Accelerated Molecular Dynamics Applied to the Peptaibol Folding Problem


Abstract
:1. Introduction
2. Results and Discussion
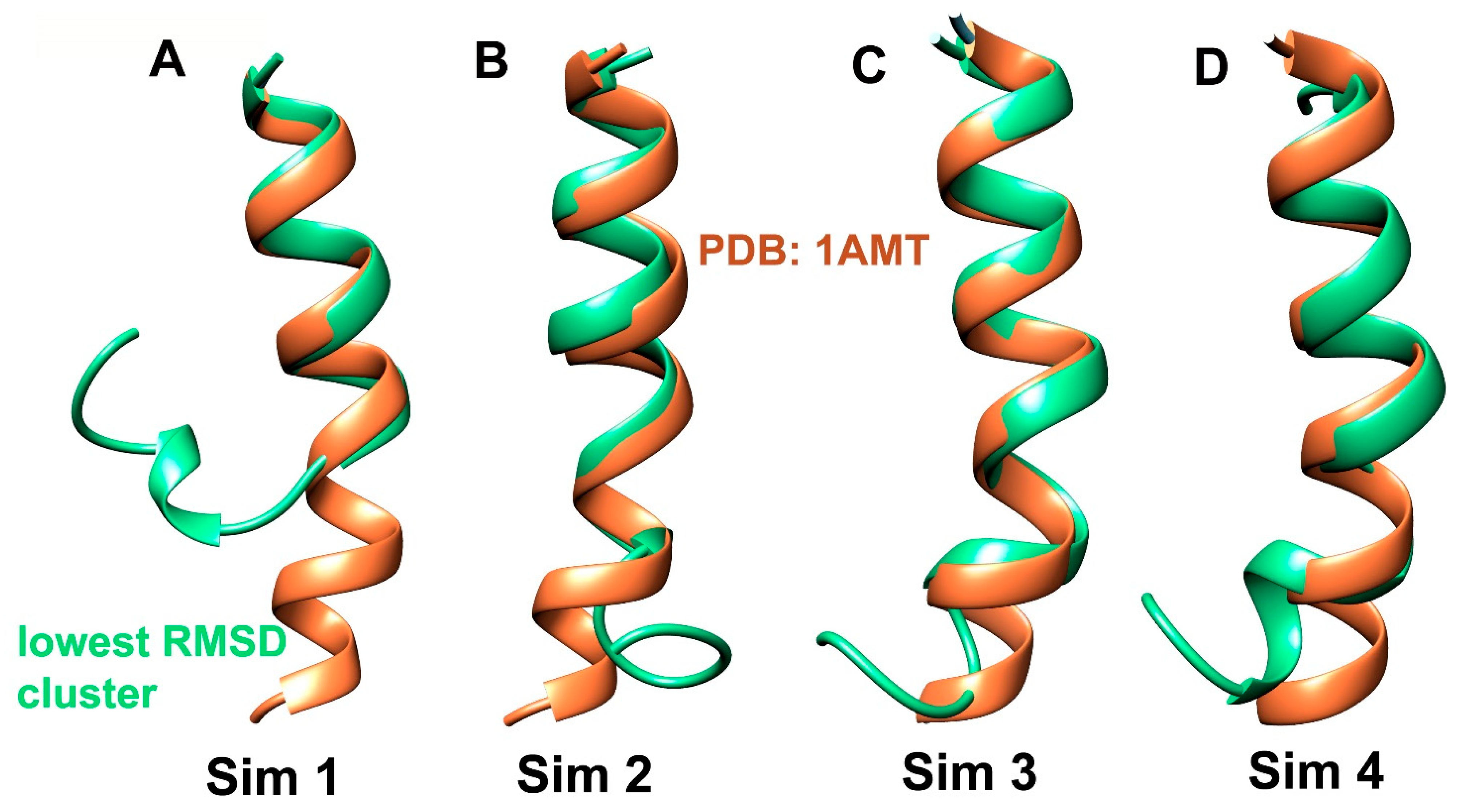
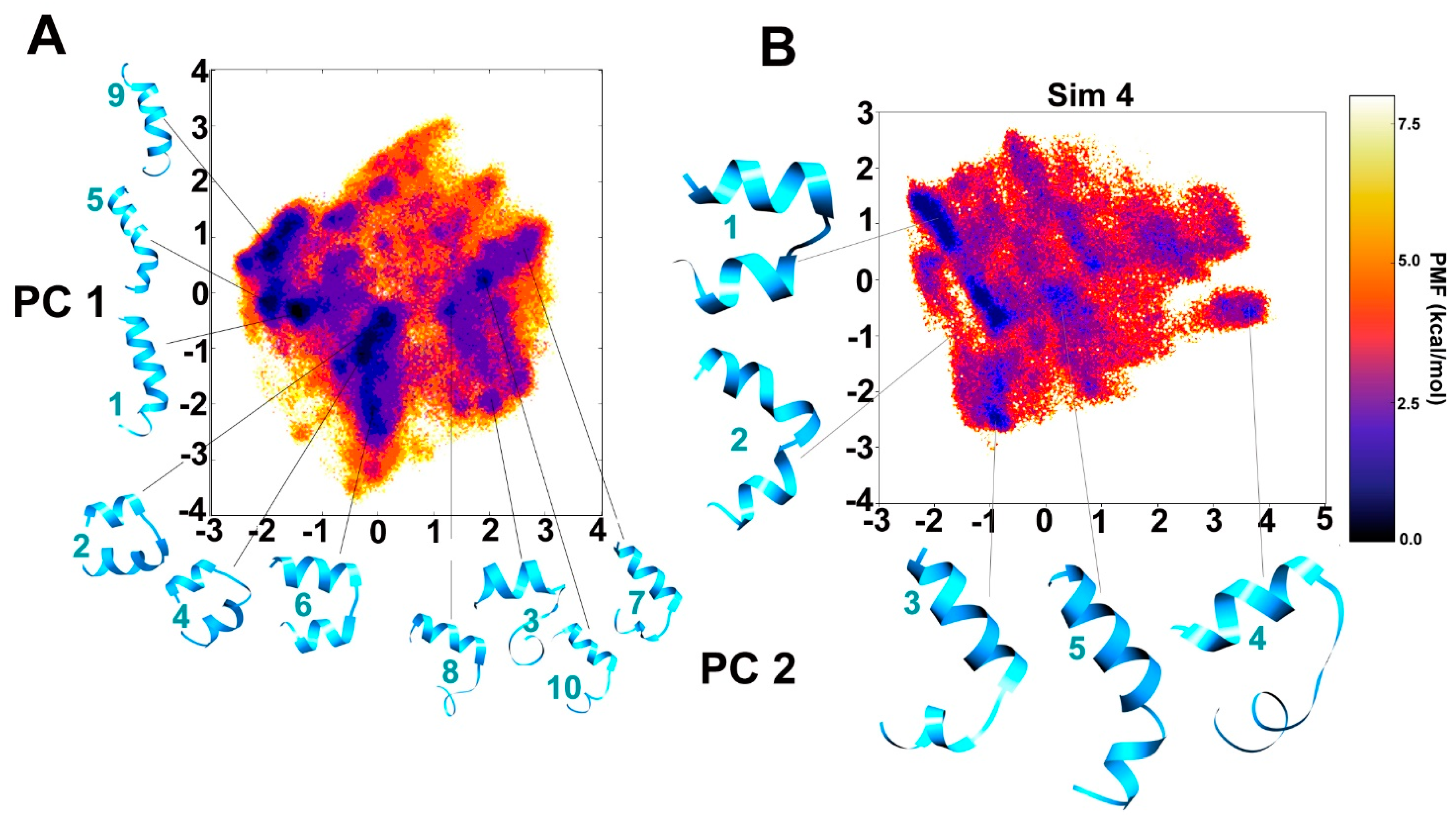
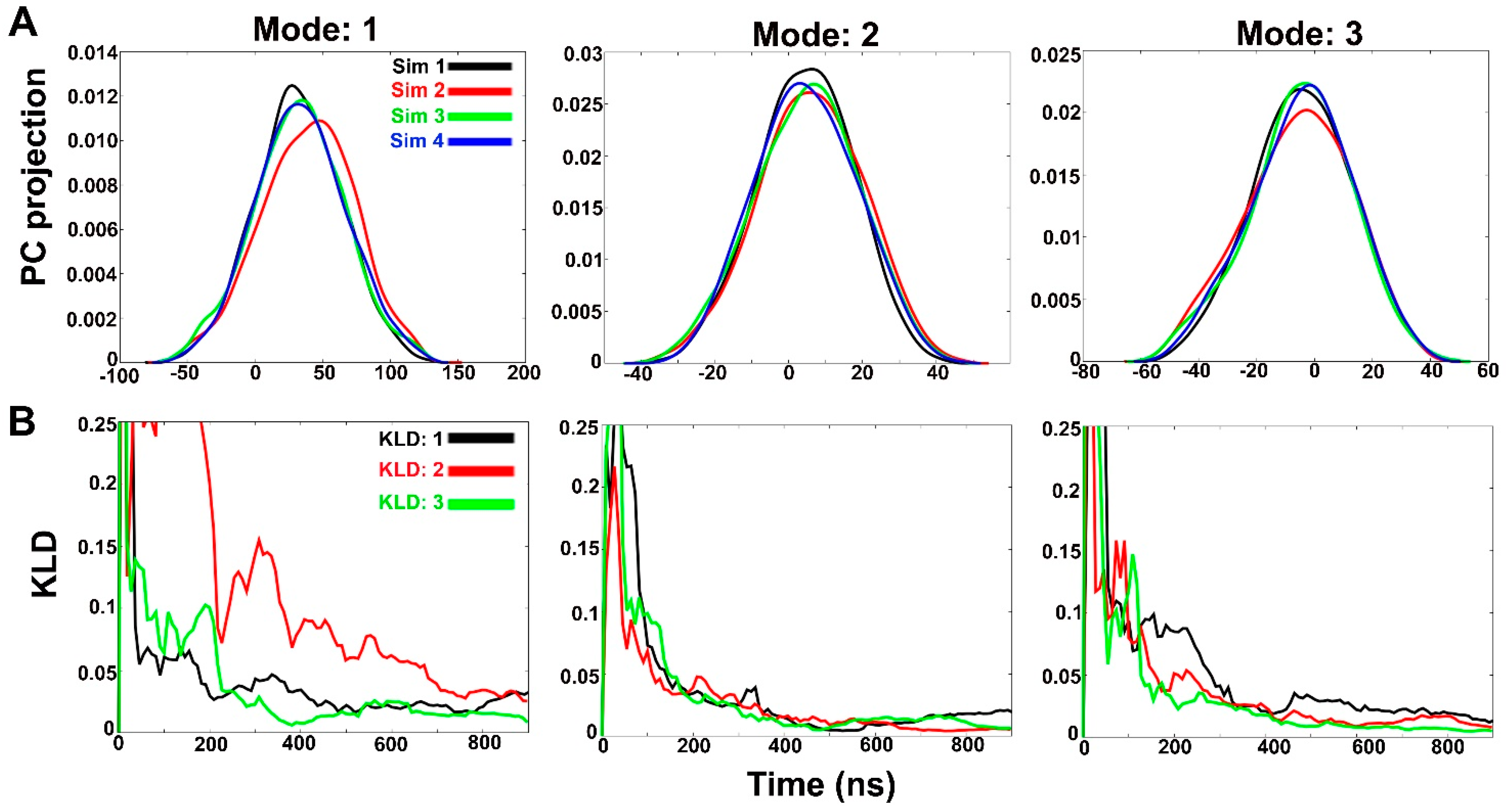
2.1. Clustering of Three Consecutive aMD Simulations and Comparison with Fourth Simulation: Free Energy Landscapes
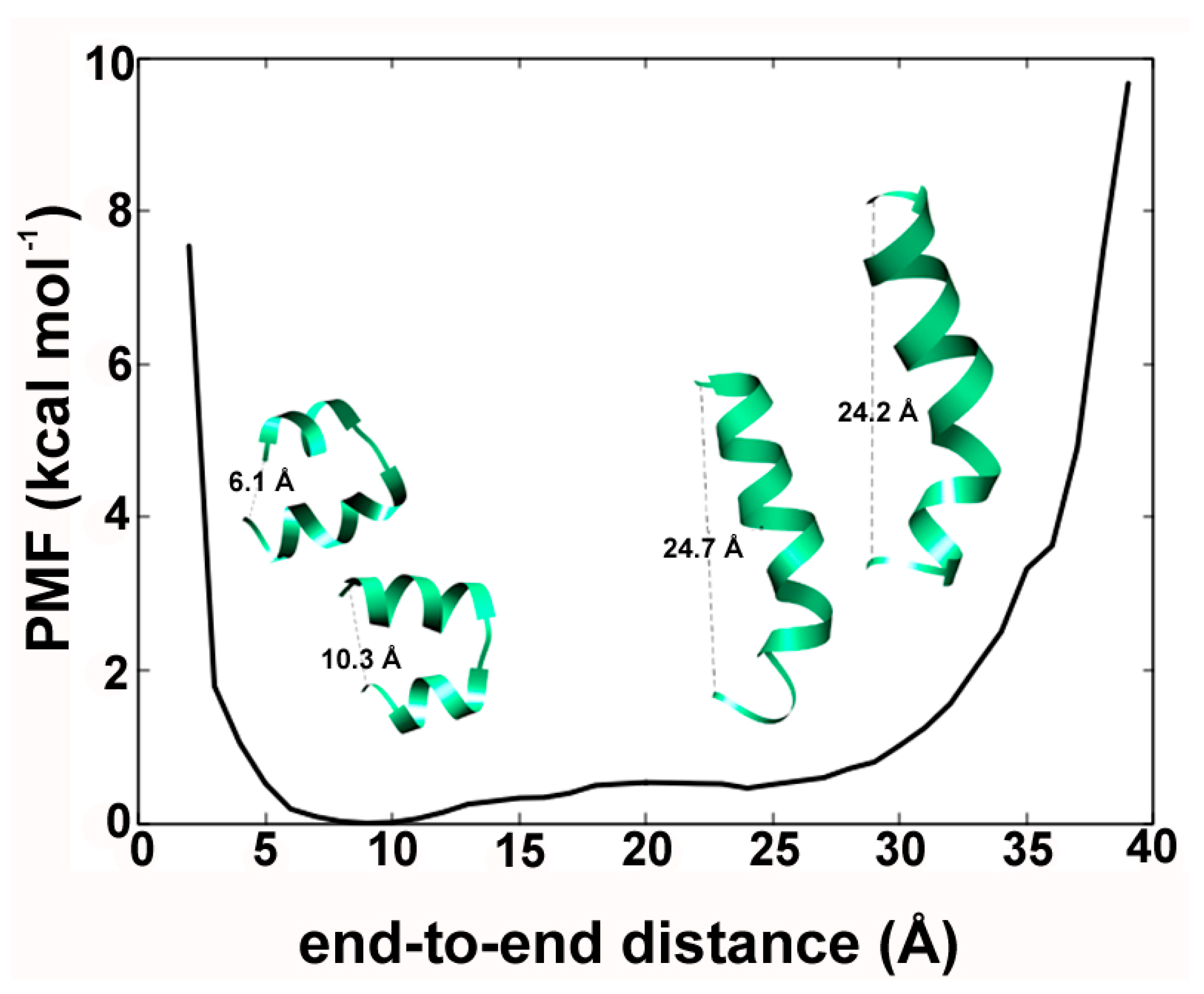
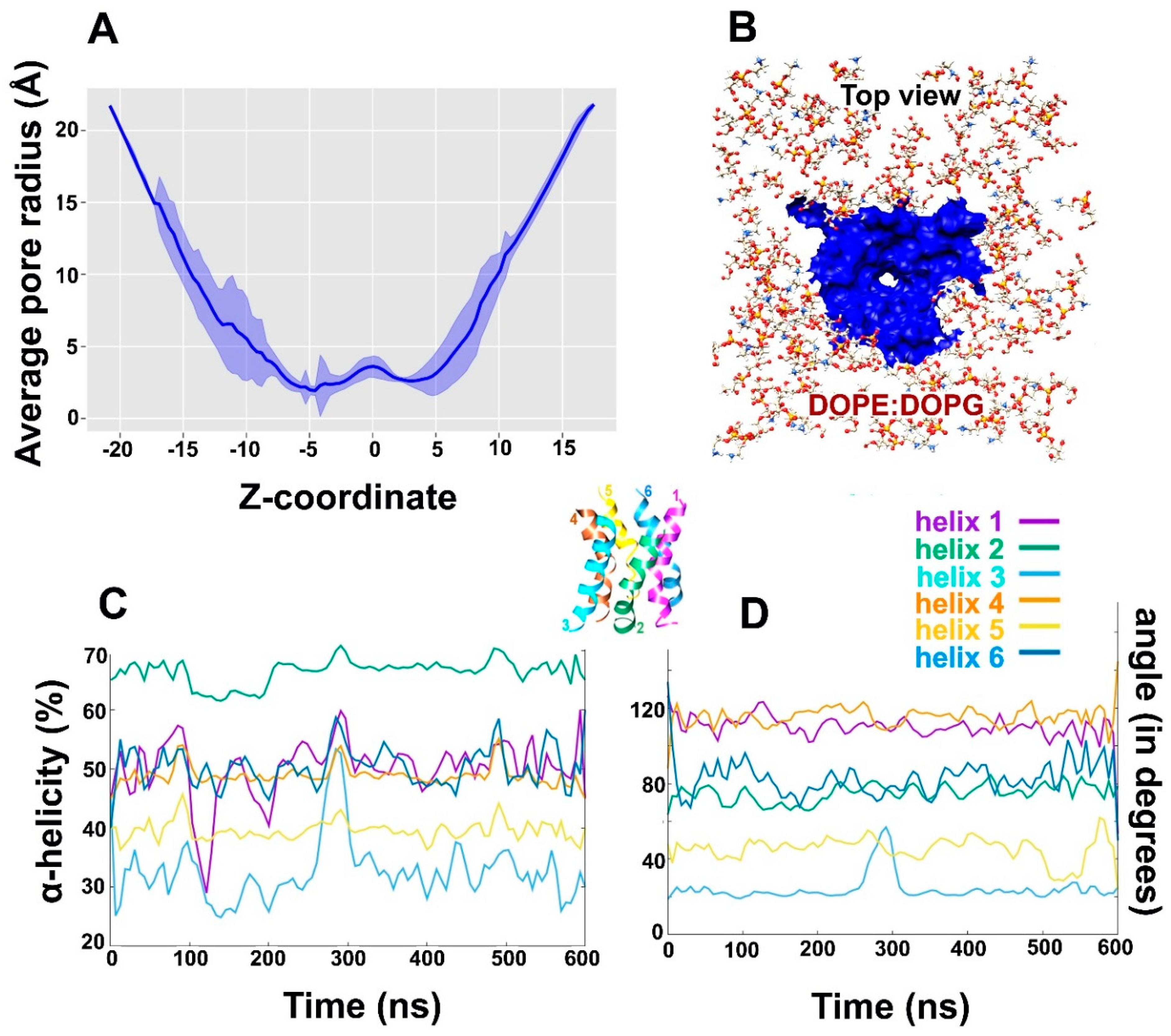
2.2. Alamethicin Backbone Bending: Functional Importance?
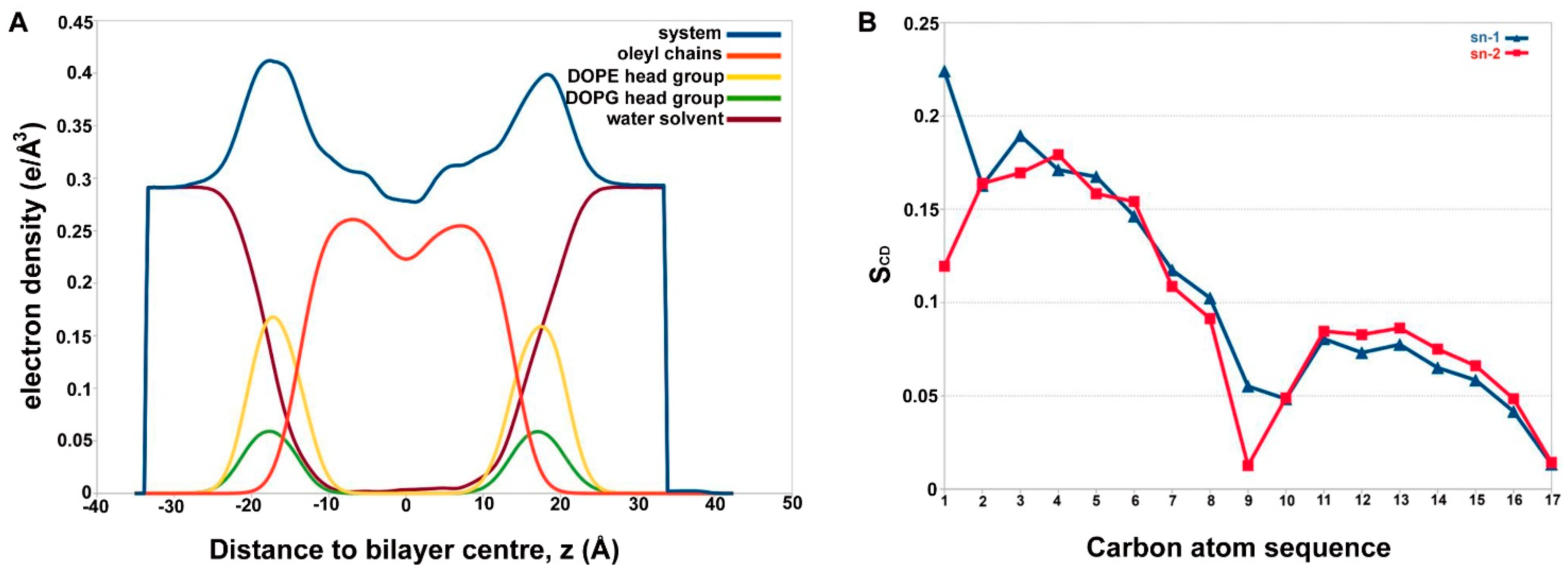
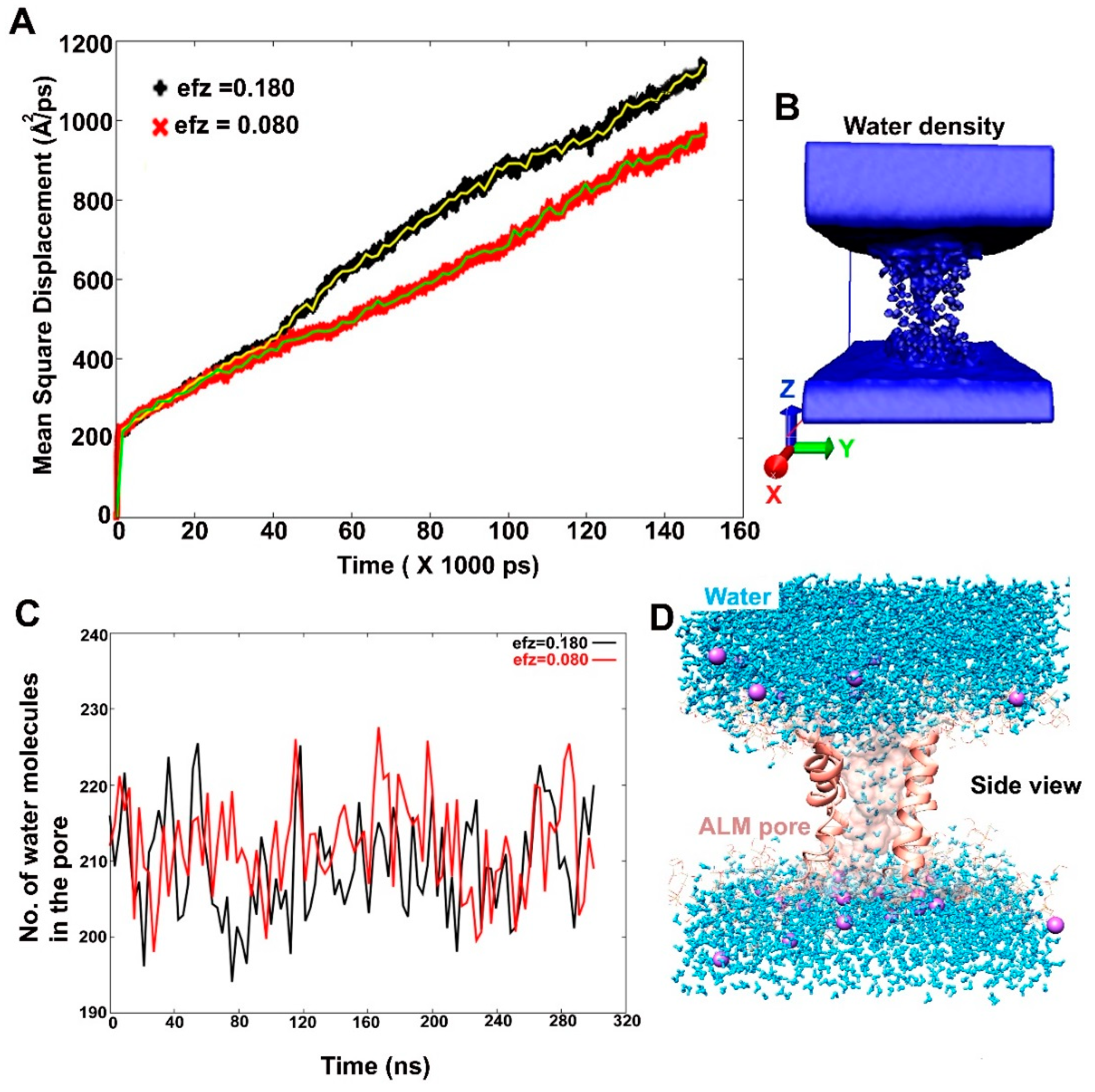
2.3. Alamethicin Hexamer Pore in a Bacterial Mimicking Bilayer Membrane
3. Materials and Methods
3.1. Partial Charge Calculation and Force Field Library Generation for Non-standard Residues
3.2. Accelerated Molecular Dynamics Simulations of Alamethicin
3.3. Accelerated Molecular Dynamics Simulations of Alamethicin Pore in DOPC Bilayer Membrane
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamelberg, D.; de Oliveira, C.A.; McCammon, J.A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 155102. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.E.; Reusser, F. A polypeptide antibacterial agent isolated from Trichoderma viride. Experientia 1967, 23, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Degenkolb, T.; Dieckmann, R.; Nielsen, K.F.; Gräfenhan, T.; Theis, C.; Zafari, D.; Chaverri, P.; Ismaiel, A.; Brückner, H.; von Döhren, H.; et al. The Trichoderma brevicompactum clade: A separate lineage with new species, new peptaibiotics, and mycotoxins. Mycol. Prog. 2008, 7, 177–219. [Google Scholar] [CrossRef]
- Payne, J.W.; Jakes, R.; Hartley, B.S. The primary structure of alamethicin. Biochem. J. 1970, 117, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.R.; Williams, R.J.P. Chemical nature and sequence of alamethicin. Biochem. J. 1976, 153, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.O., Jr.; Richards, F.M. A voltage-gated ion channel model inferred from the crystal structure of alamethicin at 1.5-Å resolution. Nature 1982, 300, 325. [Google Scholar] [CrossRef]
