Vanin 1: Its Physiological Function and Role in Diseases


Abstract
:
1. Function, Expression, and Regulation of Vanin 1 Under Physiological Conditions
2. Vanin 1 Expression and Its Role in Diseases
2.1. Liver
2.1.1. Vanin 1 in Steatosis, NAFLD, and NASH Models
2.1.2. Vanin 1 and Hepatotoxicity
2.2. Kidney
2.2.1. Vanin 1 in Acute Kidney Injury and Drug-Induced Renal Injury
2.2.2. Vanin 1 in a Unilateral Ureteral Obstruction Model
2.2.3. Vanin 1 in Diabetic Nephropathy
2.3. Intestine
2.3.1. Vanin 1 in Intestinal Inflammation Models
2.3.2. Vanin 1 in a Colitis Mouse Model
2.3.3. Vanin 1 in Human Inflammatory Bowel Disease
2.4. Lungs
Vanin 1 in Asthma
3. Conclusions
Author Contributions
Conflicts of Interest
References
- Aurrand-Lions, M.; Galland, F.; Bazin, H.; Zakharyev, V.M.; Imhof, B.A.; Naquet, P. Vanin-1, a novel GPI-linked perivascular molecule involved in thymus homing. Immunity 1996, 5, 391–405. [Google Scholar] [CrossRef]
- Galland, F.; Malergue, F.; Bazin, H.; Mattei, M.G.; Aurrand-Lions, M.; Theillet, C.; Naquet, P. Two Human Genes Related to Murine Vanin-1 Are Located on the Long Arm of Human Chromosome. Genomics 1998, 53, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Boersma, Y.L.; Newman, J.; Adams, T.E.; Cowieson, N.; Krippner, G.; Bozaoglu, K.; Peat, T.S. The structure of vanin 1: A key enzyme linking metabolic disease and inflammation. Acta. Cryst. Sect. D Biol. Cryst. 2014, 70, 3320–3329. [Google Scholar] [CrossRef] [PubMed]
- Rommelaere, S.; Millet, V.; Gensollen, T.; Bourges, C.; Eeckhoute, J.; Hennuyer, N.; Baugé, E.; Chasson, L.; Cacciatore, I.; Staels, B.; et al. PPARalpha regulates the production of serum Vanin-1 by liver. FEBS Lett. 2013, 587, 3742–3748. [Google Scholar] [Green Version]
- Comelli, E.M.; Lariani, S.; Zwahlen, M.-C.; Fotopoulos, G.; Holzwarth, J.A.; Cherbut, C.; Dorta, G.; Corthésy-Theulaz, I.; Grigorov, M. Biomarkers of human gastrointestinal tract regions. Mamm. Genome 2009, 20, 516–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, P.A.M.; Kamsteeg, M.; Rodijk-Olthuis, D.; van Vlijmen-Willems, I.M.J.J.; de Jongh, G.J.; Bergers, M.; Tjabringa, G.S.; Zeeuwen, P.L.J.M.; Schalkwijk, J. Expression of the vanin gene family in normal and inflamed human skin: Induction by proinflammatory cytokines. J. Invest. Derm. 2009, 129, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Pitari, G.; Malergue, F.; Martin, F.; Philippe, J.M.; Massucci, M.T.; Chabret, C.; Maras, B.; Duprè, S.; Naquet, P.; Galland, F. Pantetheinase activity of membrane-bound Vanin-1: Lack of free cysteamine in tissues of Vanin-1 deficient mice. FEBS Lett. 2000, 483, 149–154. [Google Scholar] [CrossRef]
