The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia


Abstract
:1. Introduction
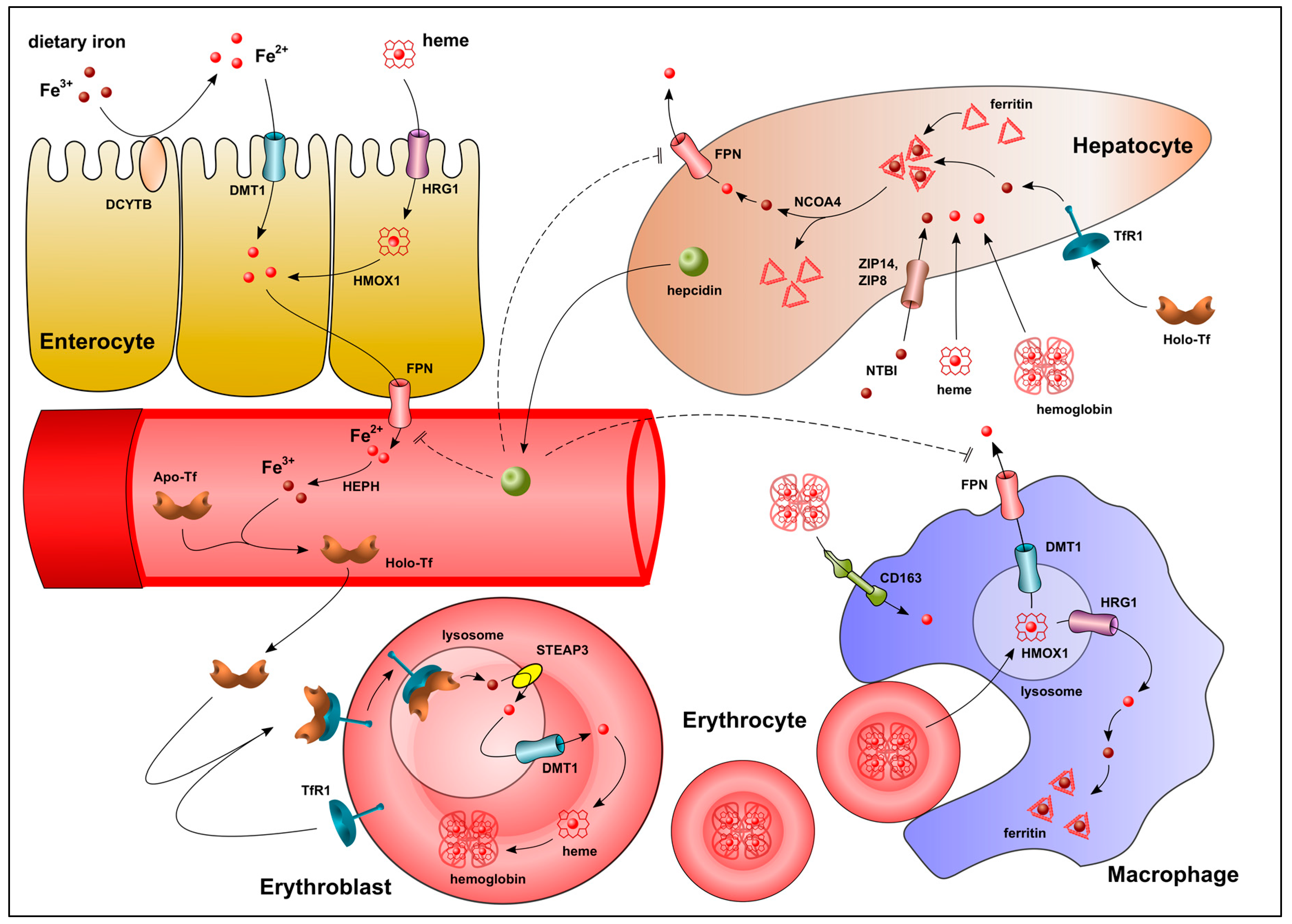
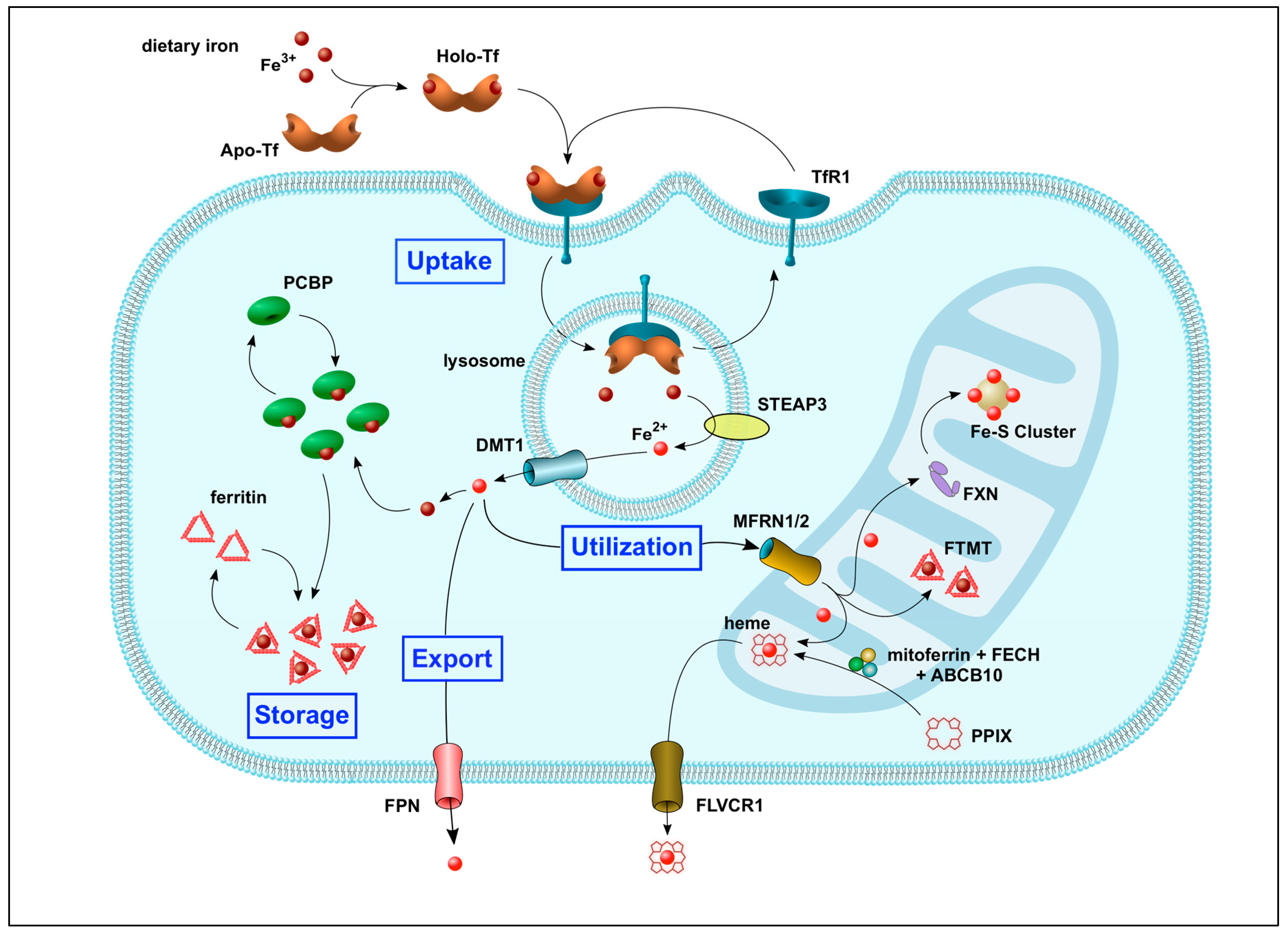
2. Systemic and Intracellular Iron Homeostasis
2.1. Regulation of Systemic Iron Homeostasis
2.2. Regulation of Intracellular Iron Trafficking
3. Impact of Dysregulated Iron Homeostasis on Reproductive Outcomes
3.1. Elevated Maternal Serum Iron/Hemoglobin Levels Are Associated with Adverse Pregnancy Outcomes
- In iron-replete women, prenatal iron supplementation throughout pregnancy may suppress maternal hepcidin production and/or activity [81], leading to continuous absorption of dietary iron, higher hemoglobin concentrations, and increased blood viscosity, ultimately compromising placental blood flow [79].
