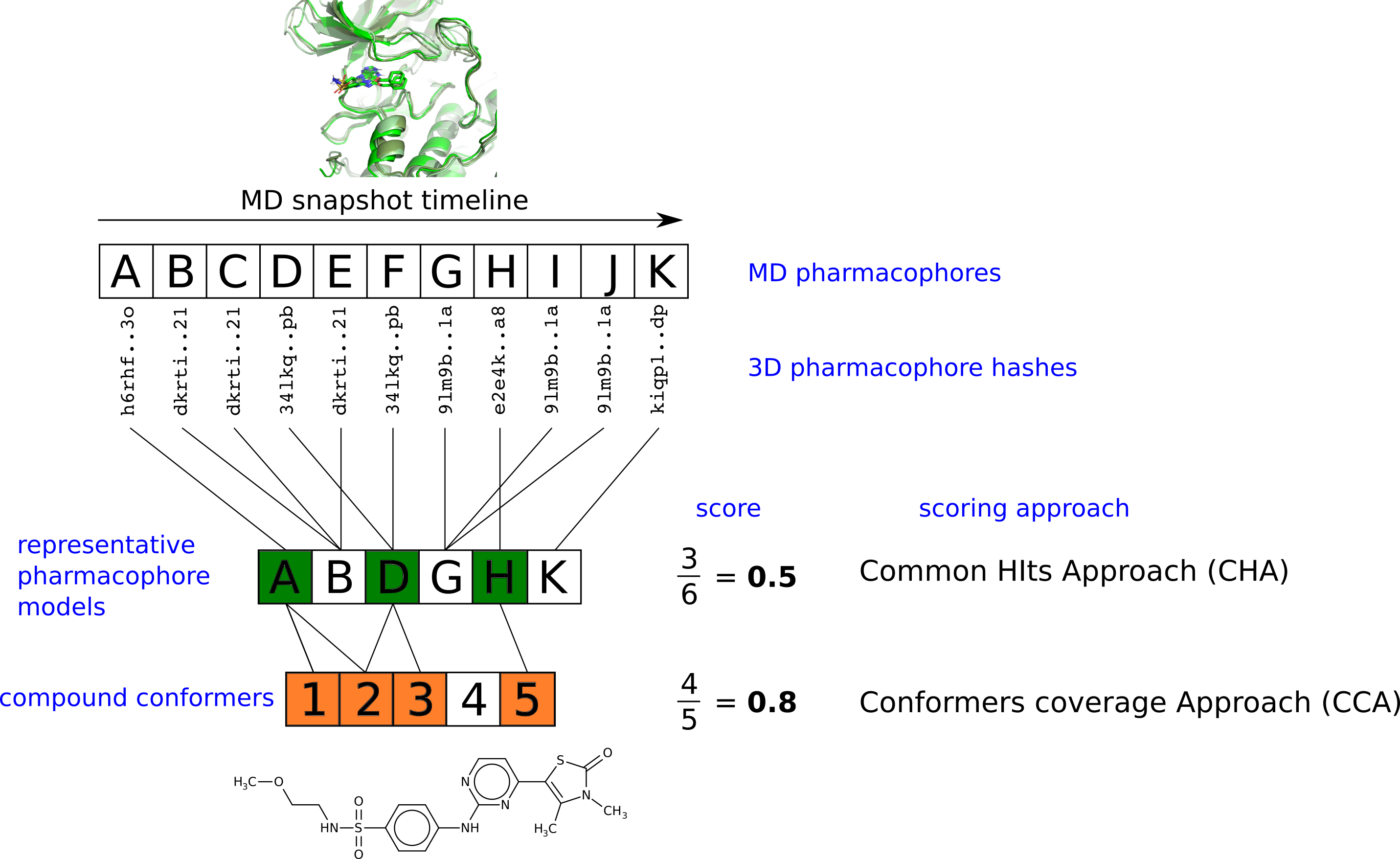
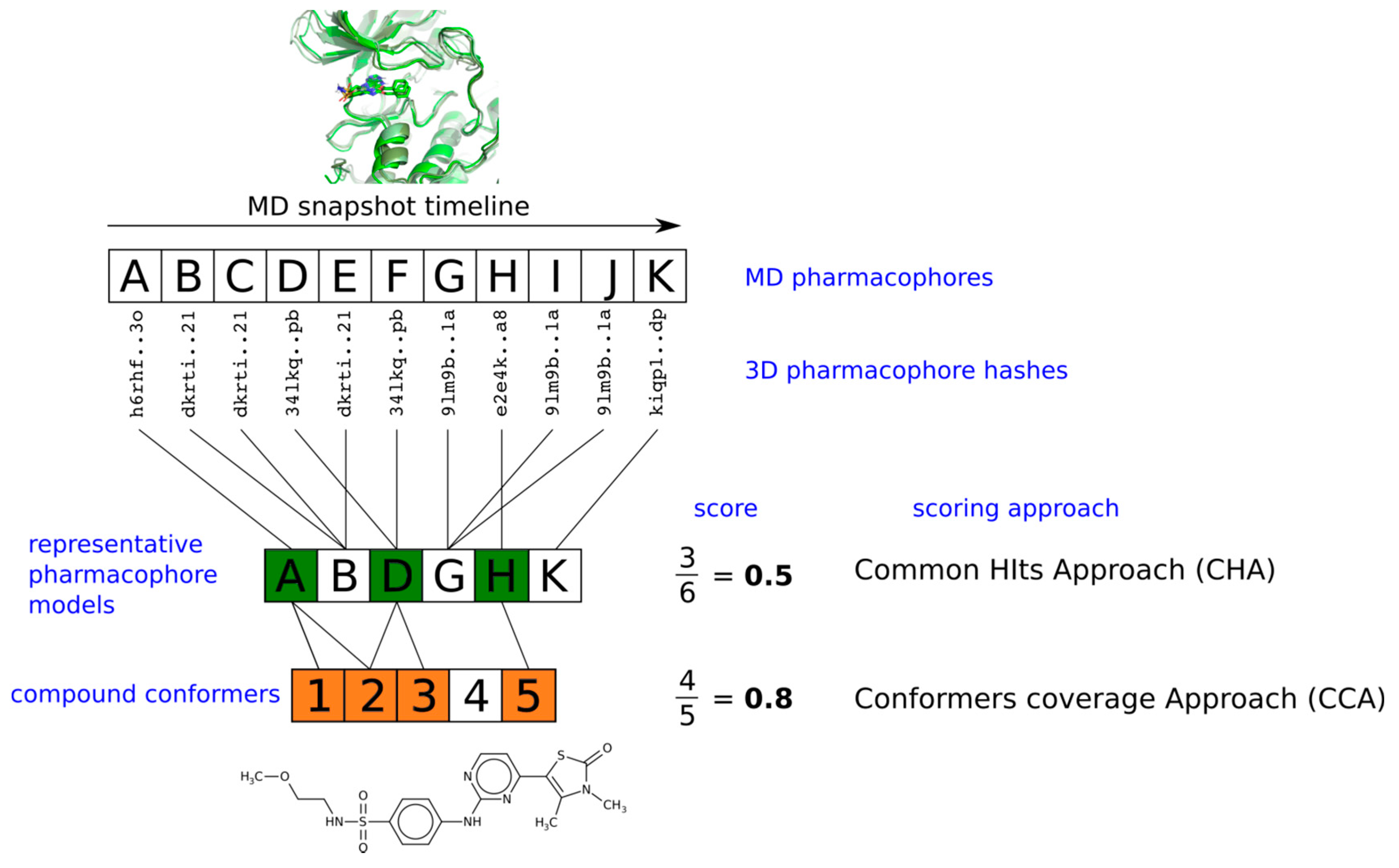
:
1. Introduction
2. Materials and Methods
2.1. Protein Target and Compound Dataset
2.2. Molecular Dynamic Simulations
2.3. Pharmacophore Model Retrieval
2.4. Virtual Screening With Ensembles of MD-Based Pharmacophore Models
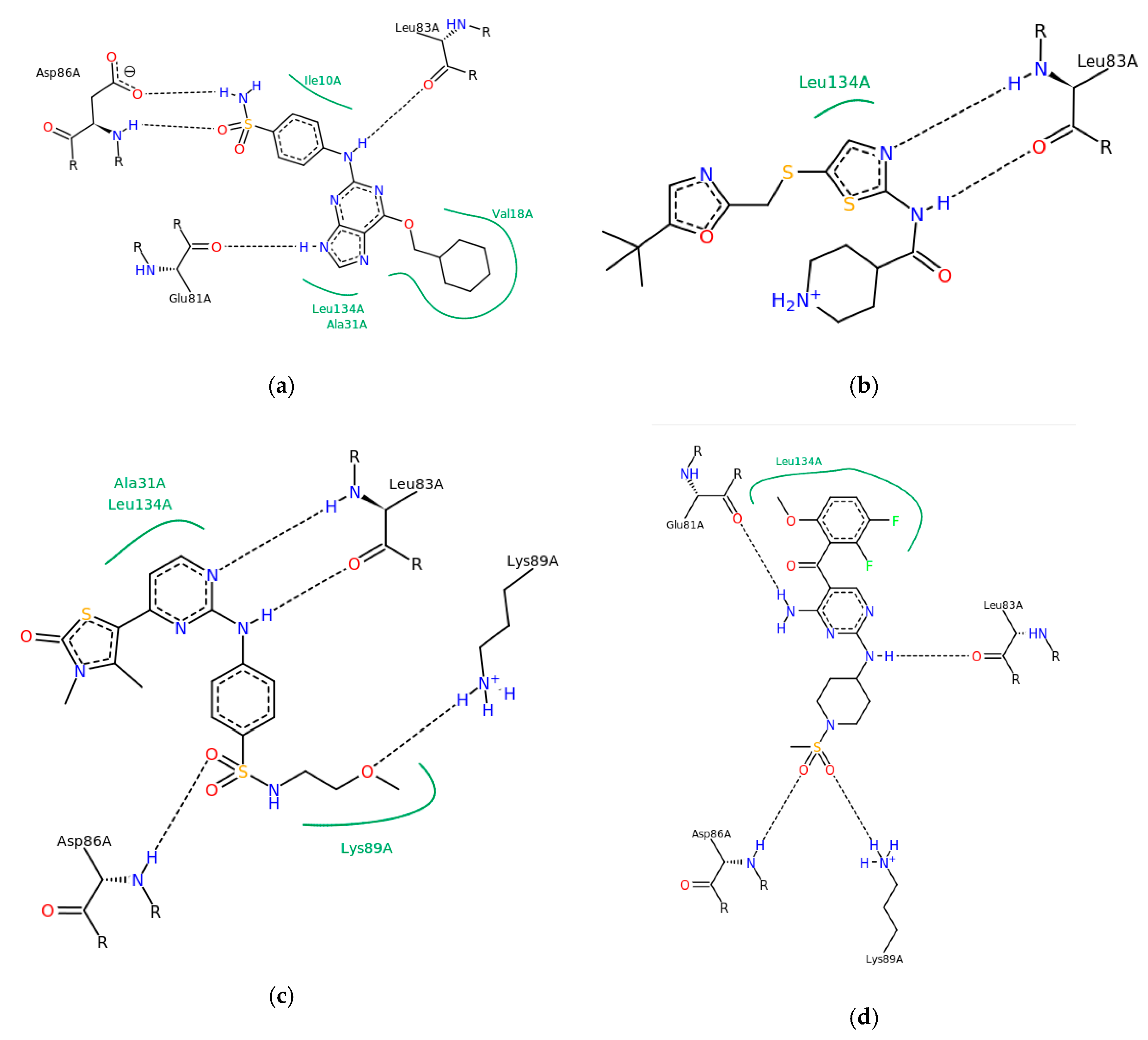
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Hritz, J.; de Ruiter, A.; Oostenbrink, C. Impact of Plasticity and Flexibility on Docking Results for Cytochrome P450 2D6: A Combined Approach of Molecular Dynamics and Ligand Docking. J. Med. Chem. 2008, 51, 7469–7477. [Google Scholar] [CrossRef]
- Campbell, A.J.; Lamb, M.L.; Joseph-McCarthy, D. Ensemble-Based Docking Using Biased Molecular Dynamics. J. Chem. Inf. Modeling 2014, 54, 2127–2138. [Google Scholar] [CrossRef]