- Kaskow, B.J.; Michael Proffit, J.; Blangero, J.; Moses, E.K.; Abraham, L.J. Diverse biological activities of the vascular non-inflammatory molecules—The Vanin pantetheinases. Biochem. Biophys. Res. Commun. 2012, 417, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Watanabe, T.; Sakurai, S.; Ohtake, K.; Kinoshita, T.; Araki, A.; Fujita, T.; Takei, H.; Takeda, Y.; Sato, Y.; et al. A novel glycosylphosphatidyl inositol-anchored protein on human leukocytes: A possible role for regulation of neutrophil adherence and migration. J. Immunol. 1999, 162, 4277–4284. [Google Scholar] [PubMed]
- Martin, F.; Malergue, F.; Pitari, G.; Philippe, J.M.; Philips, S.; Chabret, C.; Granjeaud, S.; Mattei, M.G.; Mungall, A.J.; Naquet, P.; et al. Vanin genes are clustered (human 6q22-24 and mouse 10A2B1) and encode isoforms of pantetheinase ectoenzymes. Immunogenetics 2001, 53, 296–306. [Google Scholar] [CrossRef]
- Granjeaud, S.; Naquet, P.; Galland, F. An ESTs description of the new Vanin gene family conserved from fly to human. Immunogenetics 1999, 49, 964–972. [Google Scholar] [CrossRef]
- Sobolesky, P.; Parry, C.; Boxall, B.; Wells, R.; Venn-Watson, S.; Janech, M.G. Proteomic Analysis of Non-depleted Serum Proteins from Bottlenose Dolphins Uncovers a High Vanin-1 Phenotype. Sci. Rep. 2016, 6, 33879. [Google Scholar] [CrossRef]
- Naquet, P.; Pitari, G.; Duprè, S.; Galland, F. Role of the Vnn1 pantetheinase in tissue tolerance to stress. Biochem. Soc. Trans. 2014, 42, 1094–1100. [Google Scholar] [CrossRef]
- Theodoulou, F.L.; Sibon, O.C.M.; Jackowski, S.; Gout, I. Coenzyme A and its derivatives: Renaissance of a textbook classic. Biochem. Soc. Trans. 2014, 42, 1025–1032. [Google Scholar] [CrossRef]
- Leonardi, R.; Zhang, Y.; Rock, C.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T.; Schweitzer, C.; Pearson, J.; Song, W.O.; Windham, C.T.; Wyse, B.W.; Hansen, R.G. Enzymes for liberation of pantothenic acid in blood: Use of plasma pantetheinase. Am. J. Clin. Nutr. 1989, 50, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, D.; Dupre, S.; Graziani, M.T.; Tinti, M.G. Identification of pantethinase in horse kidney extract. FEBS Lett. 1968, 1, 119–121. [Google Scholar] [CrossRef] [Green Version]
- Dupre, S.; Graziani, M.T.; Rosei, M.A.; Fabi, A.; Grosso, E. The Enzymatic Breakdown of Pantethine to Pantothenic Acid and Cystamine. Eur. J. Biochem. 1970, 16, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Maras, B.; Barra, D.; Duprè, S.; Pitari, G. Is pantetheinase the actual identity of mouse and human vanin-1 proteins? FEBS Lett. 1999, 461, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Kessler, A.; Biasibetti, M.; da Silva Melo, D.A.; Wajner, M.; Dutra-Filho, C.S.; de Souza Wyse, Â.T.; Wannmacher, C.M.D. Antioxidant Effect of Cysteamine in Brain Cortex of Young Rats. Neurochem. Res. 2008, 33, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Kavian, N.; Mehlal, S.; Marut, W.; Servettaz, A.; Giessner, C.; Bourges, C.; Nicco, C.; Chéreau, C.; Lemaréchal, H.; Dutilh, M.-F.; et al. Imbalance of the Vanin-1 Pathway in Systemic Sclerosis. J. Immunol. 2016, 197, 3326–3335. [Google Scholar] [CrossRef] [Green Version]