- Excess iron may impair the maternal systemic response to inflammation and infection leading to adverse birth outcome [84].
3.2. Maternal Iron Status and Preeclampsia
4. Molecular Basis for Dysregulated Iron Homeostasis in Preeclampsia
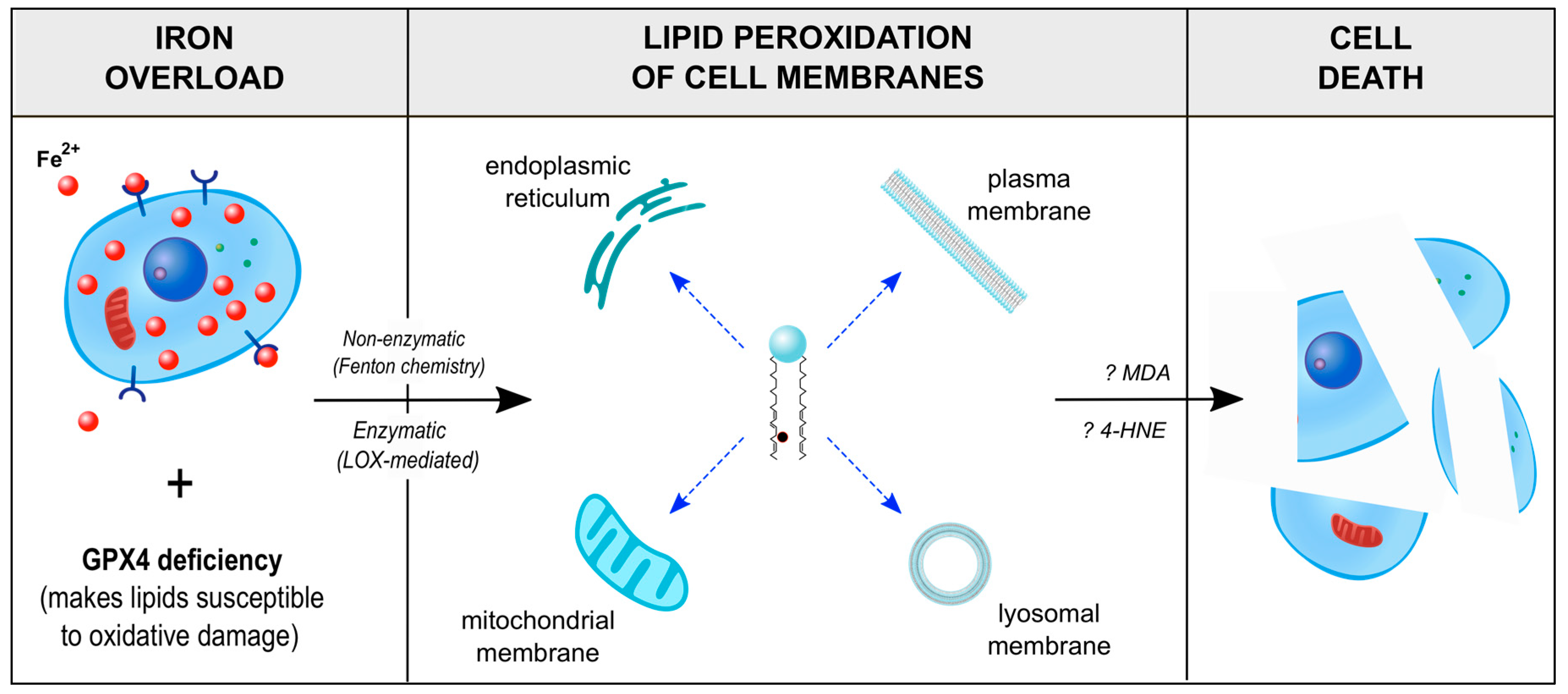
4.1. Ferroptosis, an Iron-Dependent Mechanism of Programmed Cell Death
4.2. Expression of Key Iron Homeostatic and Ferroptosis Regulatory Genes at the Maternal-Fetal Interface—Analysis of a Single-Cell RNA-Seq Dataset
- Ferritin genes, in particular Ftl, are highly expressed in almost all cell types, suggesting that intracellular iron storage and prevention of iron toxicity is likely of high importance at the maternal-fetal interface. The Fpn gene is highly expressed in trophoblasts and decidual cells relative to endometrial stromal fibroblasts. The hepcidin-FPN feedback loop is known to operate in placental syncytiotrophoblast to control iron trafficking from the placenta to the fetus [14]. Studies have also shown that FPN function varies in different organs. Deletion of the Fpn gene in mice is embryonic lethal due to a lack of iron transfer to the embryo [14]. In contrast, conditional Fpn gene deletion in intestinal enterocytes causes severe anemia. Similarly, conditional deletion in macrophages or hepatocytes is not lethal and only causes anemia when dietary iron is restricted [41]. Conditional Fpn gene deletion in cardiomyocytes causes premature cell death, suggesting that FPN may have a specific role in the heart to protect these cells from iron toxicity [131]. Whether FPN has a similar protective function in decidual cells is not known. Cellular iron homeostasis in decidual cells is likely regulated by IRP2, since the Irp2 gene is highly expressed in this cell type.
