Genetics and Extracellular Vesicles of Pediatrics Sleep Disordered Breathing and Epilepsy


Abstract
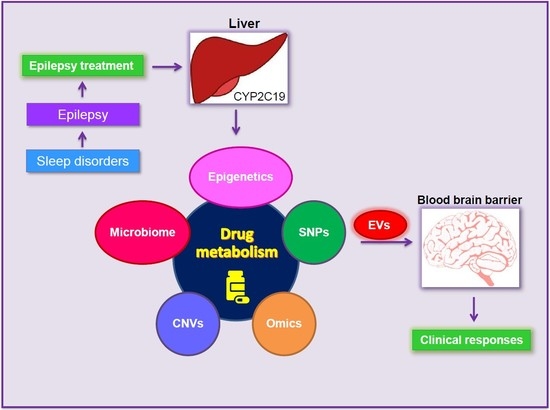
:
1. Sleep Disordered Breathing (SDB)
2. Pediatrics Sleep Disorders and Epilepsy
3. Epilepsy
3.1. Epilepsy Treatments
3.2. Pediatrics Sleep Disorders and Epilepsy Treatment
3.3. Phenobarbital
4. Gamma-Aminobutyric Acid (GABA) Receptors
5. Identifying GABAA Receptor Gene Networks
6. Cytochromes P450 (CYP450) and CYP2C19 Gene
7. Mechanism of Drug Metabolism
8. Functional Genomics and Precision Medicine
9. Extracellular Vesicles (EVs)
10. Sleep Disordered Breathing (SDB), Epilepsy and EVs
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luzzi, V.; Ierardo, G.; Di Carlo, G.; Saccucci, M.; Polimeni, A. Obstructive sleep apnea syndrome in the pediatric age: The role of the dentist. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9–14. [Google Scholar] [PubMed]
- Economou, N.T.; Ferini-Strambi, L.; Steiropoulos, P. Sleep-Related Drug Therapy in Special Conditions: Children. Sleep Med. Clin. 2018, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.L.; Storfer-Isser, A.; Taylor, H.G.; Kirchner, H.L.; Emancipator, J.L.; Redline, S. Increased behavioral morbidity in school-aged children with sleep-disordered breathing. Pediatrics 2004, 114, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.L.; Brooks, L.J.; Draper, K.A.; Gozal, D.; Halbower, A.C.; Jones, J.; Schechter, M.S.; Sheldon, S.H.; Spruyt, K.; Ward, S.D.; et al. Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics 2012, 130, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.L. Sleep-disordered breathing in children. Am. J. Respir. Crit. Care Med. 2001, 164, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Avis, K.T.; Gamble, K.L.; Schwebel, D.C. Effect of positive airway pressure therapy in children with obstructive sleep apnea syndrome: Does positive airway pressure use reduce pedestrian injury risk? Sleep Health 2019, 5, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Lee, W.T.; Jeng, S.F.; Lee, C.C.; Weng, W.C. Sleep and Behavior Problems in Children with Epilepsy. J. Pediatr. Health Care 2019, 33, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.C.; Yang, W.W.; Wong, S.W.; AlSiddiq, F.; Khu, Y.S. Sleep habits and disturbances in Malaysian children with epilepsy. J. Paediatr. Child Health 2010, 46, 80–84. [Google Scholar] [CrossRef]
- Gogou, M.; Haidopoulou, K.; Eboriadou, M.; Pavlou, E. Sleep Disturbances in Children with Rolandic Epilepsy. Neuropediatrics 2017, 48, 30–35. [Google Scholar]
- Tolaymat, A.; Liu, Z. Sleep Disorders in Childhood Neurological Diseases. Child (Basel) 2017, 4, 84. [Google Scholar] [CrossRef]
- Ellingson, R.J.; Wilken, K.; Bennett, D.R. Efficacy of sleep deprivation as an activation procedure in epilepsy patients. J. Clin. Neurophysiol. 1984, 1, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Aneja, S.; Gupta, M. Sleep and childhood epilepsy. Indian J. Pediatr. 2005, 72, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, M.; Fayyazi, A.; Cheraghi, F.; Hakimi, H.; Mosazadeh, S.; Almasi, S. The Relationship Between Severity of Epilepsy and Sleep Disorder in Epileptic Children. Iran J. Child Neurol. 2019, 13, 77–88. [Google Scholar] [PubMed]
- Jain, S.V.; Simakajornboon, S.; Shapiro, S.M.; Morton, L.D.; Leszczyszyn, D.J.; Simakajornboon, N. Obstructive sleep apnea in children with epilepsy: Prospective pilot trial. Acta Neurol. Scand. 2012, 125, e3–e6. [Google Scholar] [CrossRef]
- Larson, A.M.; Ryther, R.C.; Jennesson, M.; Geffrey, A.L.; Bruno, P.L.; Anagnos, C.J.; Shoeb, A.H.; Thibert, R.L.; Thiele, E.A. Impact of pediatric epilepsy on sleep patterns and behaviors in children and parents. Epilepsia 2012, 53, 1162–1169. [Google Scholar] [CrossRef]
- Gutter, T.; de Weerd, A.W. Effects of daytime secondarily generalized epileptic seizures on sleep during the following night. Epilepsy Behav. 2012, 25, 289–294. [Google Scholar] [CrossRef]
- Accardo, J.A.; Malow, B.A. Sleep, epilepsy, and autism. Epilepsy Behav. 2015, 47, 202–206. [Google Scholar] [CrossRef]
- Jain, S.V.; Simakajornboon, N.; Glauser, T.A. Provider practices impact adequate diagnosis of sleep disorders in children with epilepsy. J. Child Neurol. 2013, 28, 589–595. [Google Scholar] [CrossRef]
- Aurora, R.N.; Lamm, C.I.; Zak, R.S.; Kristo, D.A.; Bista, S.R.; Rowley, J.A.; Casey, K.R. Practice parameters for the non-respiratory indications for polysomnography and multiple sleep latency testing for children. Sleep 2012, 35, 1467–1473. [Google Scholar] [CrossRef]
- Sajatovic, M.; Tatsuoka, C.; Welter, E.; Friedman, D.; Spruill, T.M.; Stoll, S.; Sahoo, S.S.; Bukach, A.; Bamps, Y.A.; Valdez, J.; et al. Correlates of quality of life among individuals with epilepsy enrolled in self-management research: From the US Centers for Disease Control and Prevention Managing Epilepsy Well Network. Epilepsy Behav. 2017, 69, 177–180. [Google Scholar] [CrossRef]
- Hauser, W.A.; Lee, J.R. Do seizures beget seizures? Prog. Brain Res. 2002, 135, 215–219. [Google Scholar] [PubMed]