- Choudhury, C.; Priyakumar, U.D.; Sastry, G.N. Dynamics Based Pharmacophore Models for Screening Potential Inhibitors of Mycobacterial Cyclopropane Synthase. J. Chem. Inf. Modeling 2015, 55, 848–860. [Google Scholar] [CrossRef]
- Sohn, Y.-s.; Park, C.; Lee, Y.; Kim, S.; Thangapandian, S.; Kim, Y.; Kim, H.-H.; Suh, J.-K.; Lee, K.W. Multi-conformation dynamic pharmacophore modeling of the peroxisome proliferator-activated receptor γ for the discovery of novel agonists. J. Mol. Graph. Model. 2013, 46, 1–9. [Google Scholar] [CrossRef]
- Spyrakis, F.; Benedetti, P.; Decherchi, S.; Rocchia, W.; Cavalli, A.; Alcaro, S.; Ortuso, F.; Baroni, M.; Cruciani, G. A Pipeline To Enhance Ligand Virtual Screening: Integrating Molecular Dynamics and Fingerprints for Ligand and Proteins. J. Chem. Inf. Modeling 2015, 55, 2256–2274. [Google Scholar] [CrossRef] [PubMed]
- Wieder, M.; Garon, A.; Perricone, U.; Boresch, S.; Seidel, T.; Almerico, A.M.; Langer, T. Common Hits Approach: Combining Pharmacophore Modeling and Molecular Dynamics Simulations. J. Chem. Inf. Modeling 2017, 57, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Kutlushina, A.; Khakimova, A.; Madzhidov, T.; Polishchuk, P. Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules 2018, 23, 3094. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org (accessed on 2 November 2019).
- Sayle, K.L.; Bentley, J.; Boyle, F.T.; Calvert, A.H.; Cheng, Y.; Curtin, N.J.; Endicott, J.A.; Golding, B.T.; Hardcastle, I.R.; Jewsbury, P.; et al. Structure-Based design of 2-Arylamino-4-cyclohexylmethyl-5-nitroso-6-aminopyrimidine inhibitors of cyclin-Dependent kinases 1 and 2. Bioorganic Med. Chem. Lett. 2003, 13, 3079–3082. [Google Scholar] [CrossRef]