- Nagao, T.; Mishima, D.; Javkhlantugs, N.; Wang, J.; Ishioka, D.; Yokota, K.; Norisada, K.; Kawamura, I.; Ueda, K.; Naito, A. Structure and orientation of antibiotic peptide alamethicin in phospholipid bilayers as revealed by chemical shift oscillation analysis of solid state nuclear magnetic resonance and molecular dynamics simulation. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2789–2798. [Google Scholar] [CrossRef] [Green Version]
- Haris, P.I.; Chapman, D. Fourier transform infrared spectra of the polypeptide alamethicin and a possible structural similarity with bacteriorhodopsin. Biochim. Biophys. Acta Biomembr. 1988, 943, 375–380. [Google Scholar] [CrossRef]
- Leitgeb, B.; Szekeres, A.; Manczinger, L.; Vágvölgyi, C.; Kredics, L. The history of alamethicin: A review of the most extensively studied peptaibol. Chem. Biodivers. 2007, 4, 1027–1051. [Google Scholar] [CrossRef] [PubMed]
- Kredics, L.; Szekeres, A.; Czifra, D.; Vágvölgyi, C.; Leitgeb, B. Recent results in alamethicin research. Chem. Biodivers. 2013, 10, 744–771. [Google Scholar] [CrossRef] [PubMed]
- Pieta, P.; Mirza, J.; Lipkowski, J. Direct visualization of the alamethicin pore formed in a planar phospholipid matrix. Proc. Natl. Acad. Sci. USA. 2012, 109, 21223–21227. [Google Scholar] [CrossRef] [Green Version]
- Clejan, S.; Krulwich, T.A.; Mondrus, K.R.; Seto-Young, D. Membrane lipid composition of obligately and facultatively alkalophilic strains of Bacillus spp. J. Bacteriol. 1986, 168, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Salditt, T.; Li, C.; Spaar, A. Structure of antimicrobial peptides and lipid membranes probed by interface-sensitive X-ray scattering. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, A.; Lazaridis, T. A thermodynamic approach to alamethicin pore formation. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1440–1447. [Google Scholar] [CrossRef]