- Brodie, M.J.; Barry, S.J.; Bamagous, G.A.; Norrie, J.D.; Kwan, P. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012, 78, 1548–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donohoe, N.V. What should the child with epilepsy be allowed to do? Arch. Dis. Child 1983, 58, 934–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persenius, M.; Rystedt, I.; Wilde-Larsson, B.; Baath, C. Quality of life and sense of coherence in young people and adults with uncomplicated epilepsy: A longitudinal study. Epilepsy Behav. 2015, 47, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Howard, B.V.; Cowan, L.D.; Yeh, J.; Schaefer, C.F.; Wild, R.A.; Wang, W.; Lee, E.T. The effect of estrogen use on levels of glucose and insulin and the risk of type 2 diabetes in American Indian postmenopausal women: The strong heart study. Diabetes Care 2002, 25, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Coorg, R.; Weisenberg, J.L.; Wong, M. Clinical neurogenetics: Recent advances in the genetics of epilepsy. Neurol Clin. 2013, 31, 891–913. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, G.; Perucca, E. Interactions between antiepileptic drugs, and between antiepileptic drugs and other drugs. Epileptic Disord. 2014, 16, 409–431. [Google Scholar] [CrossRef]
- Perucca, E. Clinically relevant drug interactions with antiepileptic drugs. Br. J. Clin. Pharmacol. 2006, 61, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Kotsopoulos, I.A.; van Merode, T.; Kessels, F.G.; de Krom, M.C.; Knottnerus, J.A. Systematic review and meta-analysis of incidence studies of epilepsy and unprovoked seizures. Epilepsia 2002, 43, 1402–1409. [Google Scholar] [CrossRef]
- Forsgren, I.; Beghi, E.; Ekman, M. Cost of epilepsy in Europe. Eur. J. Neurol. 2005, 12 (Suppl. 1), 54–58. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Walia, K.S.; Khan, E.A.; Ko, D.H.; Raza, S.S.; Khan, Y.N. Side effects of antiepileptics—A review. Pain Pr. 2004, 4, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hao, Z.; Yu, D.; Xu, Q.; Zhang, L. The prevalence rates of medication adherence and factors influencing adherence to antiepileptic drugs in children with epilepsy: A systematic review and meta analysis. Epilepsy Res. 2018, 142, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Getnet, A.; Woldeyohannes, S.M.; Bekana, L.; Mekonen, T.; Fekadu, W.; Menberu, M.; Yimer, S.; Assaye, A.; Belete, A.; Belete, H. Antiepileptic Drug Nonadherence and Its Predictors among People with Epilepsy. Behav. Neurol. 2016, 2016, 3189108. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chen, H.; Pakpour, A.H. Correlation between adherence to antiepileptic drugs and quality of life in patients with epilepsy: A longitudinal study. Epilepsy Behav. 2016, 63, 103–108. [Google Scholar] [CrossRef]
- Koh, S.; Ward, S.L.; Lin, M.; Chen, L.S. Sleep apnea treatment improves seizure control in children with neurodevelopmental disorders. Pediatr. Neurol. 2000, 22, 36–39. [Google Scholar] [CrossRef]
- Sheldon, S.H. Pro-convulsant effects of oral melatonin in neurologically disabled children. Lancet 1998, 351, 1254. [Google Scholar] [CrossRef]
- Elkhayat, H.A.; Hassanein, S.M.; Tomoum, H.Y.; Abd-Elhamid, I.A.; Asaad, T.; Elwakkad, A.S. Melatonin and sleep-related problems in children with intractable epilepsy. Pediatr. Neurol. 2010, 42, 249–254. [Google Scholar] [CrossRef]
- Legros, B.; Bazil, C.W. Effects of antiepileptic drugs on sleep architecture: A pilot study. Sleep Med. 2003, 4, 51–55. [Google Scholar] [CrossRef]
- Hindmarch, I.; Dawson, J.; Stanley, N. A double-blind study in healthy volunteers to assess the effects on sleep of pregabalin compared with alprazolam and placebo. Sleep 2005, 28, 187–193. [Google Scholar] [CrossRef]
- Bazil, C.W. Sleep and Epilepsy. Semin Neurol. 2017, 37, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Ilangaratne, N.B.; Mannakkara, N.N.; Bell, G.S.; Sander, J.W. Phenobarbital: Missing in action. Bull. World Health Organ. 2012, 90, 871-A. [Google Scholar] [CrossRef] [PubMed]
- Falco-Walter, J.J.; Bleck, T. Treatment of Established Status Epilepticus. J. Clin. Med. 2016, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Ichikura, K.; Okumura, Y.; Takeuchi, T. Associations of Adverse Clinical Course and Ingested Substances among Patients with Deliberate Drug Poisoning: A Cohort Study from an Intensive Care Unit in Japan. PLoS ONE 2016, 11, e0161996. [Google Scholar] [CrossRef] [PubMed]
- van de Vrie-Hoekstra, N.W.; de Vries, T.W.; van den Berg, P.B.; Brouwer, O.F.; de Jong-van den Berg, L.T. Antiepileptic drug utilization in children from 1997–2005—A study from the Netherlands. Eur. J. Clin. Pharm. 2008, 64, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Dorks, M.; Langner, I.; Timmer, A.; Garbe, E. Treatment of paediatric epilepsy in Germany: Antiepileptic drug utilisation in children and adolescents with a focus on new antiepileptic drugs. Epilepsy Res. 2013, 103, 45–53. [Google Scholar] [CrossRef]
- Srivastava, M.C.; Salgia, K.M.; Arora, M.M. Systemic lupus erythematosus with renal and lymph node changes. J. Indian. Med. Assoc. 1965, 45, 298–302. [Google Scholar]