- Weimann; Hermann. Studies on Wound Healing: Effects of Calcium D-Pantothenate on the Migration, Proliferation and Protein Synthesis of Human Dermal Fibroblasts in Culture. Int. J. Vitam. Nutr. Res. 1999, 69, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Kusama, M.; Onda, M.; Nakahata, N. The effect of pantothenic acid deficiency on keratinocyte proliferation and the synthesis of keratinocyte growth factor and collagen in fibroblasts. J. Pharm. Sci. 2011, 115, 230–234. [Google Scholar] [CrossRef]
- Giessner, C.; Millet, V.; Mostert, K.J.; Gensollen, T.; Vu Manh, T.-P.; Garibal, M.; Dieme, B.; Attaf-Bouabdallah, N.; Chasson, L.; Brouilly, N.; et al. Vnn1 pantetheinase limits the Warburg effect and sarcoma growth by rescuing mitochondrial activity. Life Sci. Alliance 2018, 1, e201800073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, D.W.; Naquet, P.; Manautou, J.E. Influence of Vanin-1 and Catalytic Products in Liver During Normal and Oxidative Stress Conditions. Curr. Med. Chem. 2015, 22, 2407–2416. [Google Scholar] [CrossRef] [PubMed]
- O’Brian, C.A.; Chu, F. ReviewPost-translational disulfide modifications in cell signaling—Role of inter-protein, intra-protein, S-glutathionyl, and S-cysteaminyl disulfide modifications in signal transmission. Free Radic. Res. 2005, 39, 471–480. [Google Scholar]
- Besouw, M.; Masereeuw, R.; van den Heuvel, L.; Levtchenko, E. Cysteamine: An old drug with new potential. Drug Discov. Today 2013, 18, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Lawrence, D.A. Mechanisms for the cytotoxicity of cysteamine. Toxicol. Sci. Off. J. Soc. Toxicol. 2001, 63, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Lebo, R.V.; Kredich, N.M. Inactivation of human gamma-glutamylcysteine synthetase by cystamine. Demonstration and quantification of enzyme-ligand complexes. J. Biol. Chem. 1978, 253, 2615–2623. [Google Scholar]
- Berruyer, C.; Martin, F.M.; Castellano, R.; Macone, A.; Malergue, F.; Garrido-Urbani, S.; Millet, V.; Imbert, J.; Dupre, S.; Pitari, G.; et al. Vanin-1-/- Mice Exhibit a Glutathione-Mediated Tissue Resistance to Oxidative Stress. Mol. Cell. Biol. 2004, 24, 7214–7224. [Google Scholar] [CrossRef]
- Seelig, G.F.; Meister, A. Cystamine-Sepharose. A probe for the active site of gamma-glutamylcysteine synthetase. J. Biol. Chem. 1982, 257, 5092–5096. [Google Scholar]
- Straus, D.S.; Glass, C.K. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007, 28, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Van Diepen, J.A.; Jansen, P.A.; Ballak, D.B.; Hijmans, A.; Hooiveld, G.J.; Rommelaere, S.; Galland, F.; Naquet, P.; Rutjes, F.P.J.T.; Mensink, R.P.; et al. PPAR-alpha dependent regulation of vanin-1 mediates hepatic lipid metabolism. J. Hepatol. 2014, 61, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; Li, M.; Pritchard, P.H.; Wasan, K.M. Peroxisome proliferator-activated receptor (PPAR)-alpha: A pharmacological target with a promising future. Pharm. Res. 2004, 21, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Kuromitsu, J.; Tanaka, I. Microarray Analysis of Gene Expression Changes in Mouse Liver Induced by Peroxisome Proliferator- Activated Receptor α Agonists. Biochem. Biophys. Res. Commun. 