- Castro, T.G.; Micaélo, N.M. Conformational and thermodynamic properties of non-canonical α, α-dialkyl glycines in the peptaibol alamethicin: Molecular dynamics studies. J. Phys. Chem. B 2014, 118, 9861–9870. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.S.; Raya, J.; De Zotti, M.; Zaitseva, E.; Peggion, C.; Ballano, G.; Toniolo, C.; Raap, J.; Bechinger, B. Alamethicin supramolecular organization in lipid membranes from 19F solid-state NMR. Biophys. J. 2016, 111, 2450–2459. [Google Scholar] [CrossRef]
- Perrin Jr, B.S.; Pastor, R.W. Simulations of membrane-disrupting peptides I: Alamethicin pore stability and spontaneous insertion. Biophys. J. 2016, 111, 1248–1257. [Google Scholar] [CrossRef]
- Madsen, J.L.; Hjørringgaard, C.U.; Vad, B.S.; Otzen, D.; Skrydstrup, T. Incorporation of β-silicon-β3-amino acids in the antimicrobial peptide alamethicin provides a 20-fold increase in membrane permeabilization. Chem. Eur. J. 2016, 22, 8358–8367. [Google Scholar] [CrossRef]
- Das, S.; Ben Haj Salah, K.; Wenger, E.; Martinez, J.; Kotarba, J.; Andreu, V.; Ruiz, N.; Savini, F.; Stella, L.; Didierjean, C.; et al. Enhancing the antimicrobial activity of alamethicin f50/5 by incorporating n-terminal hydrophobic triazole substituents. Chem. Eur. J. 2017, 23, 17964–17972. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, E.F.; Syryamina, V.N.; Dzuba, S.A. Communication: Alamethicin can capture lipid-like molecules in the membrane. J. Chem. Phys. 2017, 146, 011103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Shodiev, M.; Leitch, J.J.; Abbasi, F.; Lipkowski, J. In situ electrochemical and PM-IRRAS studies of alamethicin ion channel formation in model phospholipid bilayers. J. Electroanal. Chem. 2018, 819, 251–259. [Google Scholar] [CrossRef]
