RFAmyloid: A Web Server for Predicting Amyloid Proteins


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Measurement
2.2. Performance of Different Features on Cross-Validation
2.3. Performance of Different Features on External Validation
2.4. Comparison with Other Classifiers
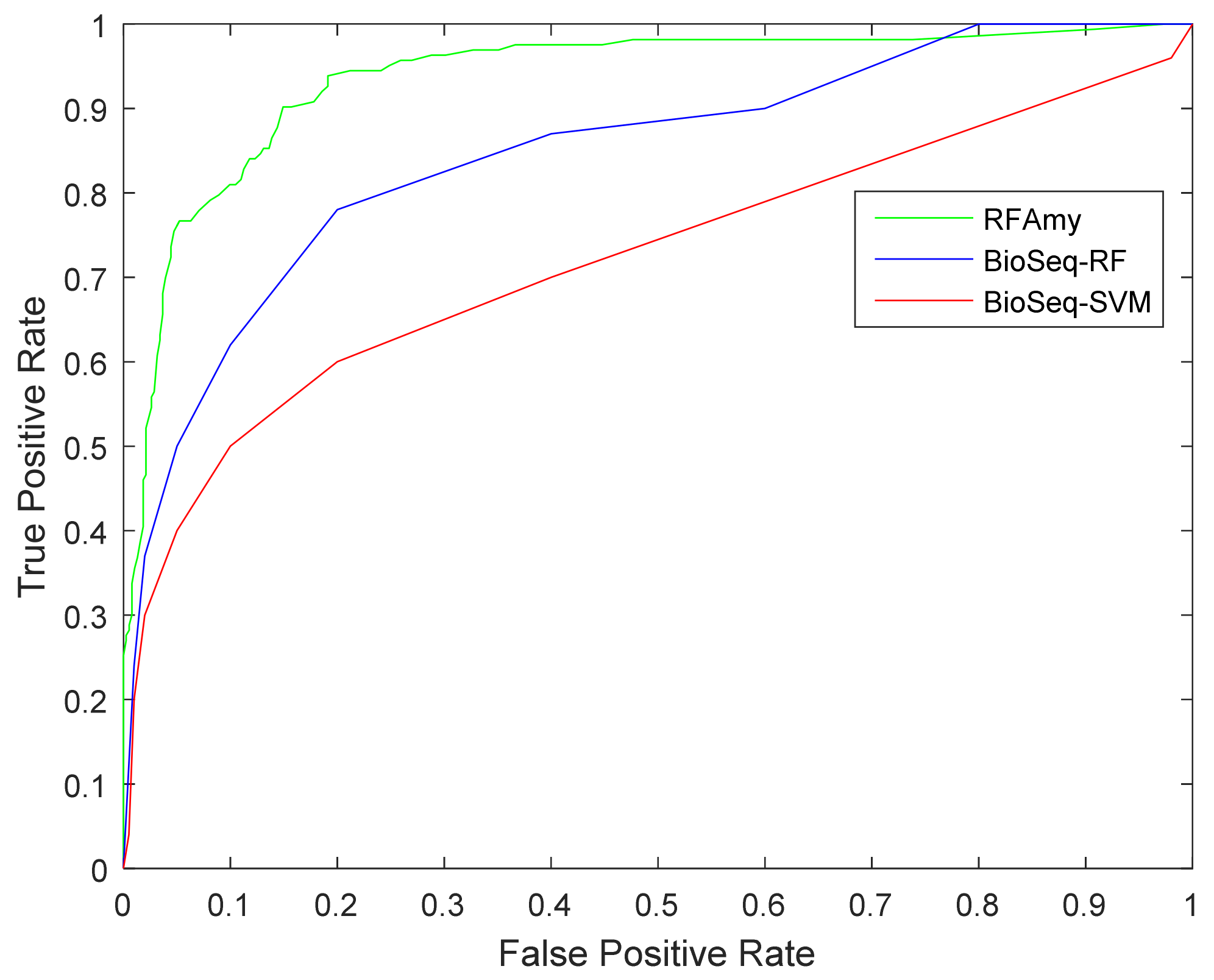
2.5. Comparison with Other Predictors
2.6. Comparison with Balanced Dataset
3. Methods
3.1. Dataset
3.2. Feature Extraction
3.3. Classifier
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beerten, J.; Van Durme, J.; Gallardo, R.; Capriotti, E.; Serpell, L.; Rousseau, F.; Schymkowitz, J. WALTZ-DB: A benchmark database of amyloidogenic hexapeptides. Bioinformatics 2015, 31, 1698–1700. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.-I. Localized amyloidogenic immunoglobulin light chain-derived amyloidosis in a young boy and an adolescent girl. Amyloid 2017, 24, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Louros, N.N.; Iconomidou, V.A.; Giannelou, P.; Hamodrakas, S.J. Structural analysis of peptide-analogues of human zona pellucida ZP1 protein with amyloidogenic properties: Insights into mammalian zona pellucida formation. PLoS ONE 2013, 8, e73258. [Google Scholar] [CrossRef] [PubMed]
- Gour, S.; Kaushik, V.; Kumar, V.; Bhat, P.; Yadav Subhash, C.; Yadav Jay, K. Antimicrobial peptide (Cn-AMP2) from liquid endosperm of cocos nucifera forms amyloid-like fibrillar structure. J. Pept. Sci. 2016, 22, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Rochet, J.-C.; Lansbury, P.T. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Bio. 2000, 10, 60–68. [Google Scholar] [CrossRef]
- Kallberg, Y.; Gustafsson, M.; Persson, B.; Thyberg, J.; Johansson, J. Prediction of amyloid fibril-forming proteins. J. Biol. Chem. 2001, 276, 12945–12950. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B 2001, 356, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.-I.; Merlini, G.; Saraiva, M.J.M.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification international society of amyloidosis 2016 nomenclature guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Dubchak, I.; Muchnik, I.; Holbrook, S.R.; Kim, S.-H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 8700–8704. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.B.; Znassi, N.; Château, M.-T.; Kajava, A.V. A structure-based approach to predict predisposition to amyloidosis. Alzheimers Dement. 2015, 11, 681–690. [Google Scholar] [CrossRef] [PubMed]
- De Groot, N.S.; Pallarés, I.; Avilés, F.X.; Vendrell, J.; Ventura, S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct. Biol. 2005, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. Foldamyloid: A method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2010, 26, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Paladin, L.; Piovesan, D.; Tosatto, S.C.E. Soda: Prediction of protein solubility from disorder and aggregation propensity. Nucleic Acids Res. 2017, 45, W236–W240. [Google Scholar] [CrossRef] [PubMed]
- Makin, O.S.; Atkins, E.; Sikorski, P.; Johansson, J.; Serpell, L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA 2005, 102, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.P.C.; Concepcion, G.P.; Padlan, E.A. Using simple artificial intelligence methods for predicting amyloidogenesis in antibodies. BMC Bioinform. 2010, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Frousios, K.K.; Iconomidou, V.A.; Karletidi, C.-M.; Hamodrakas, S.J. Amyloidogenic determinants are usually not buried. BMC Struct. Biol. 2009, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wu, N.; Guo, J.; Fan, Y. Prediction of amyloid fibril-forming segments based on a support vector machine. BMC Bioinform. 2009, 10, S45. [Google Scholar] [CrossRef] [PubMed]