- Jain, S.V.; Glauser, T.A. Effects of epilepsy treatments on sleep architecture and daytime sleepiness: An evidence-based review of objective sleep metrics. Epilepsia 2014, 55, 26–37. [Google Scholar] [CrossRef]
- Czapinski, P.; Blaszczyk, B.; Czuczwar, S.J. Mechanisms of action of antiepileptic drugs. Curr. Top Med. Chem. 2005, 5, 3–14. [Google Scholar] [CrossRef]
- Nimaga, K.; Desplats, D.; Doumbo, O.; Farnarier, G. Treatment with phenobarbital and monitoring of epileptic patients in rural Mali. Bull. World Health Organ. 2002, 80, 532–537. [Google Scholar]
- Bernus, I.; Dickinson, R.G.; Hooper, W.D.; Eadie, M.J. Urinary excretion of phenobarbitone and its metabolites in chronically treated patients. Eur. J. Clin. Pharmacol. 1994, 46, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.B.; Adams, N. Phenobarbital; StatPearls: Treasure Island, FL, USA, 2018. [Google Scholar]
- Zheleznova, N.N.; Sedelnikova, A.; Weiss, D.S. Function and modulation of delta-containing GABA(A) receptors. Psychoneuroendocrinology 2009, 34 (Suppl. 1), S67–S73. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.S.; Smart, T.G. Binding, activation and modulation of Cys-loop receptors. Trends Pharm. Sci. 2010, 31, 161–174. [Google Scholar] [CrossRef]
- Sieghart, W.; Ramerstorfer, J.; Sarto-Jackson, I.; Varagic, Z.; Ernst, M. A novel GABA(A) receptor pharmacology: Drugs interacting with the alpha(+) beta(−) interface. Br. J. Pharmacol. 2012, 166, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Baumann, S.W.; Baur, R.; Sigel, E. Subunit arrangement of gamma-aminobutyric acid type A receptors. J Biol. Chem. 2001, 276, 36275–36280. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Berkovic, S.F.; Petrou, S. Mechanisms of human inherited epilepsies. Prog. Neurobiol. 2009, 87, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, R.; Kumari, R.; Misra, U.K.; Kalita, J.; Pradhan, S.; Mittal, B. Differential role of sodium channels SCN1A and SCN2A gene polymorphisms with epilepsy and multiple drug resistance in the north Indian population. Br. J. Clin. Pharmacol. 2009, 68, 214–220. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, X. Genes associated with idiopathic epilepsies: A current overview. Neurol. Res. 2009, 31, 135–143. [Google Scholar] [CrossRef]
- Chen, X.; Durisic, N.; Lynch, J.W.; Keramidas, A. Inhibitory synapse deficits caused by familial alpha1 GABAA receptor mutations in epilepsy. Neurobiol. Dis. 2017, 108, 213–224. [Google Scholar] [CrossRef]
- Sigel, E.; Steinmann, M.E. Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef]
- Minuk, G.Y.; Zhang, M.; Gong, Y.; Minuk, L.; Dienes, H.; Pettigrew, N.; Kew, M.; Lipschitz, J.; Sun, D. Decreased hepatocyte membrane potential differences and GABAA-beta3 expression in human hepatocellular carcinoma. Hepatology 2007, 45, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Laughton, D.L.; Walding, A.; Wolstenholme, A.J. Human peripheral blood mononuclear cells express GABAA receptor subunits. Mol. Immunol. 2006, 43, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Bjurstom, H.; Wang, J.; Ericsson, I.; Bengtsson, M.; Liu, Y.; Kumar-Mendu, S.; Issazadeh-Navikas, S.; Birnir, B. GABA, a natural immunomodulator of T lymphocytes. J. Neuroimmunol. 2008, 205, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kittler, J.T.; Moss, S.J. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: Implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol. 2003, 13, 341–347. [Google Scholar] [CrossRef]
- Berka, K.; Hendrychova, T.; Anzenbacher, P.; Otyepka, M. Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site. J. Phys. Chem. A 2011, 115, 11248–11255. [Google Scholar] [CrossRef]
- Zanger, U.M.; Turpeinen, M.; Klein, K.; Schwab, M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 2008, 392, 1093–1108. [Google Scholar] [CrossRef]
- Ghosh, C.; Hossain, M.; Solanki, J.; Dadas, A.; Marchi, N.; Janigro, D. Pathophysiological implications of neurovascular P450 in brain disorders. Drug Discov. Today 2016, 21, 1609–1619. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005, 352, 2211–2221. [Google Scholar] [CrossRef]
- Shorvon, S.D. The temporal aspects of prognosis in epilepsy. J. Neurol. Neurosurg. Psychiatry 1984, 47, 1157–1165. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Drug treatment of epilepsy: When does it fail and how to optimize its use? CNS Spectr. 2004, 9, 110–119. [Google Scholar] [CrossRef]
- Mohanraj, R.; Brodie, M.J. Diagnosing refractory epilepsy: Response to sequential treatment schedules. Eur. J. Neurol. 2006, 13, 277–282. [Google Scholar] [CrossRef]
- Klose, T.S.; Blaisdell, J.A.; Goldstein, J.A. Gene structure of CYP2C8 and extrahepatic distribution of the human CYP2Cs. J. Biochem. Mol. Toxicol. 1999, 13, 289–295. [Google Scholar] [CrossRef]
- Franco, V.; Perucca, E. CYP2C9 polymorphisms and phenytoin metabolism: Implications for adverse effects. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1269–1279. [Google Scholar] [CrossRef]