- Forbrig, E.; Staffa, J.K.; Salewski, J.; Mroginski, M.A.; Hildebrandt, P.; Kozuch, J. Monitoring the orientational changes of alamethicin during incorporation into bilayer lipid membranes. Langmuir 2018, 34, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, F.; Alvarez-Malmagro, J.; Su, Z.; Leitch, J.J.; Lipkowski, J. Pore forming properties of alamethicin in negatively charged floating bilayer lipid membranes supported on gold electrodes. Langmuir 2018, 34, 13754–13765. [Google Scholar] [CrossRef]
- Syryamina, V.N.; De Zotti, M.; Toniolo, C.; Formaggio, F.; Dzuba, S.A. Alamethicin self-assembling in lipid membranes: Concentration dependence from pulsed EPR of spin labels. Phys. Chem. Chem. Phys. 2018, 20, 3592–3601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.K.; Lyu, Y.; Zhu, X.; Wang, J.P.; Jin, Z.Y.; Narsimhan, G. Enhanced solubility and antimicrobial activity of alamethicin in aqueous solution by complexation with γ-cyclodextrin. J. Funct. Foods 2018, 40, 700–706. [Google Scholar] [CrossRef]
- Abbasi, F.; Su, Z.; Alvarez-Malmagro, J.; Leitch, J.J.; Lipkowski, J. Effects of amiloride, an ion channel blocker, on alamethicin pore formation in negatively charged, gold-supported, phospholipid bilayers: A molecular view. Langmuir 2019, 35, 5060–5068. [Google Scholar] [CrossRef]
- Taylor, G.; Nguyen, M.A.; Koner, S.; Freeman, E.; Collier, C.P.; Sarles, S.A. Electrophysiological interrogation of asymmetric droplet interface bilayers reveals surface-bound alamethicin induces lipid flip-flop. Biochim. Biophys. Acta Biomembr. 2019, 1861, 335–343. [Google Scholar] [CrossRef]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; McCammon, J.A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 2677–2689. [Google Scholar] [CrossRef]