- López de la Paz, M.; Serrano, L. Sequence determinants of amyloid fibril formation. Proc. Natl. Acad. Sci. USA 2004, 101, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; de la Paz, M.L.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237. [Google Scholar] [CrossRef] [PubMed]
- Caflisch, A. Computational models for the prediction of polypeptide aggregation propensity. Curr. Opin. Chem. Biol. 2006, 10, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Sievers, S.A.; Karanicolas, J.; Ivanova, M.I.; Baker, D.; Eisenberg, D. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4074–4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Welsh, W.J. Detecting hidden sequence propensity for amyloid fibril formation. Protein Sci. 2009, 13, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, W.; Unold, O. Use of a novel grammatical inference approach in classification of amyloidogenic hexapeptides. Comput. Math. Methods Med. 2016, 2016, 1782732. [Google Scholar] [CrossRef] [PubMed]
- Emily, M.; Talvas, A.; Delamarche, C. Metamyl: A meta-predictor for amyloid proteins. PLoS ONE 2013, 8, e79722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otoo, H.N.; Lee, K.G.; Qiu, W.; Lipke, P.N. Candida albicans als adhesins have conserved amyloid-forming sequences. Eukaryot. Cell 2008, 7, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Liaw, C.; Tung, C.-W.; Ho, S.-Y. Prediction and analysis of antibody amyloidogenesis from sequences. PLoS ONE 2013, 8, e53235. [Google Scholar] [CrossRef] [PubMed]
- Lembre, P.; Vendrely, C.; Di Martino, P. Identification of an amyloidogenic peptide from the bap protein of staphylococcus epidermidis. Protein Pept. Lett. 2014, 21, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, G.G.; Cavalli, A.; Pellarin, R.; Caflisch, A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. 2009, 14, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Trovato, A.; Seno, F.; Tosatto, S.C.E. The pasta server for protein aggregation prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.-I.; Merlini, G.; Saraiva, M.J.M.; Westermark, P. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014, 21, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Louros, N.N.; Petronikolou, N.; Karamanos, T.; Cordopatis, P.; Iconomidou, V.A.; Hamodrakas, S.J. Structural studies of “aggregation-prone” peptide-analogues of teleostean egg chorion zpb proteins. Pept. Sci. 2014, 102, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yuan, S.; Huang, X.; Zou, Q. Identification of cytokine via an improved genetic algorithm. Front. Comput. Sci. 2015, 9, 643–651. [Google Scholar] [CrossRef]