- Lopez-Garcia, M.A.; Feria-Romero, I.A.; Serrano, H.; Rayo-Mares, D.; Fagiolino, P.; Vazquez, M.; Escamilla-Nunez, C.; Grijalva, I.; Escalante-Santiago, D.; Orozco-Suarez, S. Influence of genetic variants of CYP2D6, CYP2C9, CYP2C19 and CYP3A4 on antiepileptic drug metabolism in pediatric patients with refractory epilepsy. Pharmacol. Rep. 2017, 69, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Pastinen, T.; Hudson, T.J. Cis-acting regulatory variation in the human genome. Science 2004, 306, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Arefayene, M.; Skaar, T.C.; Zhao, X.; Rae, J.M.; Tanus-Santos, J.E.; Brinkmann, U.; Brehm, I.; Salat, U.; Nguyen, A.; Desta, Z.; et al. Sequence diversity and functional characterization of the 5′-regulatory region of human CYP2C19. Pharmacogenetics 2003, 13, 199–206. [Google Scholar] [CrossRef]
- McPherson, J.D.; Marra, M.; Hillier, L.; Waterston, R.H.; Chinwalla, A.; Wallis, J.; Sekhon, M.; Wylie, K.; Mardis, E.R.; Wilson, R.K.; et al. A physical map of the human genome. Nature 2001, 409, 934–941. [Google Scholar] [PubMed] [Green Version]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–933. [Google Scholar] [PubMed] [Green Version]
- Kirchheiner, J.; Seeringer, A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim. Biophys. Acta 2007, 1770, 489–494. [Google Scholar] [CrossRef]
- Fleming, I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol. Rev. 2014, 66, 1106–1140. [Google Scholar] [CrossRef]
- Spector, A.A.; Kim, H.Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Biophys. Acta 2015, 1851, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Sangkuhl, K.; Gardner, E.E.; Stein, C.M.; Hulot, J.S.; Johnson, J.A.; Roden, D.M.; Klein, T.E.; Shuldiner, A.R. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin. Pharmacol. Ther. 2011, 90, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Rosemary, J.; Surendiran, A.; Rajan, S.; Shashindran, C.H.; Adithan, C. Influence of the CYP2C9 AND CYP2C19 polymorphisms on phenytoin hydroxylation in healthy individuals from south India. Indian J. Med. Res. 2006, 123, 665–670. [Google Scholar] [PubMed]
- van der Weide, J.; Steijns, L.S.; van Weelden, M.J.; de Haan, K. The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogenetics 2001, 11, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Sadee, W. Pharmacogenomic biomarkers: Validation needed for both the molecular genetic mechanism and clinical effect. Pharmacogenomics 2011, 12, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Ensom, M.H.; Chang, T.K. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharm. 2005, 106, 97–132. [Google Scholar] [CrossRef]
- Brodie, M.J.; Mintzer, S.; Pack, A.M.; Gidal, B.E.; Vecht, C.J.; Schmidt, D. Enzyme induction with antiepileptic drugs: Cause for concern? Epilepsia 2013, 54, 11–27. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef]
- Wu, A.H. Drug metabolizing enzyme activities versus genetic variances for drug of clinical pharmacogenomic relevance. Clin. Proteom. 2011, 8, 12. [Google Scholar] [CrossRef]
- Tannenbaum, C.; Sheehan, N.L. Understanding and preventing drug-drug and drug-gene interactions. Expert Rev. Clin. Pharmacol. 2014, 7, 533–544. [Google Scholar] [CrossRef]
- Tod, M.; Goutelle, S.; Gagnieu, M.C.; Genophar, I.I.W.G. Genotype-based quantitative prediction of drug exposure for drugs metabolized by CYP2D6. Clin. Pharmacol. Ther. 2011, 90, 582–587. [Google Scholar] [CrossRef]
- Castellan, A.C.; Tod, M.; Gueyffier, F.; Audars, M.; Cambriels, F.; Kassai, B.; Nony, P.; Genophar Working, G. Quantitative prediction of the impact of drug interactions and genetic polymorphisms on cytochrome P450 2C9 substrate exposure. Clin. Pharmacokinet. 2013, 52, 199–209. [Google Scholar] [CrossRef]
- Goutelle, S.; Bourguignon, L.; Bleyzac, N.; Berry, J.; Clavel-Grabit, F.; Tod, M. In vivo quantitative prediction of the effect of gene polymorphisms and drug interactions on drug exposure for CYP2C19 substrates. AAPS J. 2013, 15, 415–426. [Google Scholar] [CrossRef]
- Romkes, M.; Faletto, M.B.; Blaisdell, J.A.; Raucy, J.L.; Goldstein, J.A. Cloning and expression of complementary DNAs for multiple members of the human cytochrome P450IIC subfamily. Biochemistry 1991, 30, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Gray, I.C.; Nobile, C.; Muresu, R.; Ford, S.; Spurr, N.K. A 2.4-megabase physical map spanning the CYP2C gene cluster on chromosome 10q24. Genomics 1995, 28, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Uehara, S.; Murayama, N.; Yamazaki, H. Cytochrome P450 1A1, 2C9, 2C19, and 3A4 Polymorphisms Account for Interindividual Variability of Toxicological Drug Metabolism in Cynomolgus Macaques. Chem. Res. Toxicol. 2018, 31, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Di Carlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef] [PubMed]
- Kasperaviciute, D.; Sisodiya, S.M. Epilepsy pharmacogenetics. Pharmacogenomics 2009, 10, 817–836. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, S.; Sisodiya, S.M. Pharmacogenomics in epilepsy. Neurosci. Lett. 2018, 667, 27–39. [Google Scholar] [CrossRef]
- Patsalos, P.N. Drug interactions with the newer antiepileptic drugs (AEDs)—Part 1: Pharmacokinetic and pharmacodynamic interactions between AEDs. Clin. Pharmacokinet. 2013, 52, 927–966. [Google Scholar] [CrossRef]
