Comparative Transcriptome Analysis Reveals the Cause for Accumulation of Reactive Oxygen Species During Pollen Abortion in Cytoplasmic Male-Sterile Kenaf Line 722HA

 ,
,
Abstract
:1. Introduction
2. Results
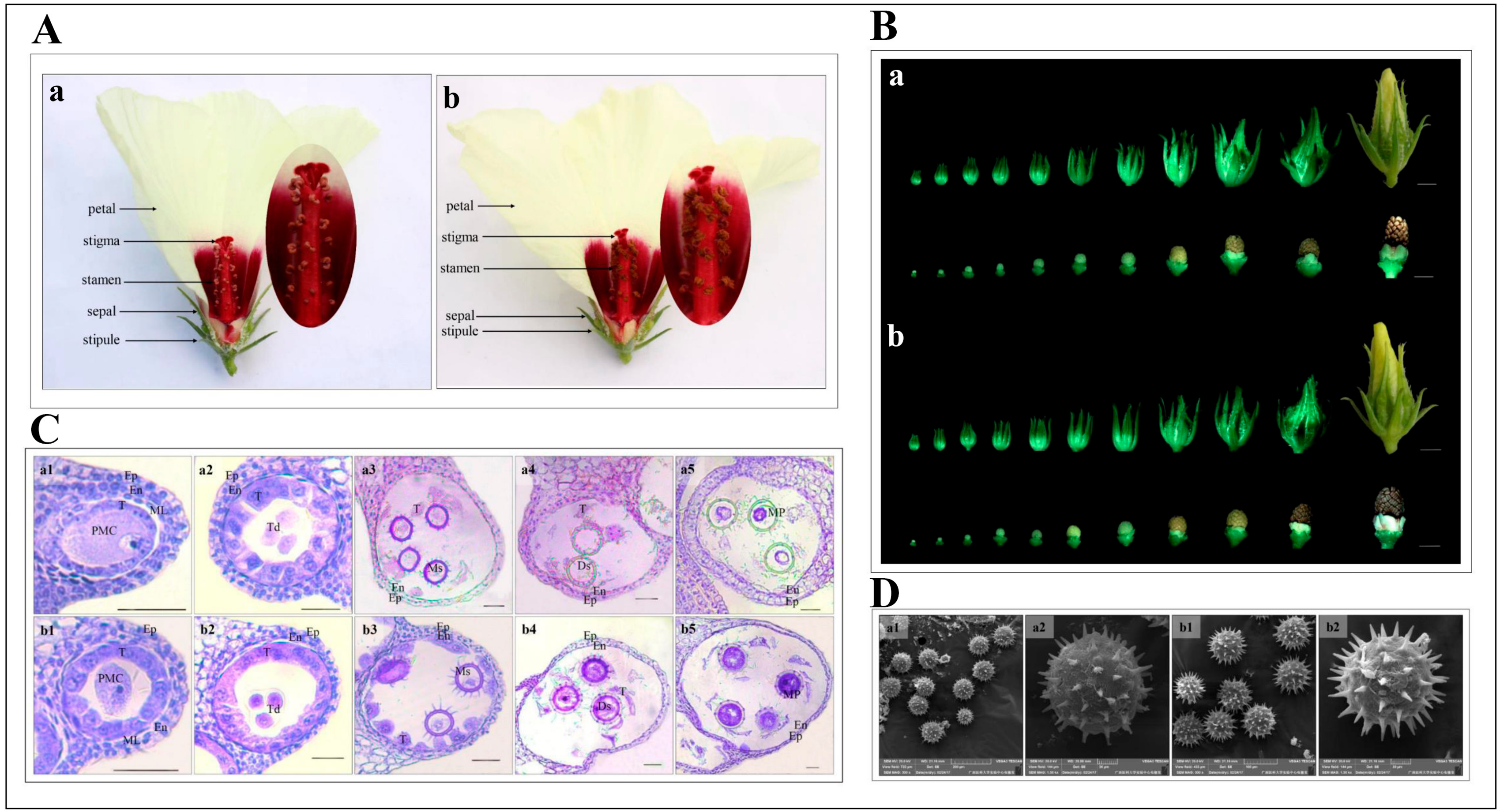
2.1. Morphological and Cytological Characterization of 722HA and 722HB
2.2. De Novo Assembly and Sequence Annotation
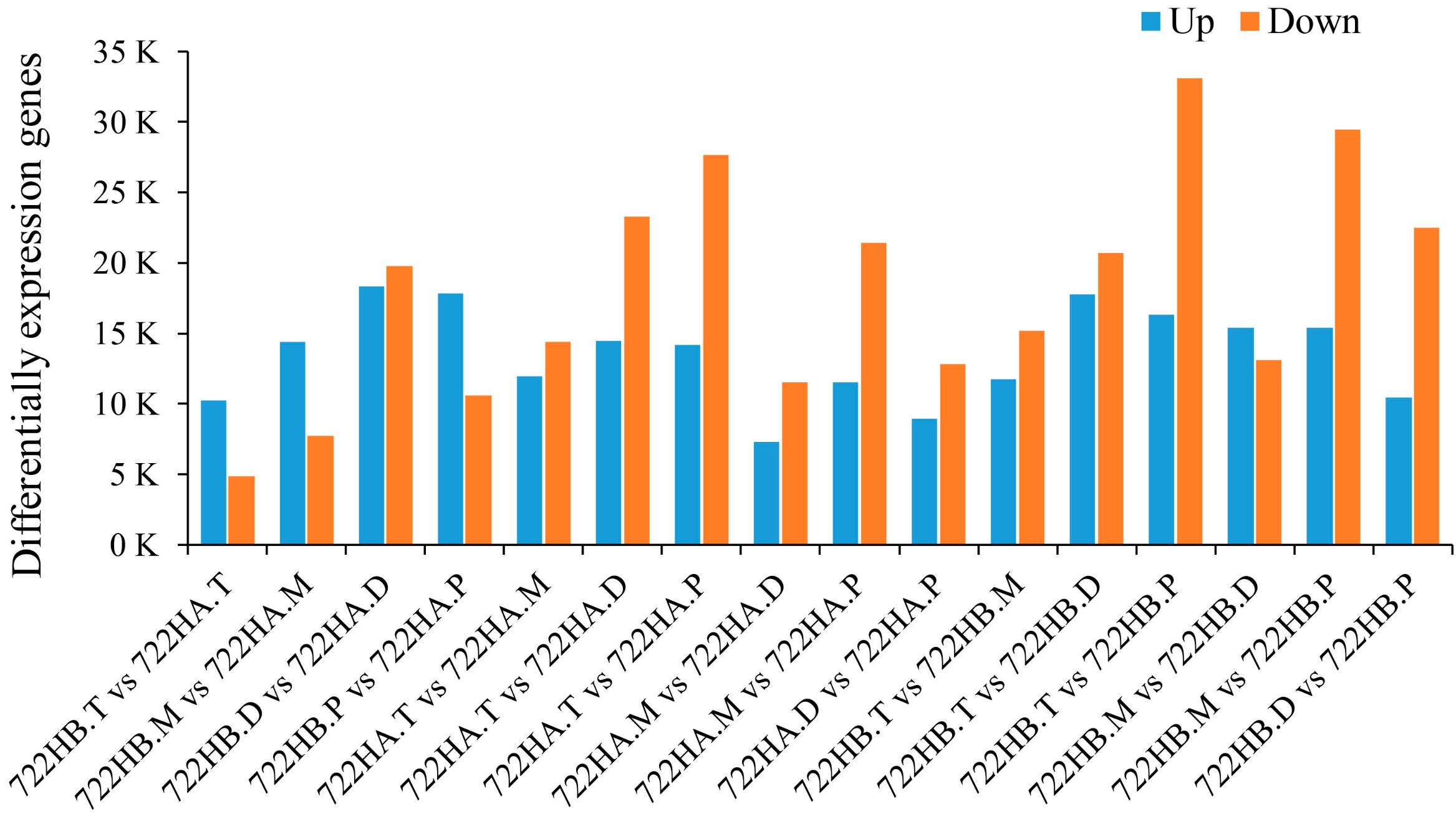
2.3. Detection of Differential Gene Expression between 722HA and 722HB
2.4. Hierarchical Clustering and Principal Component Analysis (PCA)
2.5. Gene Expression Pattern in 722HA and 722HB
2.6. Malondialdehyde (MDA) Content and SOD Activity Analysis
2.7. RT-qPCR Verification
3. Discussion
3.1. Premature Tapetum Degradation is Related to Pollen Abortion in 722HA
3.2. DEGs Involved in Electron Transfer Chain and F1F0-ATPase Proton Transport
3.3. DEGs Involved in ROS Scavenging
3.4. ROS Accumulation May Cause Premature Tapetum Degradation
4. Materials and Methods
4.1. Plant Materials
4.2. Morphological and Cytological Observations
4.3. Total RNA Extraction, cDNA Library Construction, and Deep Sequencing
4.4. De Novo Assembly and Functional Annotation
4.5. DEG Analysis
4.6. Quantitative Real-Time PCR for RNA-seq Validation
4.7. Enzyme Activity and MDA Content Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CMS | Cytoplasmic male sterility |
| ROS | Reactive oxygen species |
| DEGs | Differentially expressed genes |
| SOD | Superoxide dismutase |
| MDA | Malondialdehyde |
References
- Wang, S.; Wang, C.; Zhang, X.; Chen, X.; Liu, J.; Jia, X.; Jia, S. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol. Biochem. 2016, 105, 224–232. [Google Scholar] [CrossRef]
- Budar, F.; Pelletier, G. Male sterility in plants: Occurrence, determinism, significance and use. C. R. Acad. Sci. III 2001, 324, 543–550. [Google Scholar] [CrossRef]
- Lee, Y.; Park, S.; Lim, C.; Kim, H.; Lim, H.; Ahn, Y.; Sung, S.; Yoon, M.; Kim, S. Discovery of a novel cytoplasmic male-sterility and its restorer lines in radish (Raphanus sativus L.). Theor. Appl. Genet. 2008, 117, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, D.; Zhu, Y. Characterization and Use of Male Sterility in Hybrid Rice Breeding. J. Intergr. Plant Biol. 2010, 49, 791–804. [Google Scholar] [CrossRef]
