Non-Lytic Antibacterial Peptides That Translocate Through Bacterial Membranes to Act on Intracellular Targets

 , , and
, , and
Abstract
:
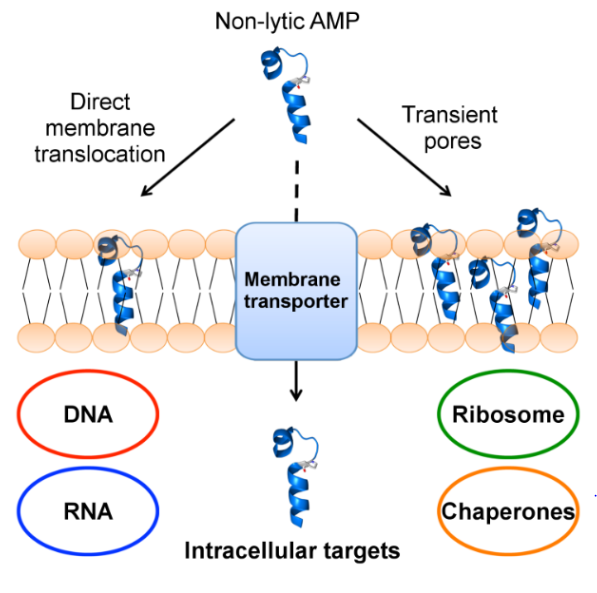
1. Introduction
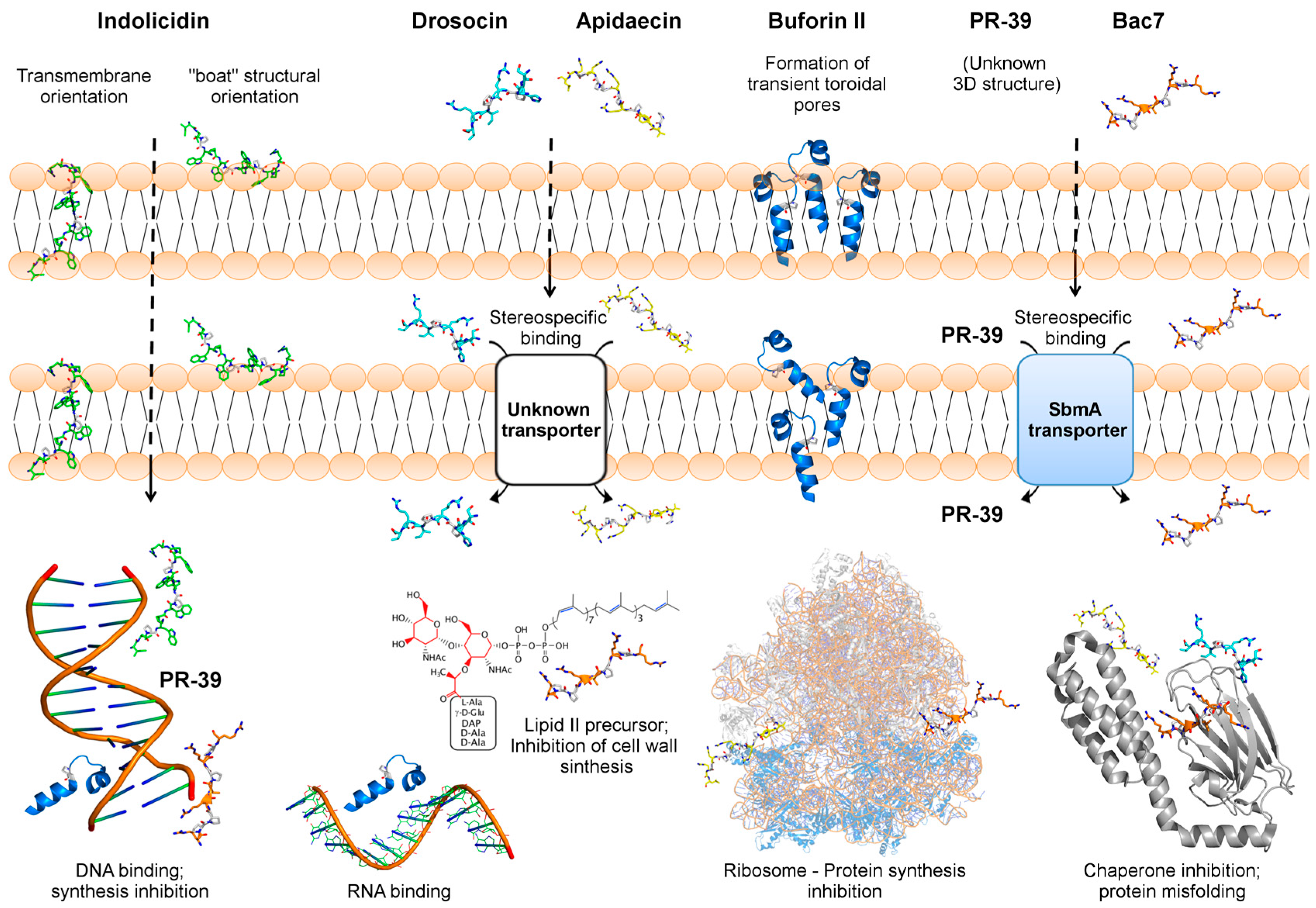
2. Indolicidin—A Tryptophan/Proline-Rich Peptide
2.1. Indolicidin Interacts with and Translocates through Bacterial Membranes
2.2. Indolicidin Antibacterial Properties
2.3. Indolicidin Targets Bacterial DNA
3. Buforin II—A Frog-Derived Peptide that Internalizes Bacterial Cells
3.1. Buforin II Translocates Membranes by the Formation Of Transient Toroidal Pores
3.2. Buforin II as a Promising Scaffold for Antibacterial Therapies
3.3. Buforin II Targets DNA and RNA
4. PR-39 and bac7—Two Proline/Arginine-Rich Peptides
4.1. PR-39 and bac7 Membrane Translocation Require an Inner Membrane Transporter
4.2. PR-39 Antibacterial Properties
4.3. Bac7 Antibacterial Properties
4.4. PR-39 and bac7 Target Bacterial Protein Synthesis
5. Apidaecin and Drosocin—Two Non-Lytic AMPs Derived from Insects
5.1. Apidaecin and Drosocin Depend on Membrane Receptors to Internalize the Target Cell
5.2. Apidaecin Antibacterial Properties
5.3. Drosocin Antibacterial Properties
5.4. Apidaecin and Drosocin Interact with Bacterial Chaperones
6. Conclusions and Future Prospects
Funding
Conflicts of Interest
References
- Shrivastava, S.; Shrivastava, P.; Ramasamy, J. World health organization releases global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. J. Med. Soc. 2018, 32, 76. [Google Scholar] [CrossRef]
- El-Halfawy, O.M.; Valvano, M.A. Antimicrobial heteroresistance: An emerging field in need of clarity. Clin. Microbiol. Rev. 2015, 28, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Nicoloff, H.; Hjort, K.; Levin, B.R.; Andersson, D.I. The high prevalence of antibiotic heteroresistance in pathogenic bacteria is mainly caused by gene amplification. Nat. Microbiol. 2019, 4, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, J.P. Cationic antimicrobial peptides. BioDrugs 2003, 17, 233–240. [Google Scholar] [CrossRef]
- Izadpanah, A.; Gallo, R.L. Antimicrobial peptides. J. Am. Acad. Dermatol. 2005, 52, 381–390. [Google Scholar] [CrossRef]
- Shagaghi, N.; Palombo, E.A.; Clayton, A.H.A.; Bhave, M. Antimicrobial peptides: Biochemical determinants of activity and biophysical techniques of elucidating their functionality. World J. Microbiol. Biotechnol. 2018, 34, 62. [Google Scholar] [CrossRef]
- Henriques, S.T.; Melo, M.N.; Castanho, M.A.R.B. Cell-penetrating peptides and antimicrobial peptides: How different are they? Biochem. J. 2006, 399, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Pep. Sci. 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta Biomembr. 1998, 1376, 391–400. [Google Scholar] [CrossRef]
- Huang, H.W. Action of antimicrobial peptides: Two-state model. Biochemistry 2000, 39, 8347–8352. [Google Scholar] [CrossRef] [PubMed]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular targeting mechanisms by pntimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef] [PubMed]
- Neundorf, I. Antimicrobial and cell-penetrating peptides: How to understand two distinct functions despite similar physicochemical properties. In Antimicrobial Peptides. Advances in Experimental Medicine and Biology; Matsuzaki, K., Ed.; Springer: Singapore, 2019; Volume 1117, pp. 93–109. [Google Scholar]
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-rich antimicrobial peptides: Converging to a non-lytic mechanism of action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. The short proline-rich antibacterial peptide family. Cell. Mol. Life Sci. 2002, 59, 1138–1150. [Google Scholar] [CrossRef]
- Ulmschneider, J.P. Charged antimicrobial peptides can translocate across membranes without forming channel-like pores. Biophys. J. 2017, 113, 73–81. [Google Scholar] [CrossRef]
- Friedrich, C.L.; Rozek, A.; Patrzykat, A.; Hancock, R.E. Structure and mechanism of action of an indolicidin peptide derivative with improved activity against gram-positive bacteria. J. Biol. Chem. 2001, 276, 24015–24022. [Google Scholar] [CrossRef]