- Pratt, D.J.; Bentley, J.; Jewsbury, P.; Boyle, F.T.; Endicott, J.A.; Noble, M.E.M. Dissecting the Determinants of Cyclin-Dependent Kinase 2 and Cyclin-Dependent Kinase 4 Inhibitor Selectivity. J. Med. Chem. 2006, 49, 5470–5477. [Google Scholar] [CrossRef] [PubMed]
- Coxon, C.R.; Anscombe, E.; Harnor, S.J.; Martin, M.P.; Carbain, B.; Golding, B.T.; Hardcastle, I.R.; Harlow, L.K.; Korolchuk, S.; Matheson, C.J.; et al. Cyclin-Dependent Kinase (CDK) Inhibitors: Structure–Activity Relationships and Insights into the CDK-2 Selectivity of 6-Substituted 2-Arylaminopurines. J. Med. Chem. 2017, 60, 1746–1767. [Google Scholar] [CrossRef] [PubMed]
- Choong, I.C.; Serafimova, I.; Fan, J.; Stockett, D.; Chan, E.; Cheeti, S.; Lu, Y.; Fahr, B.; Pham, P.; Arkin, M.R.; et al. A diaminocyclohexyl analog of SNS-032 with improved permeability and bioavailability properties. Bioorganic Med. Chem. Lett. 2008, 18, 5763–5765. [Google Scholar] [CrossRef]