- Jing, Z.; Sun, H. A comment on the reweighting method for accelerated molecular dynamics simulations. J. Chem. Theory Comput. 2015, 11, 2395–2397. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.M.M.; McCammon, J.A.; Miao, Y. Replica exchange Gaussian accelerated molecular dynamics: Improved enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2018, 14, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Bucci, R.; Contini, A.; Clerici, F.; Pellegrino, S.; Gelmi, M.L. From glucose to enantiopure morpholino β-amino acid: A new tool for stabilizing γ-turns in peptides. Org. Chem. Front. 2019, 6, 972–982. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Tyagi, C.; Marik, T.; Szekeres, A.; Vágvölgyi, C.; Kredics, L.; Ötvös, F. Tripleurin XIIc: Peptide folding dynamics in aqueous and hydrophobic environment mimic using accelerated molecular dynamics. Molecules 2019, 24, 358. [Google Scholar] [CrossRef] [PubMed]
- North, C.L.; Franklin, J.C.; Bryant, R.G.; Cafiso, D.S. Molecular flexibility demonstrated by paramagnetic enhancements of nuclear relaxation. Application to alamethicin: A voltage-gated peptide channel. Biophys. J. 1994, 67, 1861–1866. [Google Scholar] [CrossRef]
- Franklin, J.C.; Ellena, J.F.; Jayasinghe, S.; Kelsh, L.P.; Cafiso, D.S. Structure of micelle-associated alamethicin from 1H NMR. Evidence for conformational heterogeneity in a voltage-gated peptide. Biochemistry 1994, 33, 4036–4045. [Google Scholar] [CrossRef]
- North, C.L.; Barranger-Mathys, M.; Cafiso, D.S. Membrane orientation of the N-terminal segment of alamethicin determined by solid-state 15N NMR. Biophys. J. 1995, 69, 2392–2397. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, N.; Sessions, R.B.; Williams, P.B.; Dempsey, C.E. Helix bending in alamethicin: Molecular dynamics simulations and amide hydrogen exchange in methanol. Biophys. J. 1997, 72, 2490–2495. [Google Scholar] [CrossRef]