- Killinger, G.B. Kenaf (Hibiscus cannabinus L.), A Multi-use Crop. Agron. J. 1969, 61, 734–736. [Google Scholar] [CrossRef]
- Bel-Berger, P.; Hoven, T.V.; Ramaswamy, G.N.; Kimmel, L.; Bolyston, E. Cotton/Kenaf fabrics: A viable natural fabric. J. Cotton Sci. 1999, 3, 60–70. [Google Scholar]
- Coetzee, R.; Labuschagne, M.T.; Hugo, A. Fatty acid and oil variation in seed from kenaf (Hibiscus cannabinus L.). Ind. Crop. Prod. 2008, 27, 104–109. [Google Scholar] [CrossRef]
- Patil, R.C.; Thombre, M.V. Heterosis and combining ability studies in Hibiscus cannabinus L. J. Maharashtra Agric. Univ. 1980, 5, 123–126. [Google Scholar]
- Qi, J.; Li, W. Breeding and popularization of Fuhong No. 2, a new kenaf variety with high yield, stress resistance and wide adaptability. J. Fujian Agric. Univ. 1999, 28, 189–385. [Google Scholar]
- Chen, A.; Li, D.; Tang, H.; Li, J. The Breeding of “China Hybrid Kenaf 316” (H316) with Super High Yield Disease-Resistance and Wide Adaptability. Plant Fiber Sci. Chin. 2009, 31, 173–178. [Google Scholar]
- Ugale, S.D.; Khuspe, S.S. Cytoplasmic genetic male sterility in Hibiscus cannabinus L. J. Maharashtra Agric. Univ. 1976, 2, 102–106. [Google Scholar]
- Zhou, R.Y. Discovery of Male Sterile Plants in Kenaf (Hibscus cannabinus L.). Sci. Agric. Sin. 2002, 35, 212. [Google Scholar]
- Li, D.; Chen, A.; Tang, H. Discovery and preliminary creation of kenaf nucleus-nuclear interaction male sterile line. Plant Fiber Sci. Chin. 2007, 2, 78. [Google Scholar]
- Chen, G.; Ye, X.; Zhang, S.; Zhu, S.; Yuan, L.; Hou, J.; Wang, C. Comparative Transcriptome Analysis between Fertile and CMS Flower Buds in Wucai (Brassica campestris L.). BMC Genom. 2018, 19, 908. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, X.; Sha, L.; Gan, H.; Zhang, L.; Song, X. Tapetal-Delayed Programmed Cell Death (PCD) and Oxidative Stress-Induced Male Sterility of Aegilops uniaristata Cytoplasm in Wheat. Int. J. Mol. Sci. 2018, 19, 1708. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Meng, Z.; Zhang, J.; Stewart, J.M.; Xing, C.; Wu, J.; Jin, S. Transcriptome, cytological and biochemical analysis of cytoplasmic male sterility and maintainer line in CMS-D8 cotton. Plant Mol. Biol. 2018, 97, 1–15. [Google Scholar]