2002, 290, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Moffit, J.; Kozataylor, P.; Holland, R.; Thibodeau, M.; Beger, R.; Lawton, M.; Manautou, J. Differential gene expression in mouse liver associated with the hepatoprotective effect of clofibrate☆. Toxicol. Appl. Pharm. 2007, 222, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Motomura, W.; Yoshizaki, T.; Takahashi, N.; Kumei, S.; Mizukami, Y.; Jang, S.-J.; Kohgo, Y. Analysis of vanin-1 upregulation and lipid accumulation in hepatocytes in response to a high-fat diet and free fatty acids. J. Clin. Biochem. Nutr. 2012, 51, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Res. 2010, 2010, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gensollen, T.; Bourges, C.; Rihet, P.; Rostan, A.; Millet, V.; Noguchi, T.; Bourdon, V.; Sobol, H.; Dubuquoy, L.; Bertin, B.; et al. Functional Polymorphisms in the Regulatory Regions of the VNN1 Gene Are Associated with Susceptibility to Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2013, 19, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Berruyer, C.; Pouyet, L.; Millet, V.; Martin, F.M.; LeGoffic, A.; Canonici, A.; Garcia, S.; Bagnis, C.; Naquet, P.; Galland, F. Vanin-1 licenses inflammatory mediator production by gut epithelial cells and controls colitis by antagonizing peroxisome proliferator-activated receptor gamma activity. J. Exp. Med. 2006, 203, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.-X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, W.; Tang, C.; Tang, X.; Liu, L.; Liu, C. Vanin-1 Is a Key Activator for Hepatic Gluconeogenesis. Diabetes 2014, 63, 2073–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutschenreuther, A.; Birkenmeier, G.; Bigl, M.; Krohn, K.; Birkemeyer, C. Glycerophosphoglycerol, Beta-Alanine, and Pantothenic Acid as Metabolic Companions of Glycolytic Activity and Cell Migration in Breast Cancer Cell Lines. Metabolites 2013, 3, 1084–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povero, D.; Eguchi, A.; Niesman, I.R.; Andronikou, N.; de Mollerat du Jeu, X.; Mulya, A.; Berk, M.; Lazic, M.; Thapaliya, S.; Parola, M.; et al. Lipid-Induced Toxicity Stimulates Hepatocytes to Release Angiogenic Microparticles That Require Vanin-1 for Uptake by Endothelial Cells. Sci. Signal. 2013, 6, ra88. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; de Araujo Horcel, L.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-Induced Hepatocyte-Derived Extracellular Vesicles Regulate Hepatic Stellate Cells via MicroRNA Targeting Peroxisome Proliferator-Activated Receptor-γ. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Katz, N.R. Metabolic Heterogeneity of Hepatocytes across the Liver Acinus. J. Nutr. 1992, 122, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARα: Energy Combustion, Hypolipidemia, Inflammation and Cancer. Nucl. Recept. Signal. 2010, 8, nrs.08002. [Google Scholar] [CrossRef] [PubMed]