- Decidual cells likely take up iron via both the Tf and NTBI routes, as these cells have high expression levels of the Tf receptor Tfrc gene (about 3-fold higher than other cell types), Dyctb ferrireductase gene, and Dmt1 gene, all critical components of the Tf uptake pathway, as well as high expression of the Zip14 (SLC39A14) gene, important for NTBI import. Trophoblast cells, on the other hand, likely import iron as heme via the heme importer protein, FLVCR2 [132], and mitoferrin (SLC25A37 gene). The iron might be used for heme biosynthesis in the fetal compartment as suggested by the high expression of the Alas1 (δ-aminolevulinate synthase 1) gene, which codes for a rate-limiting enzyme in the heme biosynthetic pathway.
- Trophoblast cells are likely more vulnerable to ferroptosis than other cell types at the maternal-fetal interface based on the high expression of two genes: (i) Lpcat3 (lysophosphatidylcholine acyl-transferase 3), whose gene product is involved in the biosynthesis and remodeling of PUFA-phosphatidyl-ethanolamine phospholipids in cell membranes and is a critical mediator of ferroptosis [126,133]; and (ii) Sat1 (spermidine/spermine N1-acetyltransferase 1), which codes for an important enzyme in polyamine metabolism that also promotes p53-dependent ferroptotic responses [134].
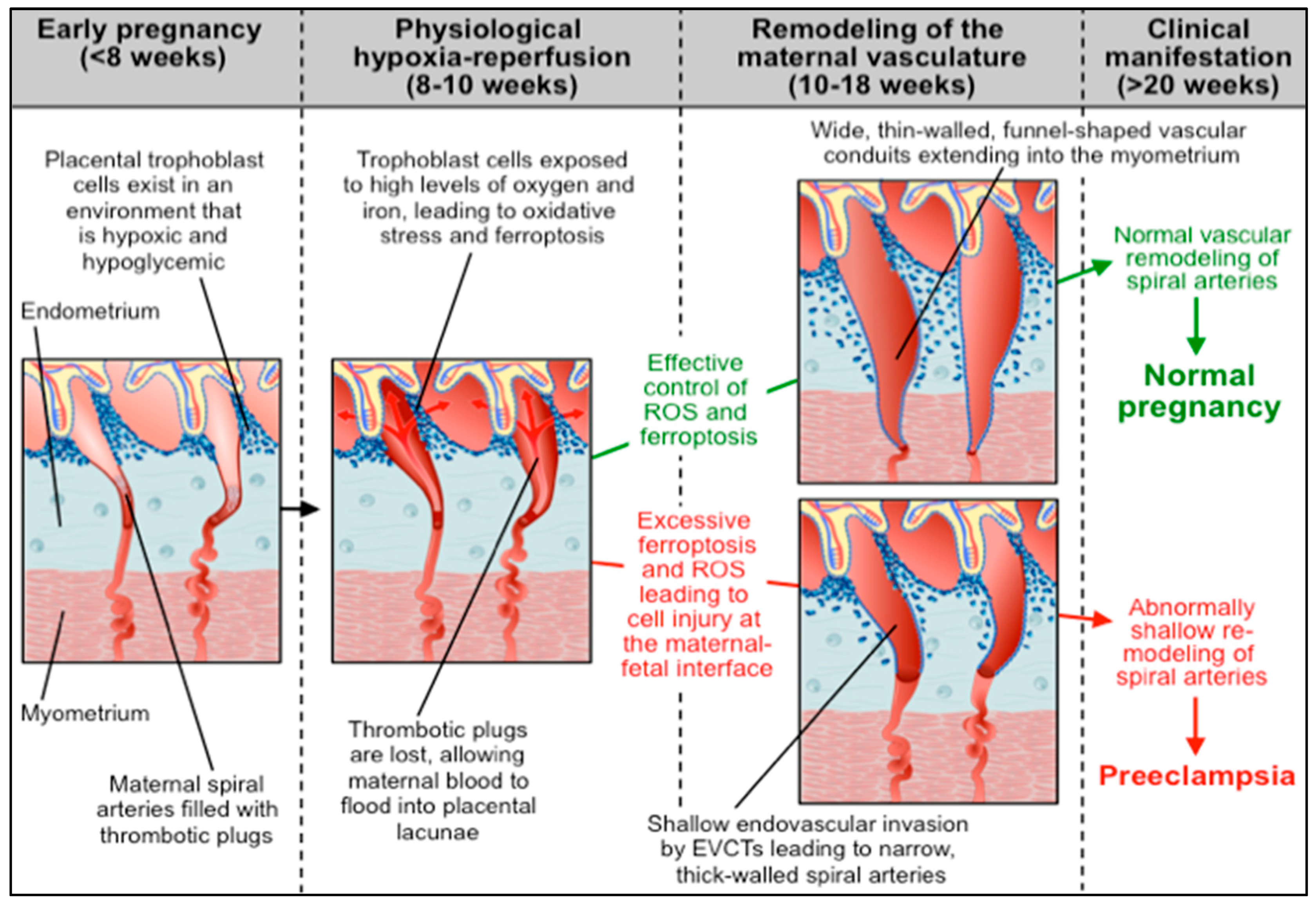
4.3. Abnormal Ferroptotic Response to Hypoxia/Reperfusion: A Novel Model for Preeclampsia
- Numerous studies investigating the association between iron status and hypertensive disorders of pregnancy have noted that levels of circulating iron and ferritin, as well as the level of Tf saturation, are significantly higher in PE patients as compared to normotensive controls [135,136,137]. Whether or not the increased serum iron in PE is due to low circulating hepcidin levels is unclear. Studies by Koenig et al. [81] and by Brunacci et al. [138] suggest that high serum iron and Tf saturation in PE women correlate with lower hepcidin levels. However, a study by Duvan et al. found no significant correlation between prohepcidin concentrations and serum iron, ferritin, or Tf levels in women with PE [139].
- A metabolomic analysis of placental mitochondria has identified that severe PE patients have significantly higher PUFA levels and other mitochondrial abnormalities relative to normotensive controls [144].
- Proteomic analysis has shown that patients with severe PE have significantly higher concentrations of free α- and β-hemoglobin in their cerebrospinal fluid (CSF) as compared to women with non-severe PE or normotensive controls [145]. Similarly, van den Berg et al. reported that CSF levels of the heme-binding protein, AMBP (α-1-microglobulin/bikunin precursor), are markedly elevated in women with severe PE relative to normotensive controls [146]. Taken together, these data suggest that patients with PE may have difficulty disposing of free hemoglobin and associated proteins.