- Marik, T.; Tyagi, C.; Racić, G.; Rakk, D.; Szekeres, A.; Vágvölgyi, C.; Kredics, L. New 19-residue peptaibols from Trichoderma clade Viride. Microorganisms 2018, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Marik, T.; Tyagi, C.; Balázs, D.; Urbán, P.; Szepesi, Á.; Bakacsy, L.; Endre, G.; Rakk, D.; Szekeres, A.; Andersson, M.A.; et al. Structural diversity and bioactivities of peptaibol compounds from the Longibrachiatum Clade of the filamentous fungal genus Trichoderma. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Högel, P.; Götz, A.; Kuhne, F.; Ebert, M.; Stelzer, W.; Rand, K.D.; Scharnagl, C.; Langosch, D. Glycine perturbs local and global conformational flexibility of a transmembrane helix. Biochemistry 2018, 57, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.; Stellato, M.I.; Oliva, R.; Falanga, A.; Galdiero, M.; Petraccone, L.; D’Errico, G.; De Santis, A.; Galdiero, S.; Del Vecchio, P. Antimicrobial peptides at work: Interaction of myxinidin and its mutant WMR with lipid bilayers mimicking the P. aeruginosa and E. coli membranes. Sci. Rep. 2017, 7, 44425. [Google Scholar] [CrossRef] [PubMed]
- Pastor, R.W.; Venable, R.M.; Karplus, M. Model for the structure of the lipid bilayer. Proc. Natl. Acad. Sci. USA 1991, 88, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Pastor, R.W.; Venable, R.M.; Karplus, M.; Szabo, A. A simulation based model of NMR T 1 relaxation in lipid bilayer vesicles. J. Chem. Phys. 1988, 89, 1128–1140. [Google Scholar] [CrossRef]
- Venable, R.M.; Brown, F.L.; Pastor, R.W. Mechanical properties of lipid bilayers from molecular dynamics simulation. Chem. Phys. Lipids 2015, 192, 60–74. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Schertzer, J.W.; Yong, X. Molecular dynamics modeling of Pseudomonas aeruginosa outer membranes. Phys. Chem. Chem. Phys. 2018, 20, 23635–23648. [Google Scholar] [CrossRef]
- Shinoda, W. Permeability across lipid membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2254–2265. [Google Scholar] [CrossRef]
- Agre, P.; Kozono, D. Aquaporin water channels: Molecular mechanisms for human diseases. FEBS Lett. 2003, 555, 72–78. [Google Scholar] [CrossRef]