- Ulvatne, H.; Samuelsen, Ø.; Haukland, H.H.; Krämer, M.; Vorland, L.H. Lactoferricin B inhibits bacterial macromolecular synthesis in Escherichia coli and Bacillus subtilis. FEMS Microbiol. Lett. 2004, 237, 377–384. [Google Scholar] [CrossRef]
- Graf, M.; Wilson, D.N. Intracellular antimicrobial peptides targeting the protein synthesis machinery. Adv. Exp. Med. Biol. 2019, 1117, 73–89. [Google Scholar] [PubMed]
- Selsted, M.E.; Novotny, M.J.; Morris, W.L.; Tang, Y.Q.; Smith, W.; Cullor, J.S. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 1992, 267, 4292–4295. [Google Scholar] [PubMed]
- Mishra, A.K.; Choi, J.; Moon, E.; Baek, K.H. Tryptophan-rich and proline-rich antimicrobial peptides. Molecules 2018, 23, 815. [Google Scholar] [CrossRef] [PubMed]
- Falla, T.J.; Karunaratne, D.N.; Hancock, R.E. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 1996, 271, 19298–19303. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Selsted, M.E.; White, S.H. Bilayer interactions of indolicidin, a small antimicrobial peptide rich in tryptophan, proline, and basic amino acids. Biophys. J. 1997, 72, 794–805. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Selsted, M.E.; White, S.H. CD spectra of indolicidin antimicrobial peptides suggest turns, not polyproline helix. Biochemistry 1999, 38, 12313–12319. [Google Scholar] [CrossRef]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Lee, A.Y.; Lin, Y.C.; Yu, Y.P.; Huang, W.T.; Wu, S.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef]
- Hsu, J.C.; Yip, C.M. Molecular dynamics simulations of indolicidin association with model lipid bilayers. Biophys. J. 2007, 92, L100–L102. [Google Scholar] [CrossRef]
- Tsai, C.W.; Lin, Z.W.; Chang, W.F.; Chen, Y.F.; Hu, W.W. Development of an indolicidin-derived peptide by reducing membrane perturbation to decrease cytotoxicity and maintain gene delivery ability. Colloids Surf. B. 2018, 165, 18–27. [Google Scholar] [CrossRef]
- Ghosh, A.; Kar, R.K.; Jana, J.; Saha, A.; Jana, B.; Krishnamoorthy, J.; Kumar, D.; Ghosh, S.; Chatterjee, S.; Bhunia, A. Indolicidin targets duplex DNA: Structural and mechanistic insight through a combination of spectroscopy and microscopy. ChemMedChem 2014, 9, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.S.; Park, C.B.; Kim, S.C.; Cheong, C. Solution structure of an antimicrobial peptide buforin II. FEBS Lett. 1996, 398, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Park, C.B.; Yi, K.S.; Matsuzaki, K.; Kim, M.S.; Kim, S.C. Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin II. Proc. Natl. Acad. Sci. USA 2000, 97, 8245–8250. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Chikushi, A.; Tougu, S.; Imura, Y.; Nishida, M.; Yano, Y.; Matsuzaki, K. Membrane translocation mechanism of the antimicrobial peptide buforin 2. Biochemistry 2004, 43, 15610–15616. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Takeshima, K.; Park, C.B.; Kim, S.C.; Matsuzaki, K. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: Proline as a translocation promoting factor. Biochemistry 2000, 39, 8648–8654. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Skerlavaj, B.; Romeo, D.; Gennaro, R. Rapid membrane permeabilization and inhibition of vital functions of gram-negative bacteria by bactenecins. Infect. Immun. 1990, 58, 3724–3730. [Google Scholar]
- Mardirossian, M.; Grzela, R.; Giglione, C.; Meinnel, T.; Gennaro, R.; Mergaert, P.; Scocchi, M. The host antimicrobial peptide Bac71–35 binds to bacterial ribosomal proteins and inhibits protein synthesis. Chem. Biol. 2014, 21, 1639–1647. [Google Scholar] [CrossRef]
- Runti, G.; Lopez Ruiz Mdel, C.; Stoilova, T.; Hussain, R.; Jennions, M.; Choudhury, H.G.; Benincasa, M.; Gennaro, R.; Beis, K.; Scocchi, M. Functional characterization of SbmA, a bacterial inner membrane transporter required for importing the antimicrobial peptide Bac7(1–35). J. Bacteriol. 2013, 195, 5343–5351. [Google Scholar] [CrossRef]