- Kahle, M.; Horsch, M.; Fridrich, B.; Seelig, A.; Schultheiß, J.; Leonhardt, J.; Irmler, M.; Beckers, J.; Rathkolb, B.; Wolf, E.; et al. Phenotypic comparison of common mouse strains developing high-fat diet-induced hepatosteatosis. Mol. Metab. 2013, 2, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.A.M.; van Diepen, J.A.; Ritzen, B.; Zeeuwen, P.L.J.M.; Cacciatore, I.; Cornacchia, C.; van Vlijmen-Willems, I.M.J.J.; de Heuvel, E.; Botman, P.N.M.; Blaauw, R.H.; et al. Discovery of Small Molecule Vanin Inhibitors: New Tools To Study Metabolism and Disease. ACS Chem. Biol. 2013, 8, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Campana, L.; Iredale, J. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults: Systematic review: Epidemiology of NAFLD and NASH. Aliment. Pharm. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Teli, M.R.; James, O.F.; Burt, A.D.; Bennett, M.K.; Day, C.P. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatol. Baltim. Md 1995, 22, 1714–1719. [Google Scholar] [CrossRef]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Hicklin, D.J.; Wu, Y.; Yanase, K.; Namisaki, T.; Yamazaki, M.; et al. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut 2003, 52, 1347–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Sukhatme, V.P. Fibrosis and angiogenesis. Curr. Opin. Nephrol. Hypertens. 2000, 9, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Cayón, A.; Crespo, J.; Guerra, A.R.; Pons-Romero, F. Gene expression in obese patients with non-alcoholic steatohepatitis. Rev. Espanola Enfermedades Dig. Organo Soc. Esp. Patol. Dig. 2008, 100, 212–218. [Google Scholar]
- Kitade, M.; Yoshiji, H.; Noguchi, R.; Ikenaka, Y.; Kaji, K.; Shirai, Y.; Yamazaki, M.; Uemura, M.; Yamao, J.; Fujimoto, M.; et al. Crosstalk between angiogenesis, cytokeratin-18, and insulin resistance in the progression of non-alcoholic steatohepatitis. World J. Gastroenterol. 2009, 15, 5193–5199. [Google Scholar] [CrossRef]
- Schumacher, J.D.; Guo, G.L. Regulation of Hepatic Stellate Cells and Fibrogenesis by Fibroblast Growth Factors. BioMed Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, E.L.; Cho, H.; Birnbaum, M.J. Role of Akt/protein kinase B in metabolism. Trends Endocrinol. Metab. TEM 2002, 13, 444–451. [Google Scholar] [CrossRef]
- Ye, H.; Nelson, L.J.; del Moral, M.G.; Martínez-Naves, E.; Cubero, F.J. Dissecting the molecular pathophysiology of drug-induced liver injury. World J. Gastroenterol. 2018, 24, 1373–1385. [Google Scholar] [CrossRef]
- Ferreira, D.W.; Goedken, M.J.; Rommelaere, S.; Chasson, L.; Galland, F.; Naquet, P.; Manautou, J.E. Enhanced hepatotoxicity by acetaminophen in Vanin-1 knockout mice is associated with deficient proliferative and immune responses. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 662–669. [Google Scholar] [CrossRef]