- Anderson, G.D.; Hakimian, S. Pharmacokinetic of antiepileptic drugs in patients with hepatic or renal impairment. Clin. Pharm. 2014, 53, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005, 5, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.C.; Kacevska, M.; Ingelman-Sundberg, M. Pharmacogenomics of drug-metabolizing enzymes: A recent update on clinical implications and endogenous effects. Pharm. J. 2013, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Preissner, S.C.; Hoffmann, M.F.; Preissner, R.; Dunkel, M.; Gewiess, A.; Preissner, S. Polymorphic cytochrome P450 enzymes (CYPs) and their role in personalized therapy. PLoS ONE 2013, 8, e82562. [Google Scholar] [CrossRef]
- Desta, Z.; Zhao, X.; Shin, J.G.; Flockhart, D.A. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet. 2002, 41, 913–958. [Google Scholar] [CrossRef]
- Rudberg, I.; Mohebi, B.; Hermann, M.; Refsum, H.; Molden, E. Impact of the ultrarapid CYP2C19*17 allele on serum concentration of escitalopram in psychiatric patients. Clin. Pharmacol. Ther. 2008, 83, 322–327. [Google Scholar] [CrossRef]
- Sibbing, D.; Koch, W.; Gebhard, D.; Schuster, T.; Braun, S.; Stegherr, J.; Morath, T.; Schomig, A.; von Beckerath, N.; Kastrati, A. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 2010, 121, 512–518. [Google Scholar] [CrossRef]
- Sibbing, D.; Gebhard, D.; Koch, W.; Braun, S.; Stegherr, J.; Morath, T.; Von Beckerath, N.; Mehilli, J.; Schomig, A.; Schuster, T.; et al. Isolated and interactive impact of common CYP2C19 genetic variants on the antiplatelet effect of chronic clopidogrel therapy. J. Thromb. Haemost. 2010, 8, 1685–1693. [Google Scholar] [CrossRef]
- Li-Wan-Po, A.; Girard, T.; Farndon, P.; Cooley, C.; Lithgow, J. Pharmacogenetics of CYP2C19: Functional and clinical implications of a new variant CYP2C19*17. Br. J. Clin. Pharmacol. 2010, 69, 222–230. [Google Scholar] [CrossRef]
- Wang, G.; Lei, H.P.; Li, Z.; Tan, Z.R.; Guo, D.; Fan, L.; Chen, Y.; Hu, D.L.; Wang, D.; Zhou, H.H. The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur. J. Clin. Pharmacol. 2009, 65, 281–285. [Google Scholar] [CrossRef]
- Sugimoto, K.; Uno, T.; Yamazaki, H.; Tateishi, T. Limited frequency of the CYP2C19*17 allele and its minor role in a Japanese population. Br. J. Clin. Pharmacol. 2008, 65, 437–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Morais, S.M.; Wilkinson, G.R.; Blaisdell, J.; Nakamura, K.; Meyer, U.A.; Goldstein, J.A. The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J. Biol. Chem. 1994, 269, 15419–15422. [Google Scholar] [PubMed]
- Rosemary, J.; Adithan, C. The pharmacogenetics of CYP2C9 and CYP2C19: Ethnic variation and clinical significance. Curr. Clin. Pharmacol. 2007, 2, 93–109. [Google Scholar] [CrossRef] [PubMed]
- De Morais, S.M.; Wilkinson, G.R.; Blaisdell, J.; Meyer, U.A.; Nakamura, K.; Goldstein, J.A. Identification of a new genetic defect responsible for the polymorphism of (S)-mephenytoin metabolism in Japanese. Mol. Pharmacol. 1994, 46, 594–598. [Google Scholar]
- van Schaik, R.H. CYP450 pharmacogenetics for personalizing cancer therapy. Drug Resist Updat. 2008, 11, 77–98. [Google Scholar] [CrossRef]
- Daly, A.K.; Brockmoller, J.; Broly, F.; Eichelbaum, M.; Evans, W.E.; Gonzalez, F.J.; Huang, J.D.; Idle, J.R.; Ingelman-Sundberg, M.; Ishizaki, T.; et al. Nomenclature for human CYP2D6 alleles. Pharmacogenetics 1996, 6, 193–201. [Google Scholar] [CrossRef]
- Ball, S.; Borman, N. Pharmacogenetics and drug metabolism. Nat. Biotechnol. 1997, 15, 925–926. [Google Scholar] [CrossRef]
- Pirmohamed, M.; Park, B.K. Genetic susceptibility to adverse drug reactions. Trends Pharmacol. Sci. 2001, 22, 298–305. [Google Scholar] [CrossRef]
- Levy, R.H. Cytochrome P450 isozymes and antiepileptic drug interactions. Epilepsia 1995, 36 (Suppl. 5), S8–S13. [Google Scholar] [CrossRef]
- Lee, C.R.; Goldstein, J.A.; Pieper, J.A. Cytochrome P450 2C9 polymorphisms: A comprehensive review of the in-vitro and human data. Pharmacogenetics 2002, 12, 251–263. [Google Scholar] [CrossRef]
- Prokop, J.W.; May, T.; Strong, K.; Bilinovich, S.M.; Bupp, C.; Rajasekaran, S.; Worthey, E.A.; Lazar, J. Genome sequencing in the clinic: The past, present, and future of genomic medicine. Physiol. Genomics 2018, 50, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Levy, S.; Huang, J.; Stockwell, T.B.; Walenz, B.P.; Li, K.; Axelrod, N.; Busam, D.A.; Strausberg, R.L.; Venter, J.C. Genetic variation in an individual human exome. PLoS Genet. 2008, 4, e1000160. [Google Scholar] [CrossRef] [PubMed]
- Shashi, A.; Kumar, M.; Bhardwaj, M. Incidence of skeletal deformities in endemic fluorosis. Trop. Doct. 2008, 38, 231–233. [Google Scholar]
- Leu, C.; Coppola, A.; Sisodiya, S.M. Progress from genome-wide association studies and copy number variant studies in epilepsy. Curr. Opin. Neurol. 2016, 29, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Mullen, S.A.; Berkovic, S.F.; Commission, I.G. Genetic generalized epilepsies. Epilepsia 2018, 59, 1148–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, S.; Johnson, M.; Bridgham, J.; Golda, G.; Lloyd, D.H.; Johnson, D.; Luo, S.; McCurdy, S.; Foy, M.; Ewan, M.; et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000, 18, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Kirkness, E.F. Whole genome sequencing. Methods Mol. Biol. 2010, 628, 215–226. [Google Scholar]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloglu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef] [Green Version]