- de Leeuw, E.; Li, C.; Zeng, P.; Li, C.; Buin, M.D.-d.; Lu, W.-Y.; Breukink, E.; Lu, W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010, 584, 1543–1548. [Google Scholar] [CrossRef] [Green Version]
- Runti, G.; Benincasa, M.; Giuffrida, G.; Devescovi, G.; Venturi, V.; Gennaro, R.; Scocchi, M. The Mechanism of killing by the proline-rich peptide bac7(1–35) against clinical strains of Pseudomonas aeruginosa differs from that against other gram-negative bacteria. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Li, W.F.; Ma, G.X.; Zhou, X.X. Apidaecin-type peptides: Biodiversity, structure-function relationships and mode of action. Peptides 2006, 27, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Bulet, P.; Dimarcq, J.L.; Hetru, C.; Lagueux, M.; Charlet, M.; Hegy, G.; Van Dorsselaer, A.; Hoffmann, J.A. A novel inducible antibacterial peptide of Drosophila carries an O-glycosylated substitution. J. Biol. Chem. 1993, 268, 14893–14897. [Google Scholar] [PubMed]
- Casteels, P.; Tempst, P. Apidaecin-type peptide antibiotics function through a non-poreforming mechanism involving stereospecificity. Biochem. Biophys. Res. Commun. 1994, 199, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Piantavigna, S.; Czihal, P.; Mechler, A.; Richter, M.; Hoffmann, R.; Martin, L.L. Cell penetrating apidaecin peptide interactions with biomimetic phospholipid membranes. I. J. P. R. Ther. 2009, 15, 139–146. [Google Scholar] [CrossRef]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial peptide structure and mechanism of action: A focus on the role of membrane structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Lele, D.S.; Talat, S.; Kumari, S.; Srivastava, N.; Kaur, K.J. Understanding the importance of glycosylated threonine and stereospecific action of Drosocin, a proline rich antimicrobial peptide. Eur. J. Med. Chem. 2015, 92, 637–647. [Google Scholar] [CrossRef]
- McManus, A.M.; Otvos, L.; Hoffmann, R.; Craik, D.J. Conformational studies by NMR of the antimicrobial peptide, drosocin, and its non-glycosylated derivative: Effects of glycosylation on solution conformation. Biochemistry 1999, 38, 705–714. [Google Scholar] [CrossRef]
- Lele, D.S.; Kaur, G.; Thiruvikraman, M.; Kaur, K.J. Comparing naturally occurring glycosylated forms of proline rich antibacterial peptide, Drosocin. Glycoconj. J. 2017, 34, 613–624. [Google Scholar] [CrossRef]
- Subbalakshmi, C.; Krishnakumari, V.; Nagaraj, R.; Sitaram, N. Requirements for antibacterial and hemolytic activities in the bovine neutrophil derived 13-residue peptide indolicidin. FEBS Lett. 1996, 395, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Subbalakshmi, C.; Bikshapathy, E.; Sitaram, N.; Nagaraj, R. Antibacterial and hemolytic activities of single tryptophan analogs of indolicidin. Biochem. Biophys. Res. Commun. 2000, 274, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Ryge, T.S.; Doisy, X.; Ifrah, D.; Olsen, J.E.; Hansen, P.R. New indolicidin analogues with potent antibacterial activity. J. Pep. Res. 2004, 64, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Frecer, V. QSAR analysis of antimicrobial and haemolytic effects of cyclic cationic antimicrobial peptides derived from protegrin-1. Bioorg. Med. Chem. 2006, 14, 6065–6074. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Hong, J.; Shai, Y. A repertoire of novel antibacterial diastereomeric peptides with selective cytolytic activity. J. Biol. Chem. 1997, 272, 14643–14649. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, J.M.; Joshi, B.P.; Cho, H.; Lee, K.H. Indolicidin-derived antimicrobial peptide analogs with greater bacterial selectivity and requirements for antibacterial and hemolytic activities. Biochim. Biophys. Acta 2009, 1794, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Jindal, H.M.; Le, C.F.; Mohd Yusof, M.Y.; Velayuthan, R.D.; Lee, V.S.; Zain, S.M.; Isa, D.M.; Sekaran, S.D. Antimicrobial activity of novel synthetic peptides derived from indolicidin and ranalexin against Streptococcus pneumoniae. PLoS ONE 2015, 10, e0128532. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.P.; Durell, S.; Maloy, W.L.; Zasloff, M. Ranalexin. A novel antimicrobial peptide from bullfrog (Rana catesbeiana) skin, structurally related to the bacterial antibiotic, polymyxin. J. Biol. Chem. 1994, 269, 10849–10855. [Google Scholar]