- Yang, Y.; Bao, S.; Zhou, X.; Liu, J.; Zhuang, Y. The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant Biol. 2018, 18, 209. [Google Scholar] [CrossRef] [PubMed]
- Wesołowski, W.; Szklarczyk, M.; Szalonek, M.; Słowińska, J. Analysis of the mitochondrial proteome in cytoplasmic male-sterile and male-fertile beets. J. Proteom. 2015, 119, 61–74. [Google Scholar] [CrossRef]
- Salminen, T.A.; Blomqvist, K.; Edqvist, J. Lipid transfer proteins: Classification, nomenclature, structure, and function. Planta 2016, 244, 971–997. [Google Scholar] [CrossRef]
- Engelke, T.; Hirsche, J.; Roitsch, T. Anther-specific carbohydrate supply and restoration of metabolically engineered male sterility. J. Exp. Bot. 2010, 61, 2693–2706. [Google Scholar] [CrossRef] [Green Version]
- Tong, Z.; Wang, T.; Xu, Y. Evidence for involvement of phytochrome regulation in male-sterility of a mutant of Oryza sativa L. Photochem. Photobiol. 1990, 52, 161–164. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Xu, B.; Zhao, N.; Yang, X.; Zhang, M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genom. 2013, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tan, M.; Yu, H.; Li, L.; Zhou, F.; Yang, M.; Zhou, T.; Zhao, Y. Comparative transcriptome profiling of the fertile and sterile flower buds of a dominant genic male sterile line in sesame (Sesamum indicum L.). BMC Plant Biol. 2016, 16, 250. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, S.; Ding, X.; He, T.; Dai, J.; Yang, S.; Gai, J. Comparative Transcriptome Analysis between the Cytoplasmic Male Sterile Line NJCMS1A and Its Maintainer NJCMS1B in Soybean (Glycine max (L.) Merr.). PLoS ONE 2015, 10, e0126771. [Google Scholar] [CrossRef]
- Chen, P.; Ran, S.; Li, R.; Huang, Z.; Qian, J.; Yu, M.; Zhou, R. Transcriptome de novo assembly and differentially expressed genes related to cytoplasmic male sterility in kenaf (Hibiscus cannabinus L.). Mol. Breed. 2014, 34, 1879–1891. [Google Scholar] [CrossRef]