- Ostrander, B.E.P.; Butterfield, R.J.; Pedersen, B.S.; Farrell, A.J.; Layer, R.M.; Ward, A.; Miller, C.; DiSera, T.; Filloux, F.M.; Candee, M.S.; et al. Whole-genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. NPJ Genom. Med. 2018, 3, 22. [Google Scholar] [CrossRef]
- Myers, K.A.; Johnstone, D.L.; Dyment, D.A. Epilepsy genetics: Current knowledge, applications, and future directions. Clin. Genet. 2019, 95, 95–111. [Google Scholar] [CrossRef]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Kasperaviciute, D.; Catarino, C.B.; Heinzen, E.L.; Depondt, C.; Cavalleri, G.L.; Caboclo, L.O.; Tate, S.K.; Jamnadas-Khoda, J.; Chinthapalli, K.; Clayton, L.M.; et al. Common genetic variation and susceptibility to partial epilepsies: A genome-wide association study. Brain 2010, 133, 2136–2147. [Google Scholar] [CrossRef] [PubMed]
- International League against Epilepsy Consortium on Complex Epilepsies. Electronic address e-auea. Genetic determinants of common epilepsies: A meta-analysis of genome-wide association studies. Lancet Neurol. 2014, 13, 893–903. [Google Scholar] [CrossRef]
- Heinzen, E.L.; Depondt, C.; Cavalleri, G.L.; Ruzzo, E.K.; Walley, N.M.; Need, A.C.; Ge, D.; He, M.; Cirulli, E.T.; Zhao, Q.; et al. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am. J. Hum. Genet. 2012, 91, 293–302. [Google Scholar] [CrossRef]
- Buono, R.J. Genome wide association studies (GWAS) and common forms of human epilepsy. Epilepsy Behav. 2013, 28 (Suppl. 1), S63–S65. [Google Scholar] [CrossRef] [Green Version]
- Mamiya, K.; Ieiri, I.; Shimamoto, J.; Yukawa, E.; Imai, J.; Ninomiya, H.; Yamada, H.; Otsubo, K.; Higuchi, S.; Tashiro, N. The effects of genetic polymorphisms of CYP2C9 and CYP2C19 on phenytoin metabolism in Japanese adult patients with epilepsy: Studies in stereoselective hydroxylation and population pharmacokinetics. Epilepsia 1998, 39, 1317–1323. [Google Scholar] [CrossRef]
- Martin, H.C.; Kim, G.E.; Pagnamenta, A.T.; Murakami, Y.; Carvill, G.L.; Meyer, E.; Copley, R.R.; Rimmer, A.; Barcia, G.; Fleming, M.R.; et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 2014, 23, 3200–3211. [Google Scholar] [CrossRef]
- Helbig, I.; Swinkels, M.E.; Aten, E.; Caliebe, A.; van’t Slot, R.; Boor, R.; von Spiczak, S.; Muhle, H.; Jahn, J.A.; van Binsbergen, E.; et al. Structural genomic variation in childhood epilepsies with complex phenotypes. Eur. J. Hum. Genet. 2014, 22, 896–901. [Google Scholar] [CrossRef]
- Magni, F.; Van Der Burgt, Y.E.; Chinello, C.; Mainini, V.; Gianazza, E.; Squeo, V.; Deelder, A.M.; Kienle, M.G. Biomarkers discovery by peptide and protein profiling in biological fluids based on functionalized magnetic beads purification and mass spectrometry. Blood Transfus. 2010, 8 (Suppl. 3), s92–s97. [Google Scholar]
- Aronica, E.; Fluiter, K.; Iyer, A.; Zurolo, E.; Vreijling, J.; van Vliet, E.A.; Baayen, J.C.; Gorter, J.A. Expression pattern of miR-146a, an inflammation-associated microRNA, in experimental and human temporal lobe epilepsy. Eur. J. Neurosci. 2010, 31, 1100–1107. [Google Scholar] [CrossRef]
- Miller-Delaney, S.F.; Bryan, K.; Das, S.; McKiernan, R.C.; Bray, I.M.; Reynolds, J.P.; Gwinn, R.; Stallings, R.L.; Henshall, D.C. Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain 2015, 138, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, H.B.; Bian, J.; Li, B.B.; Gong, T.F. Genetic association between TNF-alpha -857 C/T polymorphism and ankylosing spondylitis susceptibility: Evidence from a meta-analysis. Springerplus 2016, 5, 1930. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Tao, H.; Wang, Y.; Liu, Z.; Xu, Z.; Zhou, H.; Cai, Y.; Yao, L.; Chen, B.; Liang, W.; et al. A functional polymorphism of the microRNA-146a gene is associated with susceptibility to drug-resistant epilepsy and seizures frequency. Seizure 2015, 27, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ailhaud, G. Adipose tissue as a secretory organ: From adipogenesis to the metabolic syndrome. C. R. Biol. 2006, 329, 570–577; discussion 653–655. [Google Scholar] [CrossRef]
- Sorisky, A.; Molgat, A.S.; Gagnon, A. Macrophage-induced adipose tissue dysfunction and the preadipocyte: Should I stay (and differentiate) or should I go? Adv. Nutr. 2013, 4, 67–75. [Google Scholar] [CrossRef]
- Khalyfa, A.; Gozal, D. Exosomal miRNAs as potential biomarkers of cardiovascular risk in children. J. Transl. Med. 2014, 12, 162. [Google Scholar] [CrossRef]
- Khalyfa, A.; Gozal, D.; Kheirandish-Gozal, L. Plasma Exosomes Disrupt the Blood-Brain Barrier in Children with Obstructive Sleep Apnea and Neurocognitive Deficits. Am. J. Respir. Crit. Care Med. 2018, 197, 1073–1076. [Google Scholar] [CrossRef]
- Khalyfa, A.; Kheirandish-Gozal, L.; Gozal, D. Circulating exosomes in obstructive sleep apnea as phenotypic biomarkers and mechanistic messengers of end-organ morbidity. Respir. Physiol. Neurobiol. 2018, 256, 143–156. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef]