- Pompilio, A.; Scocchi, M.; Pomponio, S.; Guida, F.; Di Primio, A.; Fiscarelli, E.; Gennaro, R.; Di Bonaventura, G. Antibacterial and anti-biofilm effects of cathelicidin peptides against pathogens isolated from cystic fibrosis patients. Peptides 2011, 32, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Dosler, S.; Mataraci, E. In vitro pharmacokinetics of antimicrobial cationic peptides alone and in combination with antibiotics against methicillin resistant Staphylococcus aureus biofilms. Peptides 2013, 49, 53–58. [Google Scholar] [CrossRef]
- Park, C.B.; Kim, M.S.; Kim, S.C. A novel antimicrobial peptide from Bufo bufo gargarizans. Biochem. Biophys. Res. Commun. 1996, 218, 408–413. [Google Scholar] [CrossRef]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial peptides: Interaction with model and biological membranes and synergism with chemical antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Cirioni, O.; Silvestri, C.; Ghiselli, R.; Orlando, F.; Riva, A.; Gabrielli, E.; Mocchegiani, F.; Cianforlini, N.; Trombettoni, M.M.; Saba, V.; et al. Therapeutic efficacy of buforin II and rifampin in a rat model of Acinetobacter baumannii sepsis. Crit. Care Med. 2009, 37, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, Y. Synergistic effect of clinically used antibiotics and peptide antibiotics against Gram-positive and Gram-negative bacteria. Exp. Ther. Med. 2013, 6, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Shi, Y.H.; Tang, Y.L.; Le, G.W. The intracellular mechanism of action on Escherichia coli of BF2-A/C, two analogues of the antimicrobial peptide Buforin 2. J. Microbiol. 2013, 51, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.A.; Kim, H.; Lee, J.Y.; Shin, J.R.; Kim, D.J.; Cho, J.H.; Kim, S.C. Mechanism of action and specificity of antimicrobial peptides designed based on buforin IIb. Peptides 2012, 34, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.M.; Bonke, G.; Larsen, C.J.; Yavari, N.; Nielsen, P.E.; Franzyk, H. Antibacterial peptide nucleic acid-antimicrobial peptide (PNA-AMP) conjugates: Antisense targeting of fatty acid biosynthesis. Bioconjugate Chem. 2016, 27, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Veldhuizen, E.J.; Schneider, V.A.; Agustiandari, H.; van Dijk, A.; Tjeerdsma-van Bokhoven, J.L.; Bikker, F.J.; Haagsman, H.P. Antimicrobial and immunomodulatory activities of PR-39 derived peptides. PLoS ONE 2014, 9, e95939. [Google Scholar] [CrossRef]
- Shi, J.; Ross, C.R.; Leto, T.L.; Blecha, F. PR-39, a proline-rich antibacterial peptide that inhibits phagocyte NADPH oxidase activity by binding to Src homology 3 domains of p47 phox. Proc. Natl. Acad. Sci. USA 1996, 93, 6014–6018. [Google Scholar] [CrossRef]
- James, P.E.; Madhani, M.; Ross, C.; Klei, L.; Barchowsky, A.; Swartz, H.M. Tissue hypoxia during bacterial sepsis is attenuated by PR-39, an antibacterial peptide. Adv. Exp. Med. Biol. 2003, 530, 645–652. [Google Scholar]
- Ghiselli, R.; Giacometti, A.; Cirioni, O.; Circo, R.; Mocchegiani, F.; Skerlavaj, B.; D’Amato, G.; Scalise, G.; Zanetti, M.; Saba, V. Neutralization of endotoxin in vitro and in vivo by Bac7(1–35), a proline-rich antibacterial peptide. Shock 2003, 19, 577–581. [Google Scholar] [CrossRef]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar] [PubMed]
- Ho, Y.-H.; Shah, P.; Chen, Y.-W.; Chen, C.-S. Systematic analysis of intracellular-targeting antimicrobial peptides, bactenecin 7, hybrid of pleurocidin and dermaseptin, proline–arginine-rich peptide, and lactoferricin B, by using Escherichia coli proteome microarrays. Mol. Cell. Proteom. 2016, 15, 1837–1847. [Google Scholar] [CrossRef]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A. Proline-rich antimicrobial peptides targeting protein synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Berthold, N.; Czihal, P.; Fritsche, S.; Sauer, U.; Schiffer, G.; Knappe, D.; Alber, G.; Hoffmann, R. Novel apidaecin 1b analogs with superior serum stabilities for treatment of infections by gram-negative pathogens. Antimicrob. Agents Chemother. 2013, 57, 402–409. [Google Scholar] [CrossRef]