- In many cases of PE, lipid-filled ‘foam cells’ (macrophages containing low density lipoproteins) accumulate in the walls of the spiral arteries, reminiscent of the early stages of atherosclerosis [147]. Although this histologic aberration was observed decades ago, the mechanisms that contribute to this acute atherosis are largely unknown. Ferroptosis with lipid peroxidation could be a contributing factor.
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ABCB10 | ATP Binding Cassette super-family B member 10 |
| Alas1 | δ-aminolevulinate synthase 1 |
| AMBP | α-1-Microglobulin/Bikunin Precursor |
| Apo-Tf | Apo-Transferrin |
| CD163 | Cluster of Differentiation 163 |
| CI | Confidence Interval |
| CP | Ceruloplasmin |
| CSF | Cerebrospinal Fluid |
| CYT | Cytotrophoblast |
| DC | Dendritic Cell |
| DCYTB | Duodenal Cytochrome B |
| DEC | Decidual Cell |
| DMT1 | Divalent Metal Transporter 1 |
| DNA | Deoxyribonucleic Acid |
| ESF | Endometrial Stromal Fibroblast |
| EVCT | Extravillous Cytotrophoblast |
| FBXL5 | F-Box and Leucine rich repeat protein 5 |
| Fe2+ | Ferrous Iron |
| Fe3+ | Ferric Iron |
| FECH | Ferrochelatase |
| Fe-S cluster | Iron-Sulfur Cluster |
| FLVCR1 | Feline Leukemia Virus subgroup C Receptor-related protein 1 |
| FPN | Ferroportin |
| FTH1 | Ferritin Heavy chain 1 |
| FTL | Ferritin Light chain |
| FTMT | Mitoferritin |
| FXN | Frataxin |
| GPX4 | Glutathione Peroxidase 4 |
| HEPH | Hephaestin |
| HIF1α | Hypoxia-Inducible Factor 1 alpha |
| HIF2α | Hypoxia-Inducible Factor 2 alpha |
| Holo-Tf | Holo-Transferrin |
| HMOX1 | Heme Oxygenase 1 |
| HMOX2 | Heme Oxygenase 2 |
| HRG1 | Heme-Responsive Gene-1 |
| IRE | Iron-Responsive Element |
| IRP1 | Iron Regulatory Protein 1 |
| IRP2 | Iron Regulatory Protein 2 |
| JMJD6 | Jumonji Domain-containing protein 6 |
| LBW | Low Birthweight |
| LPCAT | Lysophosphatidylcholine Acyl- Transferase 3 |
| MDA | Malondialdehyde |
| MFRN1 | Mitoferrin 1 |
| MFRN2 | Mitoferrin 2 |
| NAPDH | Nicotinamide Adenine Dinucleotide Phosphate Hydrogen |
| NCOA4 | Nuclear receptor Coactivator 4 |
| NIH | National Institute of Health |
| NTBI | Non-Transferrin Bound Iron |
| OR | Odds Ratio |
| PCBP1 | Poly-(rC)-Binding Protein 1 |
| PCBP2 | Poly-(rC)-Binding Protein 2 |
| PE | Preeclampsia |
| PPIX | Protoporphyrin IX |
| PTB | Preterm Birth |
| PUFA | Polyunsaturated Fatty Acid |
| RES | Reticuloendothelial System |
| RNA | Ribonucleic Acid |
| ROS | Reactive Oxygen Species |
| SAT1 | Spermidine/spermine N1-Acetyltransferase 1 |
| SGA | Small-for-Gestational Age |
| STEAP3 | Six-Transmembrane Epithelial Antigen of Prostate 3 |
| SYN | Syncytiotrophoblast |
| Tf | Transferrin |
| TfR1 | Transferrin Receptor 1 |
| Tfrc | Transferrin Receptor C |
| TPM | Transcripts per Million |
| VHL | Von Hippel Lindau tumor suppressor gene |
| WHO | World Health Organization |
| ZIP8 | Zrt- and Irt-like Protein 8 |
| ZIP14 | Zrt- and Irt-like Protein 14 |
References
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Eschbach, J.W. Iron requirements in erythropoietin therapy. Best Pract. Res. Clin. Haematol. 2005, 18, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.L.; Nemeth, E. Iron homeostasis during pregnancy. Am. J. Clin. Nutr. 2017, 106, 1567S–1574S. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Buchanan, G.R.; Adix, L.; Zhang, S.; Gao, A.; McCavit, T.L. Effect of low-dose ferrous sulfate vs iron polysaccharide complex on hemoglobin concentration in young children with nutritional iron-deficiency anemia: A randomized clinical trial. JAMA 2017, 317, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Iron deficiency—United States, 1999–2000. MMWR 2002, 51, 897–899. [Google Scholar]
- Brannon, P.M.; Taylor, C.L. Iron supplementation during pregnancy and infancy: Uncertainties and implications for research and policy. Nutrients 2017, 9, 1327. [Google Scholar] [CrossRef]
- Raper, N.R.; Rosenthal, J.C.; Woteki, C.E. Estimates of available iron in diets of individuals 1 year old and older in the nationwide food consumption survey. J. Am. Diet. Assoc. 1984, 84, 783–787. [Google Scholar]
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; Direnzo, C.; Robine, S.; Andrews, N.C. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Investig. 2005, 115, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Shawki, A.; Anthony, S.R.; Nose, Y.; Engevik, M.A.; Niespodzany, E.J.; Barrientos, T.; Ohrvik, H.; Worrell, R.T.; Thiele, D.J.; Mackenzie, B. Intestinal dmt1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G635–G647. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of hrg-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, L.R.; Weinstein, M.B.; Huser, H.J.; Rafal, S. Absorption of hemoglobin iron: The role of a heme-splitting substance in the intestinal mucosa. J. Clin. Investig. 1968, 47, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, ireg1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Mitchell, C.J.; Shawki, A.; Ganz, T.; Nemeth, E.; Mackenzie, B. Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc. Am. J. Physiol. Cell Physiol. 2014, 306, C450–C459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [Green Version]
- Tabuchi, M.; Yoshimori, T.; Yamaguchi, K.; Yoshida, T.; Kishi, F. Human nramp2/dmt1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in hep-2 cells. J. Biol. Chem 2000, 275, 22220–22228. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Zhang, A.S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007, 110, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter flvcr1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef]
- Knutson, M.; Wessling-Resnick, M. Iron metabolism in the reticuloendothelial system. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 61–88. [Google Scholar] [CrossRef]