- Stepanian, A.; Bourguignat, L.; Hennou, S.; Coupaye, M.; Hajage, D.; Salomon, L.; Alessi, M.C.; Msika, S.; de Prost, D. Microparticle increase in severe obesity: Not related to metabolic syndrome and unchanged after massive weight loss. Obes. (Silver Spring) 2013, 21, 2236–2243. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Shearn, A.I.; Angelini, G.D.; Sahoo, S. Exosomes and exosomal miRNAs in cardiovascular protection and repair. Vascul. Pharmacol. 2015, 71, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Lakhal, S.; Wood, M.J. Exosome nanotechnology: An emerging paradigm shift in drug delivery: Exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays 2011, 33, 737–741. [Google Scholar] [CrossRef]
- Khalyfa, A.; Kheirandish-Gozal, L.; Khalyfa, A.A.; Philby, M.F.; Alonso-Alvarez, M.L.; Mohammadi, M.; Bhattacharjee, R.; Teran-Santos, J.; Huang, L.; Andrade, J.; et al. Circulating Plasma Extracellular Microvesicle MicroRNA Cargo and Endothelial Dysfunction in Children with Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2016, 194, 1116–1126. [Google Scholar] [CrossRef] [Green Version]
- Thuringer, D.; Garrido, C. Molecular chaperones in the brain endothelial barrier: Neurotoxicity or neuroprotection? Faseb. J. 2019, fj201900895R. [Google Scholar] [CrossRef]
- Marino Gammazza, A.; Colangeli, R.; Orban, G.; Pierucci, M.; Di Gennaro, G.; Lo Bello, M.; D’Aniello, A.; Bucchieri, F.; Pomara, C.; Valentino, M.; et al. Hsp60 response in experimental and human temporal lobe epilepsy. Sci. Rep. 2015, 5, 9434. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Andrews, A.M.; Paul, D.; Pachter, J.S. Extracellular vesicles: Mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS 2018, 15, 19. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, H.S. Correction to: Extracellular Vesicles in Neurodegenerative Diseases: A Double-Edged Sword. Tissue Eng. Regen. Med. 2018, 15, 127. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The Transport Mechanism of Extracellular Vesicles at the Blood-Brain Barrier. Curr. Pharm. Des. 2017, 23, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; Pereira de Almeida, L. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Lee, W.T.; Lee, C.C.; Jeng, S.F.; Weng, W.C. Sleep in children with epilepsy: The role of maternal knowledge of childhood sleep. Sleep 2018, 41, zsy157. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Wical, B.; Wical, W.; Schaffer, L.; Wical, T.; Wendorf, H.; Roiko, S. Obstructive sleep apnea in children with cerebral palsy and epilepsy. Dev. Med. Child Neurol. 2016, 58, 1057–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raoof, R.; Jimenez-Mateos, E.M.; Bauer, S.; Tackenberg, B.; Rosenow, F.; Lang, J.; Onugoren, M.D.; Hamer, H.; Huchtemann, T.; Kortvelyessy, P.; et al. Cerebrospinal fluid microRNAs are potential biomarkers of temporal lobe epilepsy and status epilepticus. Sci. Rep. 2017, 7, 3328. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, H.; Xie, W.; Meng, F.; Zhang, K.; Jiang, Y.; Zhang, X.; Zhang, J. Altered microRNA profiles in plasma exosomes from mesial temporal lobe epilepsy with hippocampal sclerosis. Oncotarget 2017, 8, 4136–4146. [Google Scholar] [CrossRef]
- Karttunen, J.; Heiskanen, M.; Lipponen, A.; Poulsen, D.; Pitkanen, A. Extracellular Vesicles as Diagnostics and Therapeutics for Structural Epilepsies. Int. J. Mol. Sci. 2019, 20, 1259. [Google Scholar] [CrossRef]
- Mehdizadeh, A.; Barzegar, M.; Negargar, S.; Yahyavi, A.; Raeisi, S. The current and emerging therapeutic approaches in drug-resistant epilepsy management. Acta Neurol. Belg. 2019, 119, 155–162. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence Homology | KEGG Pathway | Gene Ontology |
|---|---|---|---|
| GABAR1 (NM_000806.5) | GAB type B receptor subunit 1 isoform X2, (XP_006715110.1), GAB type B receptor subunit 1 isoform X3 (XP_011512755.1), GAB type B receptor subunit 1 isoform a precursor (NP_001461.1), GAB type B receptor subunit 1 isoform X1 (XP_005249039.1), GAB type B receptor subunit 1 isoform b precursor (NP_068703.1), GAB type B receptor subunit 1 isoform k (NP_001305982.1), GAB type B receptor subunit 1 isoform X4 (XP_024302160.1), GAB type B receptor subunit 1 isoform X5 (XP_011512757.1), GAB type B receptor subunit 2 precursor (NP_005449.5), and GAB type B receptor subunit 2 isoform X1 (XP_016870820.1) | GABA A receptor activation, organism-specific biosystems, ion channel transport, organism-specific biosystem, ligand-gated ion channel transport, organism-specific biosystem, morphine addiction, organism-specific biosystem, neuronal system, organism-specific biosystem, and neuroactive ligand-receptor interaction, organism-specific biosystem | Cellular components: plasma membrane (GO:0005886), integral to plasma membrane (GO:0005887), membrane (GO:0016020), integral to membrane (GO:0016021), cell junction (GO:0030054), chloride channel complex (GO:0034707), and postsynaptic membrane (GO:0045211). Biological process: transport (GO:0006810), ion transport (GO:0006811), gamma-aminobutyric acid signaling pathway (GO:0007214), synaptic transmission (GO:0007268), ion transmembrane transport (GO:0034220), synaptic transmission, GABAergic (GO:0051932), transmembrane transport (GO:0055085). Molecular function: GABAA receptor activity (GO:0004890), extracellular ligand-gated ion channel activity (GO:0005230), chloride channel activity (GO:0005254), protein binding (GO:0005515), drug binding (GO:0008144), and GABA receptor activity (GO:0016917). |