- Krizsan, A.; Volke, D.; Weinert, S.; Strater, N.; Knappe, D.; Hoffmann, R. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome. Angew. Chem. Int. Ed. Engl. 2014, 53, 12236–12239. [Google Scholar] [CrossRef]
- Scocchi, M.; Mardirossian, M.; Runti, G.; Benincasa, M. Non-membrane permeabilizing modes of action of antimicrobial peptides on bacteria. Curr. Top. Med. Chem. 2016, 16, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Castle, M.; Nazarian, A.; Yi, S.S.; Tempst, P. Lethal effects of apidaecin on Escherichia coli involve sequential molecular interactions with diverse targets. J. Biol. Chem. 1999, 274, 32555–32564. [Google Scholar] [CrossRef]
- Otvos, L.; O, I.; Rogers, M.E.; Consolvo, P.J.; Condie, B.A.; Lovas, S.; Bulet, P.; Blaszczyk-Thurin, M. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar] [CrossRef]
- Haney, E.F.; Petersen, A.P.; Lau, C.K.; Jing, W.; Storey, D.G.; Vogel, H.J. Mechanism of action of puroindoline derived tryptophan-rich antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1802–1813. [Google Scholar] [CrossRef] [Green Version]
- Yonezawa, A.; Kuwahara, J.; Fujii, N.; Sugiura, Y. Binding of tachyplesin I to DNA revealed by footprinting analysis: Significant contribution of secondary structure to DNA binding and implication for biological action. Biochemistry 1992, 31, 2998–3004. [Google Scholar] [CrossRef]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Sung, B.H.; Kim, S.C. Buforins: Histone H2A-derived antimicrobial peptides from toad stomach. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, D.E. Insights into buforin II membrane translocation from molecular dynamics simulations. Peptides 2012, 38, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Agerberth, B.; Lee, J.Y.; Bergman, T.; Carlquist, M.; Boman, H.G.; Mutt, V.; Jornvall, H. Amino acid sequence of PR-39. Isolation from pig intestine of a new member of the family of proline-arginine-rich antibacterial peptides. Eur. J. Biochem. 1991, 202, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, G.H.; Magnusson, K.P.; Chowdhary, B.P.; Johansson, M.; Andersson, L.; Boman, H.G. Structure of the gene for porcine peptide antibiotic PR-39, a cathelin gene family member: Comparative mapping of the locus for the human peptide antibiotic FALL-39. Proc. Natl. Acad. Sci. USA 1995, 92, 7085–7089. [Google Scholar] [CrossRef]
- Sang, Y.; Blecha, F. Porcine host defense peptides: Expanding repertoire and functions. Dev. Comp. Immunol. 2009, 33, 334–343. [Google Scholar] [CrossRef]
- Gallo, R.L.; Ono, M.; Povsic, T.; Page, C.; Eriksson, E.; Klagsbrun, M.; Bernfield, M. Syndecans, cell surface heparan sulfate proteoglycans, are induced by a proline-rich antimicrobial peptide from wounds. Proc. Natl. Acad. Sci. USA 1994, 91, 11035–11039. [Google Scholar] [CrossRef]
- Gennaro, R.; Skerlavaj, B.; Romeo, D. Purification, composition, and activity of two bactenecins, antibacterial peptides of bovine neutrophils. Infect. Immun. 1989, 57, 3142–3146. [Google Scholar] [Green Version]
- Litteri, L.; Romeo, D. Characterization of bovine neutrophil antibacterial polypeptides which bind to Escherichia coli. Infect. Immun. 1993, 61, 966–969. [Google Scholar]
- Price, R.; Bugeon, L.; Mostowy, S.; Makendi, C.; Wren, B.; Williams, H.; Willcocks, S. In vitro and in vivo properties of the bovine antimicrobial peptide, Bactenecin 5. PLoS ONE 2019, 14, e0210508. [Google Scholar] [CrossRef] [PubMed]