- O’Connor, M.A.; Koza-Taylor, P.; Campion, S.N.; Aleksunes, L.M.; Gu, X.; Enayetallah, A.E.; Lawton, M.P.; Manautou, J.E. Analysis of changes in hepatic gene expression in a murine model of tolerance to acetaminophen hepatotoxicity (autoprotection). Toxicol. Appl. Pharm. 2014, 274, 156–167. [Google Scholar] [CrossRef]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.-H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessely, O.; Cerqueira, D.M.; Tran, U.; Kumar, V.; Hassey, J.M.; Romaker, D. The bigger the better: Determining nephron size in kidney. Pediatr. Nephrol. 2014, 29, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kurella, M.; Beato, F.; Min, H.; Ingelfinger, J.R.; Stears, R.L.; Swinford, R.D.; Gullans, S.R.; Tang, S.-S. Monitoring changes in gene expression in renal ischemia-reperfusion in the rat. Kidney Int. 2002, 61, 1646–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosohata, K.; Ando, H.; Fujiwara, Y.; Fujimura, A. Vanin-1; a potential biomarker for nephrotoxicant-induced renal injury. Toxicology 2011, 290, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.; McMartin, K.E. Methanol and ethylene glycol poisonings. Mechanism of toxicity, clinical course, diagnosis and treatment. Med. Toxicol. 1986, 1, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Poldelski, V.; Johnson, A.; Wright, S.; Dela Rosa, V.; Zager, R.A. Ethylene glycol [ndash] mediated tubular injury: Identification of critical metabolites and injury pathways. Am. J. Kidney Dis. 2001, 38, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K.; Ando, H.; Fujimura, A. Urinary Vanin-1 As a Novel Biomarker for Early Detection of Drug-Induced Acute Kidney Injury. J. Pharm. Exp. Ther. 2012, 341, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Nigam, S.K. Acute renal failure. I. Relative importance of proximal vs. distal tubular injury. Am. J. Physiol. 1998, 275, F623–F631. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Xie, X.; Ao, Y.; Tie, C.; Song, R. Differential roles of dihydropyridine calcium antagonist nifedipine, nitrendipine and amlodipine on gentamicin-induced renal tubular toxicity in rats. Eur. J. Pharm. 2009, 620, 97–104. [Google Scholar] [CrossRef]
- Hosohata, K.; Jin, D.; Takai, S.; Iwanaga, K. Vanin-1 in Renal Pelvic Urine Reflects Kidney Injury in a Rat Model of Hydronephrosis. Int. J. Mol. Sci. 2018, 19, 3186. [Google Scholar] [CrossRef]
- Washino, S.; Hosohata, K.; Oshima, M.; Okochi, T.; Konishi, T.; Nakamura, Y.; Saito, K.; Miyagawa, T. A Novel Biomarker for Acute Kidney Injury, Vanin-1, for Obstructive Nephropathy: A Prospective Cohort Pilot Study. Int. J. Mol. Sci. 2019, 20, 899. [Google Scholar] [CrossRef] [PubMed]