- Knutson, M.D. Iron transport proteins: Gateways of cellular and systemic iron homeostasis. J. Biol. Chem. 2017, 292, 12735–12743. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. Hrg1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef] [Green Version]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Schaer, C.A.; Schoedon, G.; Imhof, A.; Kurrer, M.O.; Schaer, D.J. Constitutive endocytosis of cd163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ. Res. 2006, 99, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Carmona, U.; Li, L.; Zhang, L.; Knez, M. Ferritin light-chain subunits: Key elements for the electron transfer across the protein cage. Chem. Commun. (Camb) 2014, 50, 15358–15361. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Wright, T.L.; Ma, W.L.; Weisiger, R.A. Efficient clearance of non-transferrin-bound iron by rat liver. Implications for hepatic iron loading in iron overload states. J. Clin. Investig. 1985, 76, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- Jenkitkasemwong, S.; Wang, C.Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.S.; Hojyo, S.; Fukada, T.; Knutson, M.D. Slc39a14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective vps34 inhibitor blocks autophagy and uncovers a role for ncoa4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies ncoa4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, F.; Guo, X.; An, P.; Tao, Y.; Wang, F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 2012, 56, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D.; Oukka, M.; Koss, L.M.; Aydemir, F.; Wessling-Resnick, M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 2005, 102, 1324–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Sham, R.L.; Phatak, P.D.; Nemeth, E.; Ganz, T. Hereditary hemochromatosis due to resistance to hepcidin: High hepcidin concentrations in a family with c326s ferroportin mutation. Blood 2009, 114, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Altamura, S.; Kessler, R.; Grone, H.J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (fpn1) transcription involving bach1, nrf2 and a mare/are sequence motif at position -7007 of the fpn1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef]
- Anderson, E.R.; Taylor, M.; Xue, X.; Ramakrishnan, S.K.; Martin, A.; Xie, L.; Bredell, B.X.; Gardenghi, S.; Rivella, S.; Shah, Y.M. Intestinal hif2alpha promotes tissue-iron accumulation in disorders of iron overload with anemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4922–E4930. [Google Scholar] [CrossRef]
- Guida, C.; Altamura, S.; Klein, F.A.; Galy, B.; Boutros, M.; Ulmer, A.J.; Hentze, M.W.; Muckenthaler, M.U. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 2015, 125, 2265–2275. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of iron homeostasis: New players in metabolism, cell death, and disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each member of the poly-r(c)-binding protein 1 (pcbp) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the hif prolyl hydroxylase by the iron chaperones pcbp1 and pcbp2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and copper in mitochondrial diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the fe-s cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.M.; Greer, J.M.; Ponka, P.; Richardson, D.R. Erythroid differentiation and protoporphyrin ix down-regulate frataxin expression in friend cells: Characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood 2002, 99, 3813–3822. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Becker, E.M.; Whitnall, M.; Suryo Rahmanto, Y.; Ponka, P.; Richardson, D.R. Elucidation of the mechanism of mitochondrial iron loading in friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef]
- Ponka, P.; Wilczynska, A.; Schulman, H.M. Iron utilization in rabbit reticulocytes. A study using succinylacetone as an inhibitor or heme synthesis. Biochim. Biophys. Acta 1982, 720, 96–105. [Google Scholar] [CrossRef]
- Mochel, F.; Knight, M.A.; Tong, W.H.; Hernandez, D.; Ayyad, K.; Taivassalo, T.; Andersen, P.M.; Singleton, A.; Rouault, T.A.; Fischbeck, K.H.; et al. Splice mutation in the iron-sulfur cluster scaffold protein iscu causes myopathy with exercise intolerance. Am. J. Hum. Genet. 2008, 82, 652–660. [Google Scholar] [CrossRef]
- Wingert, R.A.; Galloway, J.L.; Barut, B.; Foott, H.; Fraenkel, P.; Axe, J.L.; Weber, G.J.; Dooley, K.; Davidson, A.J.; Schmid, B.; et al. Deficiency of glutaredoxin 5 reveals fe-s clusters are required for vertebrate haem synthesis. Nature 2005, 436, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Campanella, A.; De Falco, L.; Boschetto, L.; Merlini, R.; Silvestri, L.; Levi, S.; Iolascon, A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 2007, 110, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Lane, D.J.; Richardson, D.R. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 2011, 15, 3003–3019. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The irp/ire system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Wallander, M.L.; Leibold, E.A.; Eisenstein, R.S. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta 2006, 1763, 668–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Phillips, J.D.; Yu, Y.; Leibold, E.A. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J. Biol. Chem. 1995, 270, 21645–21651. [Google Scholar] [CrossRef] [PubMed]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Rouault, T.A. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science 2004, 306, 2087–2090. [Google Scholar] [CrossRef]
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-mediated biological control. J. Cell. Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef]
- Mole, D.R. Iron homeostasis and its interaction with prolyl hydroxylases. Antioxid. Redox Signal. 2010, 12, 445–458. [Google Scholar] [CrossRef]
- WHO. World-Wide Prevalence of Anemia 1993–2005: Who Global Database on Anemia; WHO Press: Geneva, Switzerland, 2005. [Google Scholar]
- WHO. Guideline: Daily Iron and Folic Acid Supplementation in Pregnant Women; WHO Press: Geneva, Switzerland, 2012. [Google Scholar]