| GABBR1 (NM_000812) | GAB subunit beta-1 isoform X1 (XP_024309744.1), GAB receptor subunit beta-2 isoform 2 precursor (NP_000804.1), GAB receptor subunit beta-3 isoform 1 precursor (NP_000805.1), GAB receptor subunit beta-2 isoform 1 precursor (NP_068711.1), GAB receptor subunit beta-3 isoform 2 precursor (NP_068712.1), GAB receptor subunit beta-3 isoform 4 (NP_001178250.1), GAB receptor subunit beta-3 isoform X1(XP_011519730.1), GAB receptor subunit beta-3 isoform 3 (NP_001178249.1), GAB receptor subunit beta-1 isoform X2 (XP_016863474.1), and GAB subunit theta isoform X1 (XP_011529486.1) | GABAergic synapse, morphine addiction, neuroactive ligand-receptor interaction, nicotine addiction, retrograde endocannabinoid signaling, and serotonergic synapse | Cellular components: nucleus (GO:0005634), nuclear envelope (GO:0005635), cytoplasm (GO:0005737), plasma membrane (GO:0005886), and integral component of plasma membrane (GO:0005887). Biological process: ion transport (GO:0006811), chloride transport (GO:0006821), signal transduction (GO:0007165), gamma-aminobutyric acid signaling pathway (GO:0007214), and chemical synaptic transmission (GO:0007268). Molecular functions are GABA-A receptor activity (GO:0004890), ion channel activity (GO:0005216), extracellular ligand-gated ion channel activity (GO:0005230), anion channel activity (GO:0005253), and chloride channel activity (GO:0005254). |
| GABBR2, GABA-C (NM_002043.4) | GAB receptor subunit rho-2 isoform X1 (XP_011534015.1), GAB receptor subunit rho-2 isoform X2 (XP_011534016.1), GAB receptor subunit rho-1 isoform b precursor (NP_001243632.1), GAB receptor subunit rho-1 isoform a precursor (NP_002033.2), GAB receptor subunit rho-1 isoform c (NP_001243633.1), GAB receptor subunit rho-3 precursor (NP_001099050.1), GAB receptor subunit beta-3 isoform 1 precursor (NP_000805.1), GAB receptor subunit beta-3 isoform 2 precursor (NP_068712.1), GAB receptor subunit beta-1 isoform X1 (XP_024309744.1), and GAB receptor subunit beta-1 precursor (NP_000803.2). | GABAergic synapse, morphine addiction, neuroactive ligand-receptor interaction, nicotine addiction, and retrograde endocannabinoid signaling | Cellular components: plasma membrane (GO:0005886), integral component of plasma membrane (GO:0005887), membrane (GO:0016020), integral component of membrane (GO:0016021), and cell junction (GO:0030054). Biological process: ion transport (GO:0006811), chloride transport (GO:0006821), signal transduction (GO:0007165), gamma-aminobutyric acid signaling pathway (GO:0007214), and chemical synaptic transmission (GO:0007268). Molecular function: GABA-A receptor activity (GO:0004890), ion channel activity (GO:0005216), extracellular ligand-gated ion channel activity (GO:0005230), chloride channel activity (GO:0005254), protein domain specific binding (GO:0019904). |
| exon | c.startExon | c.endExon | g.startExon | g.endExon | lengthExon | lengthIntron |
|---|---|---|---|---|---|---|
| 1 | 1 | 168 | 5001 | 5168 | 168 | 12,184 |
| 2 | 169 | 331 | 17,353 | 17,515 | 163 | 169 |
| 3 | 332 | 481 | 17,685 | 17,834 | 150 | 4959 |
| 4 | 482 | 642 | 22,794 | 22,954 | 161 | 1161 |
| 5 | 643 | 819 | 24,116 | 24,292 | 177 | 38,498 |
| 6 | 820 | 961 | 62,791 | 62,932 | 142 | 22,199 |
| 7 | 962 | 1149 | 85,132 | 85,319 | 188 | 6892 |
| 8 | 1150 | 1291 | 92,212 | 92,353 | 142 | 2674 |
| 9 | 1292 | 0 | 95028 | 95,209 | 182 |
| Chr. Position | mRNA Position | dbSNP rs# | Allele Name | Heterozygosity | MAF | Function | dbSNP Allele | Protein Residue | Amino Zcid Position |
|---|---|---|---|---|---|---|---|---|---|
| 94762706 | 1 | rs28399504 | CYP2C19*4 | 0.004 | 0.0008 | missense | G | Val [V] | 1 |
| 94775165 | 276 | rs17878459 | CYP2C19*17 | 0.046 | 0.009 | missense | C | Asp [D] | 92 |
| 94780653 | 636 | rs4986893 | CYP2C19*3 | 0.011 | 0.0142 | nonsense | A | Trp [W] | 212 |
| 94781859 | 681 | rs4244285 | CYP2C19*2 | 0.302 | 0.2214 | synonymous | A | Pro [P] | 227 |
| 94852738 | 1297 | rs56337013 | CYP2C19*5 | 0 | missense | T | Trp [W] | 433 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalyfa, A.; Sanz-Rubio, D. Genetics and Extracellular Vesicles of Pediatrics Sleep Disordered Breathing and Epilepsy. Int. J. Mol. Sci. 2019, 20, 5483. https://doi.org/10.3390/ijms20215483
Khalyfa A, Sanz-Rubio D. Genetics and Extracellular Vesicles of Pediatrics Sleep Disordered Breathing and Epilepsy. International Journal of Molecular Sciences. 2019; 20(21):5483. https://doi.org/10.3390/ijms20215483
Chicago/Turabian StyleKhalyfa, Abdelnaby, and David Sanz-Rubio. 2019. "Genetics and Extracellular Vesicles of Pediatrics Sleep Disordered Breathing and Epilepsy" International Journal of Molecular Sciences 20, no. 21: 5483. https://doi.org/10.3390/ijms20215483