- Fugmann, T.; Borgia, B.; Révész, C.; Godó, M.; Forsblom, C.; Hamar, P.; Holthöfer, H.; Neri, D.; Roesli, C. Proteomic identification of vanin-1 as a marker of kidney damage in a rat model of type 1 diabetic nephropathy. Kidney Int. 2011, 80, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roisin-Bouffay, C.; Castellano, R.; Valéro, R.; Chasson, L.; Galland, F.; Naquet, P. Mouse vanin-1 is cytoprotective for islet beta cells and regulates the development of type 1 diabetes. Diabetologia 2008, 51, 1192–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.; Penet, M.-F.; Malergue, F.; Lepidi, H.; Dessein, A.; Galland, F.; de Reggi, M.; Naquet, P.; Gharib, B. Vanin-1–/– mice show decreased NSAID- and Schistosoma-induced intestinal inflammation associated with higher glutathione stores. J. Clin. Investig. 2004, 113, 591–597. [Google Scholar] [CrossRef]
- Dessein, A.J.; Hillaire, D.; Elwali, N.E.M.A.; Marquet, S.; Mohamed-Ali, Q.; Mirghani, A.; Henri, S.; Abdelhameed, A.A.; Saeed, O.K.; Magzoub, M.M.A.; et al. Severe Hepatic Fibrosis in Schistosoma mansoni Infection Is Controlled by a Major Locus That Is Closely Linked to the Interferon-γ Receptor Gene. Am. J. Hum. Genet. 1999, 65, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Hampe, J. Genetics and inflammatory bowel disease. Curr. Opin. Gastroenterol. 1999, 15, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.A.; Itoh, J.; Fiocchi, C. Pathogenesis of inflammatory bowel disease. Curr. Opin. Gastroenterol. 1999, 15, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kawada, M. Insights from advances in research of chemically induced experimental models of human inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 5581. [Google Scholar] [CrossRef]
- Kerkel, K.; Spadola, A.; Yuan, E.; Kosek, J.; Jiang, L.; Hod, E.; Li, K.; Murty, V.V.; Schupf, N.; Vilain, E.; et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008, 40, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Tayo, B.O.; Bandyopadhyay, A.; Wang, H.; Feng, T.; Franceschini, N.; Tang, H.; Gao, J.; Sung, Y.J.; The COGENT BP consortium; et al. The Association of the Vanin-1 N131S Variant with Blood Pressure Is Mediated by Endoplasmic Reticulum-Associated Degradation and Loss of Function. PLoS Genet. 2014, 10, e1004641. [Google Scholar] [CrossRef]
- Xiao, C.; Biagini Myers, J.M.; Ji, H.; Metz, K.; Martin, L.J.; Lindsey, M.; He, H.; Powers, R.; Ulm, A.; Ruff, B.; et al. Vanin-1 expression and methylation discriminate pediatric asthma corticosteroid treatment response. J. Allergy Clin. Immunol. 2015, 136, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Min-Oo, G.; Fortin, A.; Pitari, G.; Tam, M.; Stevenson, M.M.; Gros, P. Complex genetic control of susceptibility to malaria: Positional cloning of the Char9 locus. J. Exp. Med. 2007, 204, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Pouyet, L.; Roisin-Bouffay, C.; Clément, A.; Millet, V.; Garcia, S.; Chasson, L.; Issaly, N.; Rostan, A.; Hofman, P.; Naquet, P.; et al. Epithelial vanin-1 controls inflammation-driven carcinogenesis in the colitis-associated colon cancer model. Inflamm. Bowel Dis. 2010, 16, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Dammanahalli, K.J.; Stevens, S.; Terkeltaub, R. Vanin-1 