- Steer, P.; Alam, M.A.; Wadsworth, J.; Welch, A. Relation between maternal haemoglobin concentration and birth weight in different ethnic groups. BMJ 1995, 310, 489–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garn, S.M.; Keating, M.T.; Falkner, F. Hematological status and pregnancy outcomes. Am. J. Clin. Nutr. 1981, 34, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.F.; Steenland, K.; Tapia, V. Maternal hemoglobin level and fetal outcome at low and high altitudes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1477–R1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.C.; O’Brien, K.O.; Nathanson, M.S.; Mancini, J.; Witter, F.R. Hemoglobin concentrations influence birth outcomes in pregnant african-american adolescents. J. Nutr. 2003, 133, 2348–2355. [Google Scholar] [CrossRef]
- Dewey, K.G.; Oaks, B.M. U-shaped curve for risk associated with maternal hemoglobin, iron status, or iron supplementation. Am. J. Clin. Nutr. 2017, 106, 1694S–1702S. [Google Scholar] [CrossRef] [Green Version]
- Finch, C.A.; Bellotti, V.; Stray, S.; Lipschitz, D.A.; Cook, J.D.; Pippard, M.J.; Huebers, H.A. Plasma ferritin determination as a diagnostic tool. West. J. Med. 1986, 145, 657–663. [Google Scholar]
- Ziaei, S.; Norrozi, M.; Faghihzadeh, S.; Jafarbegloo, E. A randomised placebo-controlled trial to determine the effect of iron supplementation on pregnancy outcome in pregnant women with haemoglobin > or = 13.2 g/dl. BJOG 2007, 114, 684–688. [Google Scholar] [CrossRef]
- Shastri, L.; Mishra, P.E.; Dwarkanath, P.; Thomas, T.; Duggan, C.; Bosch, R.; McDonald, C.M.; Thomas, A.; Kurpad, A.V. Association of oral iron supplementation with birth outcomes in non-anaemic south indian pregnant women. Eur. J. Clin. Nutr. 2015, 69, 609–613. [Google Scholar] [CrossRef]
- Koenig, M.D.; Tussing-Humphreys, L.; Day, J.; Cadwell, B.; Nemeth, E. Hepcidin and iron homeostasis during pregnancy. Nutrients 2014, 6, 3062–3083. [Google Scholar] [CrossRef]
- Kehrer, J.P. The haber-weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Casanueva, E.; Viteri, F.E. Iron and oxidative stress in pregnancy. J. Nutr. 2003, 133, 1700S–1708S. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Prentice, A.M. Hepcidin and the iron-infection axis. Science 2012, 338, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Jaeggi, T.; Kortman, G.A.; Moretti, D.; Chassard, C.; Holding, P.; Dostal, A.; Boekhorst, J.; Timmerman, H.M.; Swinkels, D.W.; Tjalsma, H.; et al. Iron fortification adversely affects the gut microbiome, increases pathogen abundance and induces intestinal inflammation in kenyan infants. Gut 2015, 64, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, S.; Janghorban, R.; Shariatdoust, S.; Faghihzadeh, S. The effects of iron supplementation on serum copper and zinc levels in pregnant women with high-normal hemoglobin. Int. J. Gynaecol. Obstet. 2008, 100, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, Y.F.; Hao, J.H.; Chen, Y.H.; Su, P.Y.; Wang, Y.; Yu, Z.; Fu, L.; Xu, Y.Y.; Zhang, C.; et al. Maternal zinc deficiency during pregnancy elevates the risks of fetal growth restriction: A population-based birth cohort study. Sci. Rep. 2015, 5, 11262. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecoloqists; Task Force on Hypertension in Preqnancy. Hypertension in pregnancy. Report of the american college of obstetricians and gynecologists’ task force on hypertension in pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Mongraw-Chaffin, M.L.; Cirillo, P.M.; Cohn, B.A. Preeclampsia and cardiovascular disease death: Prospective evidence from the child health and development studies cohort. Hypertension 2010, 56, 166–171. [Google Scholar] [CrossRef]
- Mol, B.W.J.; Roberts, C.T.; Thangaratinam, S.; Magee, L.A.; De Groot, C.J.M.; Hofmeyr, G.J. Pre-eclampsia. Lancet 2016, 387, 999–1011. [Google Scholar] [CrossRef]
- Pruthi, D.; Khankin, E.V.; Blanton, R.M.; Aronovitz, M.; Burke, S.D.; McCurley, A.; Karumanchi, S.A.; Jaffe, I.Z. Exposure to experimental preeclampsia in mice enhances the vascular response to future injury. Hypertension 2015, 65, 863–870. [Google Scholar] [CrossRef]
- Fox, A.; McHugh, S.; Browne, J.; Kenny, L.C.; Fitzgerald, A.; Khashan, A.S.; Dempsey, E.; Fahy, C.; O’Neill, C.; Kearney, P.M. Estimating the cost of preeclampsia in the healthcare system: Cross-sectional study using data from scope study (screening for pregnancy end points). Hypertension 2017, 70, 1243–1249. [Google Scholar] [CrossRef]
- Carter, A.M.; Pijnenborg, R. Evolution of invasive placentation with special reference to non-human primates. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, P.L. Animal models to study placental development and function throughout normal and dysfunctional human pregnancy. Semin. Reprod. Med. 2016, 34, 11–16. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Amaral, L.M.; Harmon, A.C.; Cornelius, D.C.; Faulkner, J.L.; Cunningham, M.W., Jr. Placental ischemia and resultant phenotype in animal models of preeclampsia. Curr. Hypertens. Rep. 2016, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.P.; Berkowitz, R.S. Current management of complete and partial molar pregnancy. J. Reprod. Med. 1994, 39, 139–146. [Google Scholar] [PubMed]
- Piering, W.F.; Garancis, J.G.; Becker, C.G.; Beres, J.A.; Lemann, J., Jr. Preeclampsia related to a functioning extrauterine placenta: Report of a case and 25-year follow-up. Am. J. Kidney Dis. 1993, 21, 310–313. [Google Scholar] [CrossRef]
- Ananth, C.V. Ischemic placental disease: A unifying concept for preeclampsia, intrauterine growth restriction, and placental abruption. Semin. Perinatol. 2014, 38, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.K.; Janowiak, M.; Badger, G.J.; Bernstein, I.M. Evidence for distinct preterm and term phenotypes of preeclampsia. J. Matern. Fetal Neonatal Med. 2010, 23, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Vatten, L.J.; Skjaerven, R. Is pre-eclampsia more than one disease? BJOG 2004, 111, 298–302. [Google Scholar] [CrossRef]
- Brosens, I.A.; Robertson, W.B.; Dixon, H.G. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet. Gynecol. Annu. 1972, 1, 177–191. [Google Scholar] [CrossRef]