- Giuseppina, F.; Simone, D.; Rita, B.; Laura, F.; Maja, M.; Antonella, C.; Maria Maddalena, A. Tapetum and middle layer control male fertility in Actinidia deliciosa. Ann. Bot. 2013, 112, 1045–1055. [Google Scholar] [Green Version]
- Liu, Z.; Shi, X.; Li, S.; Zhang, L.; Song, X. Oxidative Stress and Aberrant Programmed Cell Death Are Associated with Pollen Abortion in Isonuclear Alloplasmic Male-Sterile Wheat. Front. Plant Sci. 2018, 9, 595. [Google Scholar] [CrossRef]
- Du, K.; Liu, Q.; Wu, X.; Jiang, J.; Jian, W.; Fang, Y.; Li, A.; Wang, Y. Morphological Structure and Transcriptome Comparison of the Cytoplasmic Male Sterility Line in Brassica napus(SaNa-1A) Derived from Somatic Hybridization and Its Maintainer Line SaNa-1B. Front. Plant Sci. 2016, 7, 1313. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Chen, Y.; Xu, L.; Jiang, L.; Gong, Y.; Wang, L.; Liu, L. Differential gene expression profiling of Ogura CMS line and its maintainer in radish (Raphanus sativus L.). Acta. Physiol. Plant 2013, 35, 3413–3425. [Google Scholar] [CrossRef]
- Siedow, J.N.; Umbach, A.L. Plant Mitochondrial Electron Transfer and Molecular Biology. Plant Cell 1995, 7, 821–831. [Google Scholar] [CrossRef]
- Moller, I.M. Plant mitochondria and oxidative stress: Electron transport, NADPH turnover, and metabolism of reactive oxygen species [Review]. Annu. Rev. Plant Biol. 2001, 52, 561–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, G.; Zhang, Y.; Song, Q.; Chen, Z.; Wang, J.; Guo, J.; Niu, N.; Wang, J.; Ma, S. Comparative studies of mitochondrial proteomics reveal an intimate protein network of male sterility in wheat (Triticum aestivum L.). J. Exp. Bot. 2015, 66, 6191–6203. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Ye, J.; Yang, X.; Li, S.; Zhang, L.; Song, X. Identification of Proteins Involved in Carbohydrate Metabolism and Energy Metabolism Pathways and Their Regulation of Cytoplasmic Male Sterility in Wheat. Int. J. Mol. Sci. 2018, 19, 324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Chen, P.; Khan, A.; Zhao, Y.; Chen, L.; Liu, D.; Liao, X.; Kong, X.; Zhou, R. Candidate Reference Genes Selection and Application for RT-qPCR Analysis in Kenaf with Cytoplasmic Male Sterility Background. Front. Plant Sci. 2017, 8, 1520. [Google Scholar] [CrossRef]