- Zahn, M.; Kieslich, B.; Berthold, N.; Knappe, D.; Hoffmann, R.; Strater, N. Structural identification of DnaK binding sites within bovine and sheep bactenecin Bac7. Protein Pept. Lett. 2014, 21, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Le, M.T.; Ahn, B.; Cho, H.S.; Le, V.C.Q.; Yum, J.; Hong, K.; Kim, J.H.; Song, H.; Park, C. Copy number variation of PR-39 cathelicidin, and identification of PR-35, a natural variant of PR-39 with reduced mammalian cytotoxicity. Gene 2019, 692, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, M.; Scocchi, M.; Podda, E.; Skerlavaj, B.; Dolzani, L.; Gennaro, R. Antimicrobial activity of Bac7 fragments against drug-resistant clinical isolates. Peptides 2004, 25, 2055–2061. [Google Scholar] [CrossRef]
- Benincasa, M.; Pelillo, C.; Zorzet, S.; Garrovo, C.; Biffi, S.; Gennaro, R.; Scocchi, M. The proline-rich peptide Bac7(1–35) reduces mortality from Salmonella typhimurium in a mouse model of infection. BMC Microbiol. 2010, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, M.; Zahariev, S.; Pelillo, C.; Milan, A.; Gennaro, R.; Scocchi, M. PEGylation of the peptide Bac7(1–35) reduces renal clearance while retaining antibacterial activity and bacterial cell penetration capacity. Eur. J. Med. Chem. 2015, 95, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 2016, 44, 2439–2450. [Google Scholar] [CrossRef]
- Bulet, P.; Urge, L.; Ohresser, S.; Hetru, C.; Otvos, L. Enlarged scale chemical synthesis and range of activity of drosocin, an O-glycosylated antibacterial peptide of Drosophila. Eur. J. Biochem. 1996, 238, 64–69. [Google Scholar] [CrossRef]
- Bikker, F.J.; Kaman-van Zanten, W.E.; de Vries-van de Ruit, A.M.; Voskamp-Visser, I.; van Hooft, P.A.; Mars-Groenendijk, R.H.; de Visser, P.C.; Noort, D. Evaluation of the antibacterial spectrum of drosocin analogues. Chem. Biol. Drug Des. 2006, 68, 148–153. [Google Scholar] [CrossRef]
- Dutta, R.C.; Nagpal, S.; Salunke, D.M. Functional mapping of apidaecin through secondary structure correlation. Int. J. Biochem. Cell Biol. 2008, 40, 1005–1015. [Google Scholar] [CrossRef]
- Czihal, P.; Knappe, D.; Fritsche, S.; Zahn, M.; Berthold, N.; Piantavigna, S.; Muller, U.; van Dorpe, S.; Herth, N.; Binas, A.; et al. Api88 is a novel antibacterial designer peptide to treat systemic infections with multidrug-resistant Gram-negative pathogens. ACS Chem. Biol. 2012, 7, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Ostorhazi, E.; Nemes-Nikodem, E.; Knappe, D.; Hoffmann, R. In vivo activity of optimized apidaecin and oncocin peptides against a multiresistant, KPC-producing Klebsiella pneumoniae strain. Protein Pept. Lett. 2014, 21, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, M.; Benincasa, M.; Bertoloni, G.; Biondi, B.; Dosselli, R.; Papini, E.; Reddi, E.; Rocchi, R.; Tavano, R.; Gennaro, R. Substitution of the arginine/leucine residues in apidaecin Ib with peptoid residues: Effect on antimicrobial activity, cellular uptake, and proteolytic degradation. J. Med. Chem. 2009, 52, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Yamazaki, K.; Kawakami, S.; Miyoshi, D.; Ooi, T.; Hashimoto, S.; Taguchi, S. In vivo target exploration of apidaecin based on Acquired Resistance induced by Gene Overexpression (ARGO assay). Sci. Rep. 2017, 7, 12136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chairatana, P.; Nolan, E.M. Molecular basis for self-assembly of a human host-defense peptide that entraps bacterial pathogens. J. Am. Chem. Soc. 2014, 136, 13267–13276. [Google Scholar] [CrossRef] [PubMed]
- Omardien, S.; Drijfhout, J.W.; van Veen, H.; Schachtschabel, S.; Riool, M.; Hamoen, L.W.; Brul, S.; Zaat, S.A.J. Synthetic antimicrobial peptides delocalize membrane bound proteins thereby inducing a cell envelope stress response. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Xhindoli, D.; Pacor, S.; Guida, F.; Antcheva, N.; Tossi, A. Native oligomerization determines the mode of action and biological activities of human cathelicidin LL-37. Biochem. J. 2014, 457, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.L.; Hunter, H.N.; Vogel, H.J. Lactoferricin: A lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell. Mol. Life Sci. 2005, 62, 2588–2598. [Google Scholar] [CrossRef]