- Fan, J.; Fahr, B.; Stockett, D.; Chan, E.; Cheeti, S.; Serafimova, I.; Lu, Y.; Pham, P.; Walker, D.H.; Hoch, U.; et al. Modifications of the isonipecotic acid fragment of SNS-032: Analogs with improved permeability and lower efflux ratio. Bioorganic Med. Chem. Lett. 2008, 18, 6236–6239. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Griffiths, G.; Midgley, C.A.; Barnett, A.L.; Cooper, M.; Grabarek, J.; Ingram, L.; Jackson, W.; Kontopidis, G.; McClue, S.J.; et al. Discovery and Characterization of 2-Anilino-4- (Thiazol-5-yl)Pyrimidine Transcriptional CDK Inhibitors as Anticancer Agents. Chem. Biol. 2010, 17, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-J.; DePinto, W.; Bartkovitz, D.; So, S.-S.; Vu, B.T.; Packman, K.; Lukacs, C.; Ding, Q.; Jiang, N.; Wang, K.; et al. Discovery of [4-Amino-2-(1-methanesulfonylpiperidin-4-ylamino)pyrimidin-5-yl](2,3-difluoro-6- methoxyphenyl)methanone (R547), A Potent and Selective Cyclin-Dependent Kinase Inhibitor with Significant in Vivo Antitumor Activity. J. Med. Chem. 2006, 49, 6549–6560. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Abraham, M.J.; van der Spoel, D.; Lindahl, E.; Hess, B.; The GROMACS Development Team. GROMACS User Manual Version 2016. Available online: www.gromacs.org (accessed on 18 November 2019).
- Ponder, J.W.; Case, D.A. Force Fields for Protein Simulations. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 2003; Volume 66, pp. 27–85. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lemkul, J. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1. 0]. Living J. Comput. Mol. Sci. 2018, 1, 5068. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
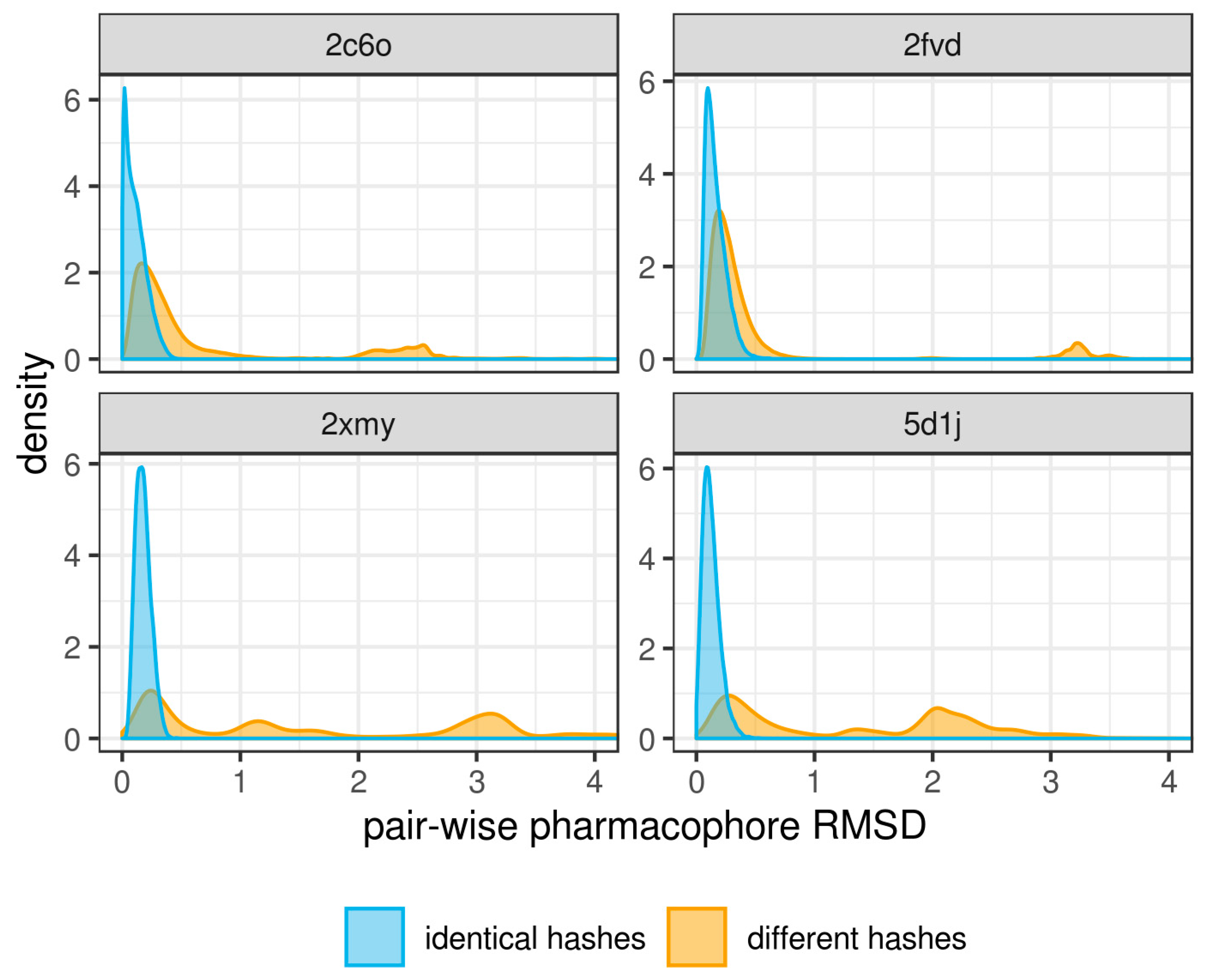{kind=link}
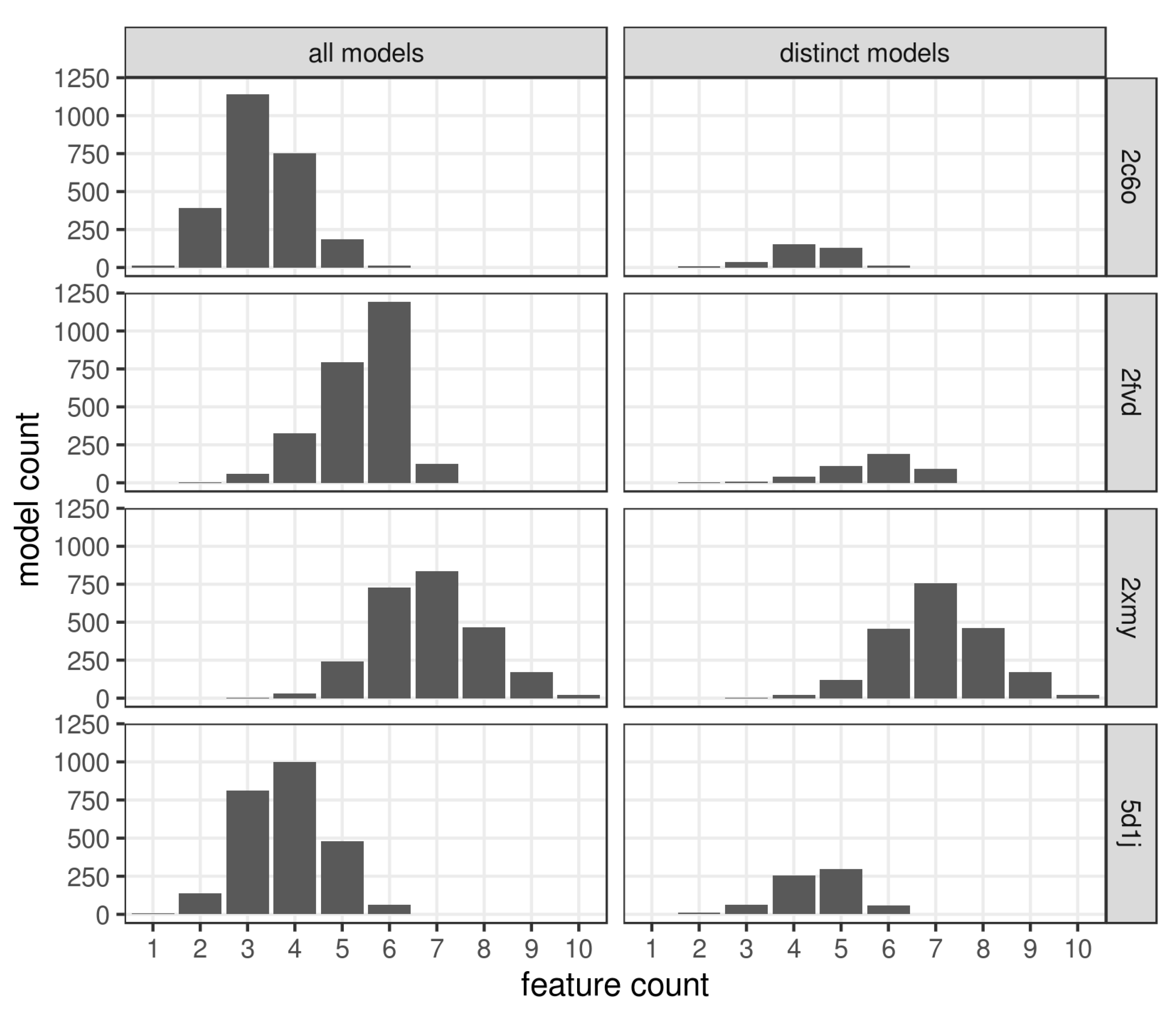{kind=link}
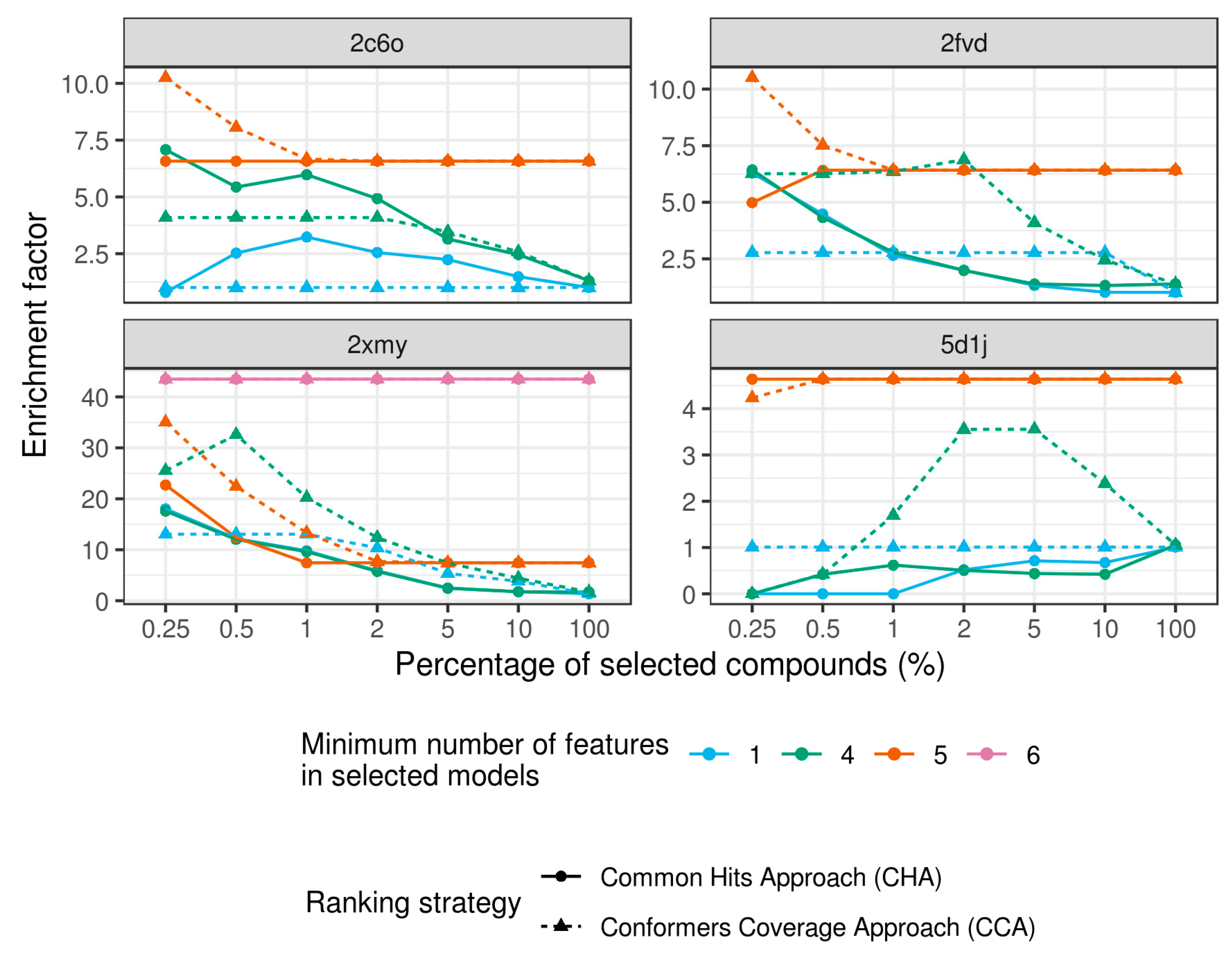{kind=link}