- Tsibris, A.M.; Hirsch, M.S. Antiretroviral therapy for human immunodeficiency virus infection. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 1st ed.; Churchill Livingstone: London, UK, 2009; pp. 1833–1854. [Google Scholar]
- Rosenthal, S.; Decano, A.G.; Bandali, A.; Lai, D.; Malat, G.E.; Bias, T.E. Oritavancin (Orbactiv): A new-generation lipoglycopeptide for the treatment of acute bacterial skin and skin structure infections. P & T. 2018, 43, 143–179. [Google Scholar]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.Y.R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucl. Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, UK.
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.C.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Proceedings of the AMBER 2018, San Francisco, CA, USA, 2018; Available online: ambermd.org/doc12/Amber18.pdf (accessed on 30 August 2019).
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.; Tong, W.; Weng, Z. M-ZDOCK: A grid-based approach for C n symmetric multimer docking. Bioinformatics 2004, 21, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Schott-Verdugo, S.; Gohlke, H. PACKMOL-Memgen: A simple-to-use generalized workflow for membrane-protein/lipid-bilayer system building. J. Chem. Inf. Model. 2019, 59, 2522–2528. [Google Scholar] [CrossRef] [PubMed]
- Nugent, T.; Jones, D.T. Membrane protein orientation and refinement using a knowledge-based statistical potential. BMC Bioinform. 2013, 14, 276. [Google Scholar] [CrossRef]
- Escalona, Y.; Garate, J.A.; Araya-Secchi, R.; Huynh, T.; Zhou, R.; Perez-Acle, T. Exploring the membrane potential of simple dual-membrane systems as models for gap-junction channels. Biophys. J. 2016, 110, 2678–2688. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Intracellular ion environment and membrane electric potential. In Molecular Cell Biology, 4th ed.; WH Freeman & Company: New York, NY, USA, 2000. [Google Scholar]