- Yesodi, V.; Hauschner, H.; Tabib, Y.; Firon, N. An intact F1ATPase-subunit gene and a pseudogene with differing genomic organization are detected in both male-fertile and CMS petunia mitochondria. Curr. Genet. 1997, 32, 348–357. [Google Scholar] [CrossRef]
- Pastore, D.; Trono, D.; Laus, M.N.; Di, F.N.; Flagella, Z. Possible plant mitochondria involvement in cell adaptation to drought stress. A case study: Durum wheat mitochondria. J. Exp. Bot. 2007, 58, 195–210. [Google Scholar] [CrossRef]
- McCormick, S. Control of Male Gametophyte Development. Plant Cell 2004, 16, S142–S153. [Google Scholar] [CrossRef]
- Li, S.; Wan, C.; Hu, C.; Feng, G.; Qi, H.; Wang, K.; Wang, T.; Zhu, Y. Mitochondrial mutation impairs cytoplasmic male sterility rice in response to H2O2 stress. Plant Sci. 2012, 195, 143–150. [Google Scholar] [CrossRef]
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef]
- Budhi Sagar, T.; Beatrice, B.; Alex, L. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002, 128, 1271–1281. [Google Scholar]
- Baxter, A.; Mittler, R.; Suzuki, N. ROS as key players in plant stress signalling. J. Exp. Bot. 2013, 65, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D. Enzymes and other agents that enhance cell wall extensibility. Annu. Rev. Plant Biol. 1999, 50, 391–417. [Google Scholar] [CrossRef]
- Hameed, A.; Goher, M.; Iqbal, N. Drought induced programmed cell death and associated changes in antioxidants, proteases, and lipid peroxidation in wheat leaves. Biol. Plant. 2013, 57, 370–374. [Google Scholar] [CrossRef]
- Wan, C.; Li, S.; Wen, L.; Kong, J.; Wang, K.; Zhu, Y. Damage of oxidative stress on mitochondria during microspores development in Honglian CMS line of rice. Plant Cell Rep. 2007, 26, 373–382. [Google Scholar] [CrossRef]
- Li, J.; Tang, H.; Chen, Y.; Chen, A.; Huang, S.; Li, D. Cytomorphological Observation on Cytoplasm Nuclear Male Sterile line of Kenaf. Plant Fiber Sci. Chin. 2015, 37, 229–232. [Google Scholar]