- Cross, J.C.; Werb, Z.; Fisher, S.J. Implantation and the placenta: Key pieces of the development puzzle. Science 1994, 266, 1508–1518. [Google Scholar] [CrossRef]
- Dekker, G.A.; Sibai, B.M. Etiology and pathogenesis of preeclampsia: Current concepts. Am. J. Obstet. Gynecol. 1998, 179, 1359–1375. [Google Scholar] [CrossRef]
- Meekins, J.W.; Pijnenborg, R.; Hanssens, M.; McFadyen, I.R.; Van Asshe, A. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br. J. Obstet. Gynaecol. 1994, 101, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, J.S.; Stanek, J.; Warshak, C.; Khoury, J.; Sibai, B. The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am. J. Obstet. Gynecol. 2003, 189, 1173–1177. [Google Scholar] [CrossRef]
- Norwitz, E.R. Defective implantation and placentation: Laying the blueprint for pregnancy complications. Reprod. Biomed. Online 2006, 13, 591–599. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 2010, 63, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.A.; Sharkey, D.J. Seminal fluid and fertility in women. Fertil. Steril. 2016, 106, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlicev, M.; Norwitz, E.R. Human parturition: Nothing more than a delayed menstruation. Reprod. Sci. 2018, 25, 166–173. [Google Scholar] [CrossRef]
- Griffith, O.W.; Wagner, G.P. The placenta as a model for understanding the origin and evolution of vertebrate organs. Nat. Ecol. Evol. 2017, 1, 72. [Google Scholar] [CrossRef]
- Goto, M.P.; Goldman, A.S. Diabetic embryopathy. Curr. Opin. Pediatr. 1994, 6, 486–491. [Google Scholar] [CrossRef]
- Norwitz, E.R.; Schust, D.J.; Fisher, S.J. Implantation and the survival of early pregnancy. N. Engl. J. Med. 2001, 345, 1400–1408. [Google Scholar] [CrossRef]
- Pringle, K.G.; Kind, K.L.; Sferruzzi-Perri, A.N.; Thompson, J.G.; Roberts, C.T. Beyond oxygen: Complex regulation and activity of hypoxia inducible factors in pregnancy. Hum. Reprod. Update 2010, 16, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Kasterstein, E.; Strassburger, D.; Komarovsky, D.; Bern, O.; Komsky, A.; Raziel, A.; Friedler, S.; Ron-El, R. The effect of two distinct levels of oxygen concentration on embryo development in a sibling oocyte study. J. Assist. Reprod. Genet. 2013, 30, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.; Li, Y.; Ai, J.; Gu, L.; Chen, W.; Liu, Q. Two different concentrations of oxygen for culturing precompaction stage embryos on human embryo development competence: A prospective randomized sibling-oocyte study. Int. J. Clin. Exp. Pathol. 2014, 7, 6191–6198. [Google Scholar] [PubMed]
- Kaser, D.J.; Bogale, B.; Sarda, V.; Farland, L.V.; Williams, P.L.; Racowsky, C. Randomized controlled trial of low (5%) versus ultralow (2%) oxygen for extended culture using bipronucleate and tripronucleate human preimplantation embryos. Fertil. Steril. 2018, 109, 1030–1037.e2. [Google Scholar] [CrossRef] [PubMed]
- Genbacev, O.; Zhou, Y.; Ludlow, J.W.; Fisher, S.J. Regulation of human placental development by oxygen tension. Science 1997, 277, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by gpx4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. Acsl4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic pes navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and aif-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Pavlicev, M.; Wagner, G.P.; Chavan, A.R.; Owens, K.; Maziarz, J.; Dunn-Fletcher, C.; Kallapur, S.G.; Muglia, L.; Jones, H. Single-cell transcriptomics of the human placenta: Inferring the cell communication network of the maternal-fetal interface. Genome Res. 2017, 27, 349–361. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Carr, C.A.; Miller, J.J.; Christian, H.C.; Ball, V.; Santos, A.; Diaz, R.; Biggs, D.; Stillion, R.; et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA 2015, 112, 3164–3169. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The fowler syndrome-associated protein flvcr2 is an importer of heme. Mol. Cell. Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of sat1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P.; Barlis, J.; Evans, R.W.; Redman, C.W.; King, L.J. Abnormal iron parameters in the pregnancy syndrome preeclampsia. Am. J. Obstet. Gynecol. 2002, 187, 412–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, I.A.; Jaleel, A.; Kadri, H.M.; Saeed, W.A.; Tamimi, W. Iron status parameters in preeclamptic women. Arch. Gynecol. Obstet. 2011, 284, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Aguirre, C.H.; Garcia-Lozano, J.A.; Trevino-Montemayor, O.R.; Iglesias-Benavides, J.L.; Cantu-Rodriguez, O.G.; Gonzalez-Llano, O.; Gomez-De Leon, A.; Salazar-Riojas, R.; Mancias-Guerra, C.; Jaime-Perez, J.C.; et al. Comparative analysis of iron status and other hematological parameters in preeclampsia. Hematology 2017, 22, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Brunacci, F.; Rocha, V.S.; De Carli, E.; Esposito, B.P.; Ruano, R.; Colli, C. Increased serum iron in preeclamptic women is likely due to low hepcidin levels. Nutr. Res. 2018, 53, 32–39. [Google Scholar] [CrossRef]
- Duvan, C.I.; Simavli, S.; Keskin, E.A.; Onaran, Y.; Turhan, N.O.; Koca, C. Is the level of maternal serum prohepcidin associated with preeclampsia? Hypertens. Pregnancy 2015, 34, 145–152. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Patil, S.B.; Kodliwadmath, M.V.; Kodliwadmath, M. Lipid peroxidation and antioxidant activity in complicated pregnancies. Clin. Exp. Obstet. Gynecol. 2009, 36, 110–112. [Google Scholar]
- Sarandol, E.; Safak, O.; Dirican, M.; Uncu, G. Oxidizability of apolipoprotein b-containing lipoproteins and serum paraoxonase/arylesterase activities in preeclampsia. Clin. Biochem. 2004, 37, 990–996. [Google Scholar] [CrossRef]