- Jeu, L.; Fung, H.B. Daptomycin: A cyclic lipopeptide antimicrobial agent. Clin. Ther. 2004, 26, 1728–1757. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Pazgier, M.; Jung, G.; Nuccio, S.P.; Castillo, P.A.; de Jong, M.F.; Winter, M.G.; Winter, S.E.; Wehkamp, J.; Shen, B.; et al. Human alpha-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 2012, 337, 477–481. [Google Scholar] [CrossRef]
- Melo, M.N.; Ferre, R.; Castanho, M.A. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Candido, E.S.; de Barros, E.; Cardoso, M.H.; Franco, O.L. Bacterial cross-resistance to anti-infective compounds. Is it a real problem? Curr. Opin. Pharmacol. 2019, 48, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; de Almeida, K.C.; Candido, E.S.; Murad, A.M.; Dias, S.C.; Franco, O.L. Comparative NanoUPLC-MS(E) analysis between magainin I-susceptible and -resistant Escherichia coli strains. Sci. Rep. 2017, 7, 4197. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Slepenkov, S.V.; Witt, S.N. The insect antimicrobial peptide, L-pyrrhocoricin, binds to and stimulates the ATPase activity of both wild-type and lidless DnaK. FEBS Lett. 2004, 565, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.N.; Lomakin, I.B.; Gagnon, M.G.; Steitz, T.A. The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin. Nat. Struct. Mol. Biol. 2015, 22, 466–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, K.M.; Spille, J.H.; Sahl, H.G.; Grein, F.; Kubitscheck, U. The lantibiotic nisin induces lipid II aggregation, causing membrane instability and vesicle budding. Biophys. J. 2015, 108, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Nishie, M.; Nagao, J.; Zendo, T.; Keller, S.; Nakayama, J.; Kohda, D.; Sahl, H.G.; Sonomoto, K. Ring A of nukacin ISK-1: A lipid II-binding motif for type-A(II) lantibiotic. J. Am. Chem. Soc. 2012, 134, 3687–3690. [Google Scholar] [CrossRef]
- Collin, F.; Thompson, R.E.; Jolliffe, K.A.; Payne, R.J.; Maxwell, A. Fragments of the bacterial toxin microcin B17 as gyrase poisons. PLoS ONE 2013, 8, e61459. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.L.; Lang, K.; Meenan, N.A.; Sharma, A.; Kelley, A.C.; Kleanthous, C.; Ramakrishnan, V. Structural basis for 16S ribosomal RNA cleavage by the cytotoxic domain of colicin E3. Nat. Struct. Mol. Biol. 2010, 17, 1241–1246. [Google Scholar] [CrossRef] [Green Version]
- Garza-Sanchez, F.; Gin, J.G.; Hayes, C.S. Amino acid starvation and colicin D treatment induce A-site mRNA cleavage in Escherichia coli. J. Mol. Biol. 2008, 378, 505–519. [Google Scholar] [CrossRef]
- Melo, M.N.; Dugourd, D.; Castanho, M.A. Omiganan pentahydrochloride in the front line of clinical applications of antimicrobial peptides. Recent Pat. Antiinfect. Drug Discov. 2006, 1, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.; Gellatly, S.L.; Hancock, R.E. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 2011, 68, 2161–2176. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Non-Lytic AMPs | Antibacterial Potential | Treatment Strategies | Design Strategies | Structural Profile | Membrane Translocation | Intracellular Target | References |
|---|---|---|---|---|---|---|---|
| Indolicidin | Bacteriostatic; bactericide; anti-bacteremia | Monotherapy; synergism between two indolicidin analogues | Amino acid substitution; Amide bond modification; Hybrid peptides | poly-l-proline type II helix; extended structures; β-turn | Transmembrane orientation followed by cell internalization | DNA binding; DNA biosynthesis inhibition | [27,28,29,30,31,32] |
| Buforin II | Bacteriostatic; bactericide; anti-sepsis | Monotherapy; synergism with rifampicin; additive effects when combined with ranalexin, amoxicillin-clavulanate, ceftriaxone, meropenem, doxycycline, and clarithromycin; conjugation with PNA | Amino acid substitution; truncated analogues | Helical-helix-propeller structure | Formation of transient toroidal pores | DNA and RNA binding | [33,34,35,36] |