- Smart, O.S.; Neduvelil, J.G.; Wang, X.; Wallace, B.A.; Sansom, M.S. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.; Melo, M.N.; Seyler, S.L.; Domański, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In Proceedings of the 15th Python in Science Conference; SciPy: Austin, TX, USA; Available online: https://www.researchgate.net/profile/Sean_Seyler2/publication/320267580_MDAnalysis_a_Python_package_for_the_rapid_analysis_of_molecular_dynamics_simulations/links/59d8dcab458515a5bc262281/MDAnalysis-a-Python-package-for-the-rapid-analysis-of-molecular-dynamics-simulations.pdf (accessed on 30 August 2019).
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulations | Starting Conformation | Time (ns) | a1,a2 Total (kcal mol−1) | b1,b2 Dihedral (kcal mol−1) | Avg Boost (kcal mol−1) |
|---|---|---|---|---|---|
| Sim 1 | Unfolded | 936 | 0.16 | 4 | 11.21 |
| Sim 2 | Folded N-terminal | 950 | 0.20 | 4.5 | 11.10 |
| Sim 3 | Folded with bent backbone | 897 | 0.20 | 4.5 | 29.85 |
| Sim 4 | unfolded | 1000 | 0.20 | 4.5 | 24.09 |
| Membrane-sim 1 | Hexamer with efz = 0.180 | 300 | 0.16 | 4 | 31.71 |
| Membrane-sim 2 | Hexamer with efz = 0.080 | 300 | 0.16 | 4 | 22.60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyagi, C.; Marik, T.; Vágvölgyi, C.; Kredics, L.; Ötvös, F. Accelerated Molecular Dynamics Applied to the Peptaibol Folding Problem. Int. J. Mol. Sci. 2019, 20, 4268. https://doi.org/10.3390/ijms20174268
Tyagi C, Marik T, Vágvölgyi C, Kredics L, Ötvös F. Accelerated Molecular Dynamics Applied to the Peptaibol Folding Problem. International Journal of Molecular Sciences. 2019; 20(17):4268. https://doi.org/10.3390/ijms20174268
Chicago/Turabian StyleTyagi, Chetna, Tamás Marik, Csaba Vágvölgyi, László Kredics, and Ferenc Ötvös. 2019. "Accelerated Molecular Dynamics Applied to the Peptaibol Folding Problem" International Journal of Molecular Sciences 20, no. 17: 4268. https://doi.org/10.3390/ijms20174268