- Ahmadi, R.; Rahimi, Z.; Vaisi-Raygani, A.; Kiani, A.; Jalilian, N.; Rahimi, Z. Apolipoprotein e genotypes, lipid peroxidation, and antioxidant status among mild and severe preeclamptic women from western iran: Protective role of apolipoprotein epsilon2 allele in severe preeclampsia. Hypertens. Pregnancy 2012, 31, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, T.L.; Chen, H.; Baker, P.N.; Qi, H.; Zhang, H. Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp. Cell Res. 2017, 359, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norwitz, E.R.; Tsen, L.C.; Park, J.S.; Fitzpatrick, P.A.; Dorfman, D.M.; Saade, G.R.; Buhimschi, C.S.; Buhimschi, I.A. Discriminatory proteomic biomarker analysis identifies free hemoglobin in the cerebrospinal fluid of women with severe preeclampsia. Am. J. Obstet. Gynecol. 2005, 193, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berg, C.B.; Duvekot, J.J.; Guzel, C.; Hansson, S.R.; De Leeuw, T.G.; Steegers, E.A.; Versendaal, J.; Luider, T.M.; Stoop, M.P. Elevated levels of protein ambp in cerebrospinal fluid of women with preeclampsia compared to normotensive pregnant women. Proteom. Clin. Appl 2017, 11, 1600082. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C.; Johnsen, G.M.; Dechend, R.; Redman, C.W.G. Preeclampsia and uteroplacental acute atherosis: Immune and inflammatory factors. J. Reprod. Immunol. 2014, 101, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed]
- Holland, O.J.; Cuffe, J.S.M.; Dekker Nitert, M.; Callaway, L.; Kwan Cheung, K.A.; Radenkovic, F.; Perkins, A.V. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis. 2018, 9, 1150. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Shao, L.; Luo, X.; Mu, Y.; Xu, W.; Gao, C.; Gao, L.; Liu, J.; Cui, Y. Ultrastructure of placenta of gravidas with gestational diabetes mellitus. Obstet. Gynecol. Int. 2015, 2015, 283124. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int. J. Mol. Sci. 2015, 16, 4600–4614. [Google Scholar] [CrossRef]
- Shi, Z.; Long, W.; Zhao, C.; Guo, X.; Shen, R.; Ding, H. Comparative proteomics analysis suggests that placental mitochondria are involved in the development of pre-eclampsia. PLoS ONE 2013, 8, e64351. [Google Scholar] [CrossRef]
- Poidatz, D.; Dos Santos, E.; Gronier, H.; Vialard, F.; Maury, B.; De Mazancourt, P.; Dieudonne, M.N. Trophoblast syncytialisation necessitates mitochondrial function through estrogen-related receptor-gamma activation. Mol. Hum. Reprod. 2015, 21, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, J.; Ramirez-Velez, R.; Czerniczyniec, A.; Cicerchia, D.; De Plata, A.C.A.; Lores-Arnaiz, S. Oxygen metabolism in human placenta mitochondria. J. Bioenerg. Biomembr. 2014, 46, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Poston, L.; Burton, G.J. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum. Reprod. Update 2006, 12, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Alahari, S.; Post, M.; Rolfo, A.; Weksberg, R.; Caniggia, I. Compromised jmjd6 histone demethylase activity affects vhl gene repression in preeclampsia. J. Clin. Endocrinol. Metab. 2018, 103, 1545–1557. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Highly expressed genes (within the top 1000 ranked highly expressed genes in any cell type) | ||||||||
| GENE | CYT1 | CYT2 | CYT3 | DC | EVCT | SYN | ESF | DEC |
| Ftl | 2807 (54) | 5427 (24) | 4936 (29) | 26753 (2) | 15921 (3) | 19339 (4) | 4849 (12) | 12331 (3) |
| Fth1 | 493 (318) | 1224 (128) | 722 (227) | 5411 (29) | 1093 (163) | 1348 (92) | 3416 (19) | 7596 (6) |
| Slc40a1 (Fpn) | 580 (289) | 326 (467) | 294 (503) | 22 (2332) | 2 (7287) | 51 (1627) | 1 (10,661) | 317 (611) |
| Tfrc (Tfr1) | 227 (633) | 217 (666) | 203 (717) | 191 (731) | 22 (3187) | 26 (2860) | 236 (800) | 591 (282) |
| Genes mostly expressed in endometrial stromal fibroblasts and decidual cells | ||||||||
| GENE | CYT1 | CYT2 | CYT3 | DC | EVCT | SYN | ESF | DEC |
| Cybrd1 (Dyctb) | 0 | 1 | 0 | 0 | 0 | 45 | 334 | 334 |
| Flvcr1 | 1 | 0 | 2 | 0 | 0 | 1 | 12 | 7 |
| Fxn | 0 | 4 | 2 | 3 | 0 | 0 | 21 | 12 |
| Ireb2 (Irp2) | 1 | 6 | 6 | 0 | 9 | 18 | 47 | 68 |
| Pcbp4 | 0 | 0 | 0 | 0 | 0 | 4 | 39 | 18 |
| Slc7a11 | 0 | 0 | 0 | 0 | 0 | 0 | 76 | 66 |
| Zip14 (Slc9a14) | 0 | 3 | 0 | 0 | 0 | 2 | 205 | 280 |
| Genes mostly expressed in trophoblasts | ||||||||
| GENE | CYT1 | CYT2 | CYT3 | DC | EVCT | SYN | ESF | DEC |
| Alas1 | 150 | 348 | 472 | 12 | 205 | 41 | 14 | 18 |
| Cp | 37 | 5 | 1 | 3 | 0 | 2 | 0 | 0 |
| Flvcr2 | 8 | 12 | 24 | 0 | 11 | 4 | 1 | 1 |
| Lpcat3 | 46 | 86 | 77 | 0 | 13 | 109 | 13 | 26 |
| Sat1 | 10937 | 3510 | 4303 | 6383 | 9724 | 548 | 97 | 629 |
| Mfrn (Slc25a37) | 12 | 88 | 122 | 0 | 493 | 19 | 57 | 36 |
| Genes expressed in both trophoblasts and stromal fibroblasts and decidual cells | ||||||||
| GENE | CYT1 | CYT2 | CYT3 | DC | EVCT | SYN | ESF | DEC |
| Abcb10 | 8 | 7 | 2 | 1 | 6 | 16 | 30 | 14 |
| Aco1 (Irp1) | 10 | 43 | 30 | 53 | 14 | 8 | 13 | 24 |
| Fech | 7 | 22 | 28 | 5 | 10 | 2 | 55 | 60 |
| Hmox1 | 111 | 249 | 82 | 2 | 53 | 25 | 114 | 113 |
| Hmox2 | 2 | 8 | 66 | 0 | 15 | 8 | 56 | 32 |
| Pcbp1 | 175 | 314 | 265 | 60 | 175 | 178 | 192 | 152 |
| Pcbp2 | 138 | 152 | 174 | 225 | 144 | 66 | 179 | 153 |
| Dmt1 (Slc11a2) | 3 | 30 | 5 | 26 | 2 | 25 | 33 | 72 |
| Mfrn2 (Slc25a28) | 27 | 34 | 53 | 0 | 10 | 4 | 40 | 25 |
| Zip8 (Slc39a8) | 0 | 4 | 7 | 25 | 48 | 3 | 22 | 14 |
| Hrg1 (Slc48a1) | 35 | 184 | 305 | 0 | 31 | 3 | 61 | 173 |
| Steap3 | 39 | 19 | 15 | 17 | 0 | 1 | 9 | 3 |
| Downregulated genes | ||||||||
| GENE | CYT1 | CYT2 | CYT3 | DC | EVCT | SYN | ESF | DEC |
| Hamp (Hepcidin) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hpx | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, S.-W.; Norwitz, S.G.; Norwitz, E.R. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3283. https://doi.org/10.3390/ijms20133283
Ng S-W, Norwitz SG, Norwitz ER. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. International Journal of Molecular Sciences. 2019; 20(13):3283. https://doi.org/10.3390/ijms20133283
Chicago/Turabian StyleNg, Shu-Wing, Sam G. Norwitz, and Errol R. Norwitz. 2019. "The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia" International Journal of Molecular Sciences 20, no. 13: 3283. https://doi.org/10.3390/ijms20133283