| PR-39 | Bacteriostatic; bactericide; anti-sepsis; toxin neutralization; wound healing | Monotherapy | Truncated analogues; amino acid substitution | Extended | Receptor-mediated (SbmA) | Protein and DNA synthesis inhibition | [37,38,39] |
| Bac7 | Bacteriostatic; bactericide; anti-sepsis; immunomodulatory | Monotherapy; synergism; association with PEG | Truncated analogues; amino acid substitution | Extended | Receptor-mediated (SbmA) | Protein and DNA synthesis inhibition; Ribosome; Binding to lipid II precursor; cell wall synthesis | [37,40,41,42,43,44] |
| Apidaecin | Bacteriostatic; bactericide | Monotherapy | Chemical modifications; amino acid substitution; peptide–peptoid hybrids | Extended | Oligomers formation (OM); Interaction with IM permeases and transporters | DnaK and GroEL, leading to bacterial protein misfolding; Protein synthesis; Ribosome | [45,46,47] |
| Drosocin | Bacteriostatic; bactericide | Monotherapy | Chemical modifications | Extended | Receptor-mediated (unknown) | DnaK and GroEL, leading to bacterial protein misfolding | [48,49,50] |
| Peptide | Organism | Source | Class | Analogues | Antibacterial Activity Spectrum | MIC Range (μM) | References |
|---|---|---|---|---|---|---|---|
| Indolicidin | Bos taurus | Neutrophils cytoplasmic granules | Tryptophan-rich | N-substituted class of non-proteogenic residues or by glycine; ID, ID-I, ID-W and ID-IW; RN7-IN6 to RN7-IN10 | E. coli, P. aeruginosa, S. aureus, S. epidermidis, S. pneumoniae, M. luteus and S. typhimurium | 0.2 to 50 | [22,51,53,56,57,58] |
| Buforin II | Bufo bufo gargarizans | Stomach tissue | Helical-helix-propeller peptide | BF2-A; BF2-C; BUF(5–21); BUF(5–13)-[RLLR]3; Buf-IIIa to Buf-IIId | A. baumannii,B. subtilis, C. neoformans, E. coli, L. monocytogene, P. putida, S. aureus, S. dysenteriae, S. hemolyticus, S. marcescens, S. mutans, S. pneumoniae, S. typhimurium and Serratia sp. | 0.2 to 3.2 | [34,35,36,61,62,63,64,65,66,67] |
| PR-39 | Sus scrofa | Porcine neutrophils | Proline/arginine-rich | PR-39 (1–26); PR-39 (1–22); PR-39 (1–18); PR-39 (1–15); PR35 | B. globigii, E. coli, S.typhimurium and S.choleraesuis | 1.25 to 20 | [68,69,70] |
| Bac7 | Bos taurus | Bovine neutrophils | Proline/arginine-rich | Bac7 (1–35); Bac7 (5.35); Bac7 (1–23); Bac7 (5–23); Bac7 (1–16); Bac7 (1–18) | A. baumannii, E. coli, K. pneumoniae, P. aeruginosa, S. aureus, S. enterica and S. maltophilia | 0.06 to 64 | [71,72,73] |
| Apidaecin | Apis mellifera | Lymph fluid | Proline/arginine-rich | api6; api7; api39; api88; api137; apidaecin Ib; | E. coli, K. pneumoniae, P. aeruginosa, S. enteritidis, S. typhimurium | 0.27 to 64 | [50,74,75,76,77] |
| Drosocin | Drosophila melanogaster | Abdomen and thoraxes | Proline/arginine-rich | Thr6-glycosylated drosocin; β-Ala drosocin; M-drosocin; Di-drosocin | E. coli, Erwinia herbicola, S. enteritidis, S. infantis, S. montevideo, S. panama, S. typhimurium, and M. luteus | 0.25 to 100 | [78,79] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, M.H.; Meneguetti, B.T.; Costa, B.O.; Buccini, D.F.; Oshiro, K.G.N.; Preza, S.L.E.; Carvalho, C.M.E.; Migliolo, L.; Franco, O.L. Non-Lytic Antibacterial Peptides That Translocate Through Bacterial Membranes to Act on Intracellular Targets. Int. J. Mol. Sci. 2019, 20, 4877. https://doi.org/10.3390/ijms20194877
Cardoso MH, Meneguetti BT, Costa BO, Buccini DF, Oshiro KGN, Preza SLE, Carvalho CME, Migliolo L, Franco OL. Non-Lytic Antibacterial Peptides That Translocate Through Bacterial Membranes to Act on Intracellular Targets. International Journal of Molecular Sciences. 2019; 20(19):4877. https://doi.org/10.3390/ijms20194877
Chicago/Turabian StyleCardoso, Marlon H., Beatriz T. Meneguetti, Bruna O. Costa, Danieli F. Buccini, Karen G. N. Oshiro, Sergio L. E. Preza, Cristiano M. E. Carvalho, Ludovico Migliolo, and Octávio L. Franco. 2019. "Non-Lytic Antibacterial Peptides That Translocate Through Bacterial Membranes to Act on Intracellular Targets" International Journal of Molecular Sciences 20, no. 19: 4877. https://doi.org/10.3390/ijms20194877