pantetheinase drives smooth muscle cell activation in post-arterial injury neointimal hyperplasia. PLoS ONE 2012, 7, e39106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lo, C.; Shen, L.; Sood, R.; Jones, C.; Cusmano-Ozog, K.; Park-Snyder, S.; Wong, W.; Jeng, M.; Cowan, T.; et al. The role of vanin-1 and oxidative stress-related pathways in distinguishing acute and chronic pediatric ITP. Blood 2011, 117, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Shah, Y.M. PPARα: Mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology 2008, 246, 2–8. [Google Scholar] [CrossRef]
- Bennett, M.R.; Devarajan, P. Characteristics of an Ideal Biomarker of Kidney Diseases. Biomarkers of Kidney Disease; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–20. ISBN 978-0-12-803014-1. [Google Scholar]
- Ruan, B.H.; Cole, D.C.; Wu, P.; Quazi, A.; Page, K.; Wright, J.F.; Huang, N.; Stock, J.R.; Nocka, K.; Aulabaugh, A.; et al. A fluorescent assay suitable for inhibitor screening and vanin tissue quantification. Anal. Biochem. 2010, 399, 284–292. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Jansen, P. Chemical biology tools to study pantetheinases of the vanin family. Biochem. Soc. Trans. 2014, 42, 1052–1055. [Google Scholar] [CrossRef]
- Wedel, J.; Jansen, P.A.M.; Botman, P.N.M.; Rutjes, F.P.J.T.; Schalkwijk, J.; Hillebrands, J.-L. Pharmacological Inhibition of Vanin Activity Attenuates Transplant Vasculopathy in Rat Aortic Allografts. Transplantation 2016, 100, 1656–1666. [Google Scholar] [CrossRef]


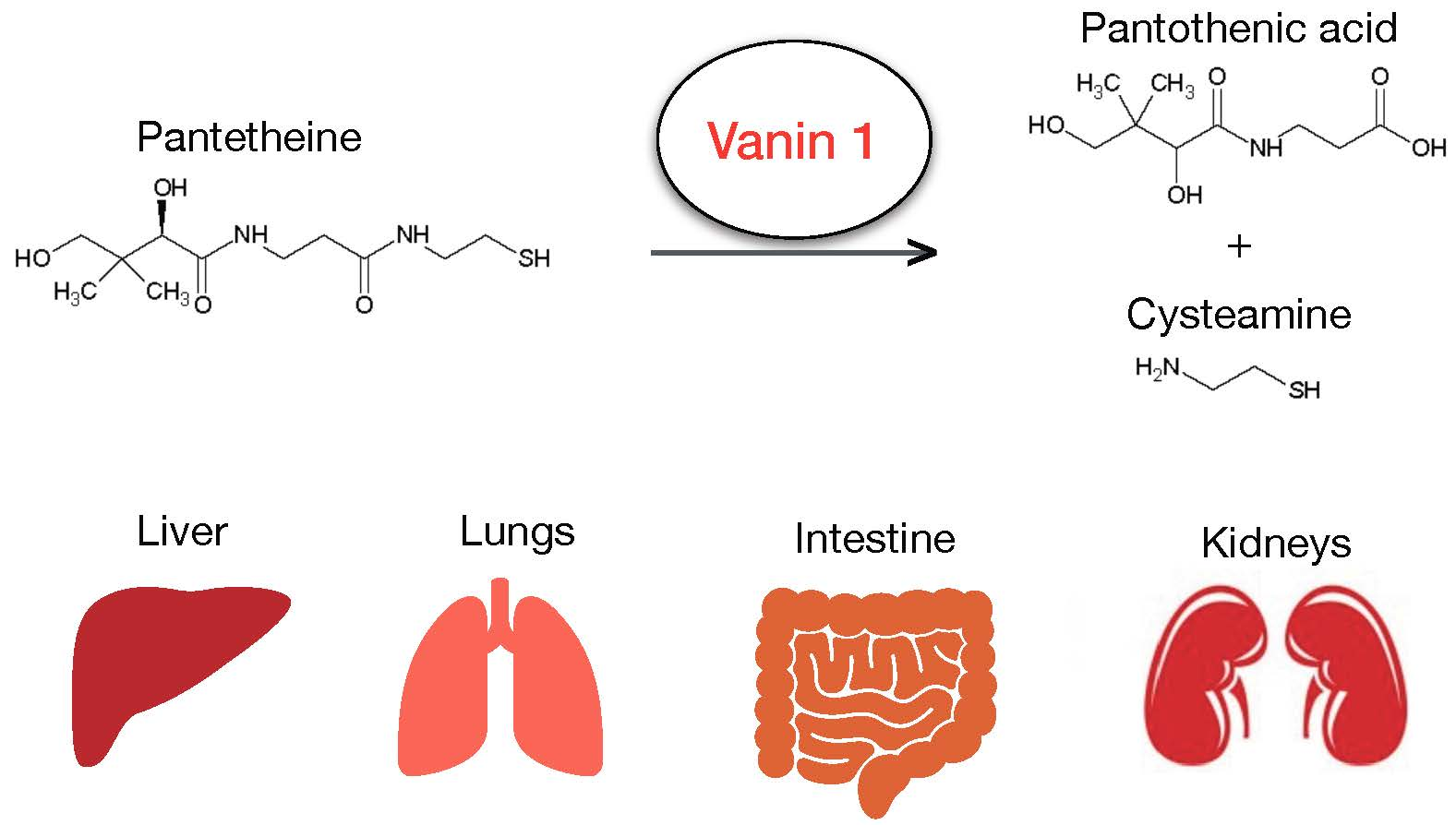{kind=link}
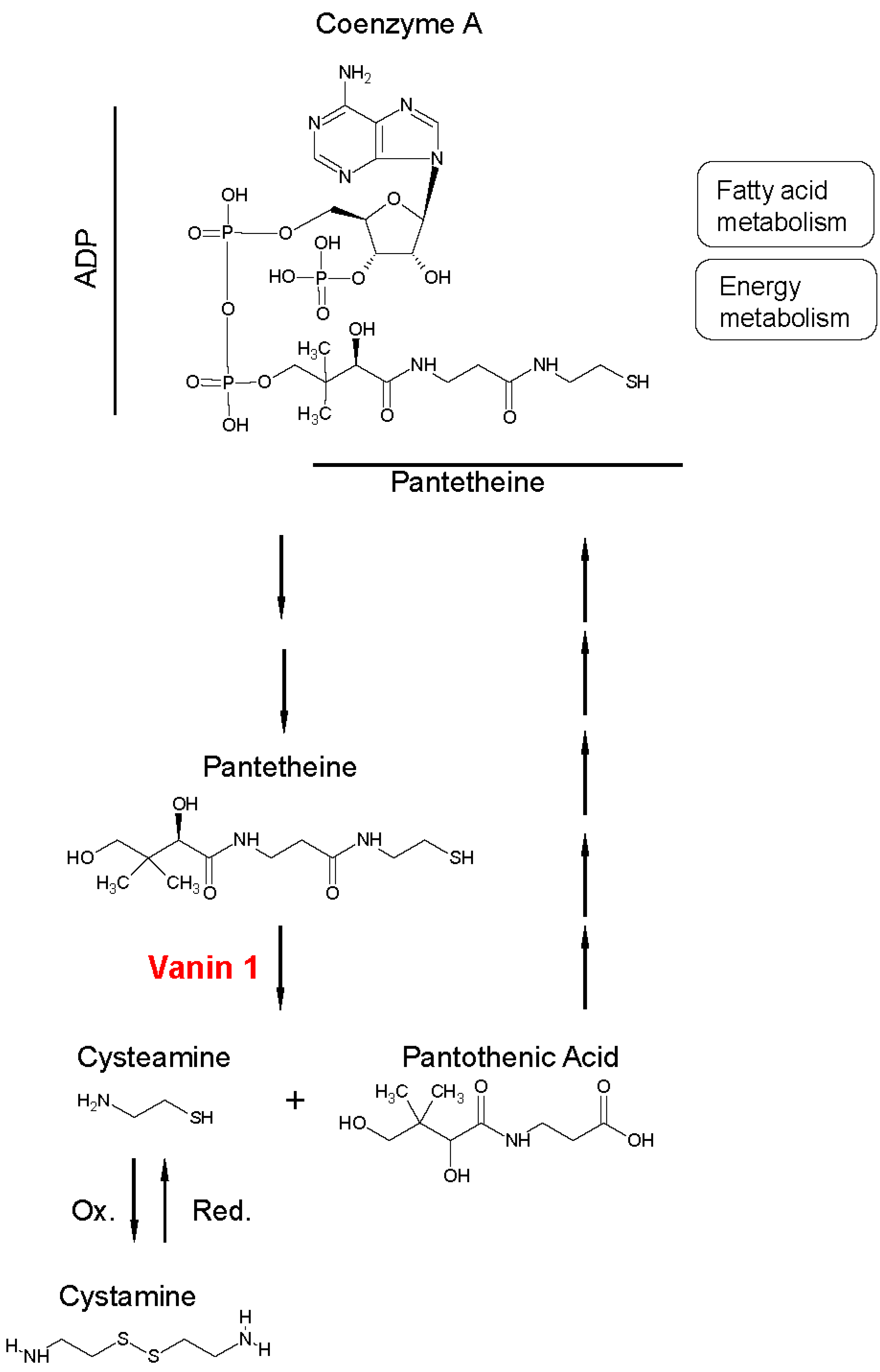{kind=link}
| Organ. | Disease | Vanin 1 |
|---|---|---|
| Liver | Steatosis | Higher transcription of gene expression [33,38,49] |
| NAFLD/NASH | Promoting MP uptake by HSCs and endothelial cells [44,45] | |
| Hepatoxicity (APAP and CCl4) | Protective role [59,60,61] | |
| Kidneys | AKI/drug-induced kidney injury/hydronephrosis and fibrosis/DN | Urinary biomarker [64,65,68,71,72] |
| Intestine | Intestinal inflammation/colitis/IBD | Pro-inflammatory role [40,75,76] Intestinal biomarker [39] |
| Lungs | Asthma | Biomarker as discriminating factor to corticosteroid treatment response [82] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartucci, R.; Salvati, A.; Olinga, P.; Boersma, Y.L. Vanin 1: Its Physiological Function and Role in Diseases. Int. J. Mol. Sci. 2019, 20, 3891. https://doi.org/10.3390/ijms20163891
Bartucci R, Salvati A, Olinga P, Boersma YL. Vanin 1: Its Physiological Function and Role in Diseases. International Journal of Molecular Sciences. 2019; 20(16):3891. https://doi.org/10.3390/ijms20163891
Chicago/Turabian StyleBartucci, Roberta, Anna Salvati, Peter Olinga, and Ykelien L. Boersma. 2019. "Vanin 1: Its Physiological Function and Role in Diseases" International Journal of Molecular Sciences 20, no. 16: 3891. https://doi.org/10.3390/ijms20163891