- Song, Q.; Wang, S.; Zhang, G.; Li, Y.; Li, Z.; Guo, J.; Niu, N.; Wang, J.; Ma, S. Comparative proteomic analysis of a membrane-enriched fraction from flag leaves reveals responses to chemical hybridization agent SQ-1 in wheat. Front. Plant Sci. 2015, 6, 669. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Sub-Categories | 722HA | 722HB | Total |
|---|---|---|---|---|
| Reads | Total raw reads (M) | 1230.85 | 1238.08 | 2468.93 |
| Clean reads (M) | 1124.69 | 1129.13 | 2253.82 | |
| Q20 (%) | 97.75 | 97.64 | 97.69 | |
| Q30 (%) | 91.15 | 90.99 | 91.01 | |
| Clean reads ratio (%) | 91.34 | 91.22 | 91.2 | |
| Transcripts | Total number | 1,410,176.00 | 1,310,102.00 | 2,720,278.00 |
| Total length (nt) | 1,330,236,352.00 | 1,215,811,030.00 | 2,546,047,382.00 | |
| Mean length (nt) | 940 | 926.08 | 933.04 | |
| N50 (nt) | 1730.33 | 1679.00 | 1704.67 | |
| GC (%) | 41.83 | 42.05 | 41.94 | |
| Unigenes | Total number | 1,011,304.00 | 937,700.00 | 1,949,004.00 |
| Total length (nt) | 1,054,727,027.00 | 949,656,291.00 | 2,004,383,318.00 | |
| Mean length (nt) | 1039.00 | 1010.33 | 1024.67 | |
| N50 (nt) | 1781.17 | 1706.17 | 1743.67 | |
| GC (%) | 41.77 | 41.99 | 41.88 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, B.; Liu, Y.; Chen, Z.; Liu, D.; Wang, Y.; Zheng, J.; Liao, X.; Zhou, a.R. Comparative Transcriptome Analysis Reveals the Cause for Accumulation of Reactive Oxygen Species During Pollen Abortion in Cytoplasmic Male-Sterile Kenaf Line 722HA. Int. J. Mol. Sci. 2019, 20, 5515. https://doi.org/10.3390/ijms20215515
Zhou B, Liu Y, Chen Z, Liu D, Wang Y, Zheng J, Liao X, Zhou aR. Comparative Transcriptome Analysis Reveals the Cause for Accumulation of Reactive Oxygen Species During Pollen Abortion in Cytoplasmic Male-Sterile Kenaf Line 722HA. International Journal of Molecular Sciences. 2019; 20(21):5515. https://doi.org/10.3390/ijms20215515
Chicago/Turabian StyleZhou, Bujin, Yiding Liu, Zhengxia Chen, Dongmei Liu, Yining Wang, Jie Zheng, Xiaofang Liao, and and Ruiyang Zhou. 2019. "Comparative Transcriptome Analysis Reveals the Cause for Accumulation of Reactive Oxygen Species During Pollen Abortion in Cytoplasmic Male-Sterile Kenaf Line 722HA" International Journal of Molecular Sciences 20, no. 21: 5515. https://doi.org/10.3390/ijms20215515