{kind=link}
| PDB | Minimum Number of Pharmacophore Features in Models | Number of Representative Models | Number of Retrieved Compounds | TP/FP | EF100% 1 |
|---|---|---|---|---|---|
| 2C6O | 1 | 338 | 27,884 (98.6%) | 471/27,413 | 1.01 |
| 4 | 295 | 8109 (28.7%) | 178/7931 | 1.31 | |
| 5 | 143 | 291 (1.03%) | 32/259 | 6.58 | |
| 2FVD | 1 | 440 | 25262 (89.3%) | 430/24,832 | 1.02 |
| 4 | 431 | 7745 (27.4%) | 180/7565 | 1.39 | |
| 5 | 390 | 205 (0.73%) | 22/183 | 6.42 | |
| 6 | 282 | 2 (0.007%) | 2/0 | 59.79 | |
| 2XMY | 1 | 2009 | 14,877 (52.6%) | 337/14,540 | 1.35 |
| 4 | 2008 | 10,470 (37.0%) | 300/10,170 | 1.71 | |
| 5 | 1988 | 707 (2.5%) | 88/619 | 7.44 | |
| 6 | 1868 | 33 (0.117%) | 24/9 | 43.48 | |
| 7 | 1411 | 1 (0.004%) | 1/0 | 59.79 | |
| 5D1J | 1 | 683 | 27,884 (98.6%) | 471/27,413 | 1.01 |
| 4 | 609 | 15,312 (54.1%) | 270/15,042 | 1.05 | |
| 5 | 356 | 116 (0.41%) | 9/107 | 4.64 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polishchuk, P.; Kutlushina, A.; Bashirova, D.; Mokshyna, O.; Madzhidov, T. Virtual Screening Using Pharmacophore Models Retrieved from Molecular Dynamic Simulations. Int. J. Mol. Sci. 2019, 20, 5834. https://doi.org/10.3390/ijms20235834
Polishchuk P, Kutlushina A, Bashirova D, Mokshyna O, Madzhidov T. Virtual Screening Using Pharmacophore Models Retrieved from Molecular Dynamic Simulations. International Journal of Molecular Sciences. 2019; 20(23):5834. https://doi.org/10.3390/ijms20235834
Chicago/Turabian StylePolishchuk, Pavel, Alina Kutlushina, Dayana Bashirova, Olena Mokshyna, and Timur Madzhidov. 2019. "Virtual Screening Using Pharmacophore Models Retrieved from Molecular Dynamic Simulations" International Journal of Molecular Sciences 20, no. 23: 5834. https://doi.org/10.3390/ijms20235834