- Qu, K.; Han, K.; Wu, S.; Wang, G.; Wei, L. Identification of DNA-binding proteins using mixed feature representation methods. Molecules 2017, 22, 1602. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting tata binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, J.; Deng, L. Prediction of lncRNA-protein interactions using hetesim scores based on heterogeneous networks. Sci. Rep. 2017, 7, 3664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qu, Q.; Zhang, Y.; Wang, W. The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing 2018, 273, 526–534. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Li, D. Drug-target interaction prediction through label propagation with linear neighborhood information. Molecules 2017, 22, 2056. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Z.; Han, L.Y.; Ji, Z.L.; Chen, X.; Chen, Y.Z. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003, 31, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liao, M.; Gao, X.; Zou, Q. An improved protein structural classes prediction method by incorporating both sequence and structure information. IEEE Trans. Nanobiosci. 2015, 14, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, N.; Ruan, J. Prediction of protein modification sites of gamma-carboxylation using position specific scoring matrices based evolutionary information. Comput. Biol. Chem. 2013, 47, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yue, X.; Huang, F.; Liu, R.; Chen, Y.; Ruan, C. Predicting drug-disease associations and their therapeutic function based on the drug-disease association bipartite network. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinform. 2017, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Huang, T.; Kong, X.; Cai, Y.D. Prediction and analysis of cell-penetrating peptides using pseudo-amino acid composition and random forest models. Amino Acids 2015, 47, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, J.; Xuan, P.; Zou, Q. Bp neural network could help improve pre-miRNA identification in various species. Biomed. Res. Int. 2016, 2016, 9565689. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Guo, J.; Ju, Y.; Wu, M.; Zeng, X.; Hong, Z. Improving tRNAscan-se annotation results via ensemble classifiers. Mol. Inform. 2015, 34, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Wang, Z.; Guan, X.; Liu, B.; Wu, Y.; Lin, Z. An approach for identifying cytokines based on a novel ensemble classifier. Biomed. Res. Int. 2013, 2013, 686090. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, Z.; Zhan, W.; Deng, L. Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 2017, 34, 1473–1480. [Google Scholar]
- Zhang, J.; Zhang, Z.; Chen, Z.; Deng, L. Integrating multiple heterogeneous networks for novel lncRNA-disease association inference. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Chen, Z. An integrated framework for functional annotation of protein structural domains. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Niu, Y.; Xiong, Y.; Zhao, M.; Yu, R.; Liu, J. Computational prediction of conformational b-cell epitopes from antigen primary structures by ensemble learning. PLoS ONE 2012, 7, e43575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Niu, Y.; Zou, H.; Luo, L.; Liu, Q.; Wu, W. Accurate prediction of immunogenic t-cell epitopes from epitope sequences using the genetic algorithm-based ensemble learning. PLoS ONE 2015, 10, e0128194. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, L.; Zhang, W.; Liu, F.; Luo, F. A genetic algorithm-based weighted ensemble method for predicting transposon-derived piRNAs. BMC Bioinform. 2016, 17, 329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zou, H.; Luo, L.; Liu, Q.; Wu, W.; Xiao, W. Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing 2016, 173, 979–987. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, J.; Tang, G.; Wu, W.; Yue, X.; Li, D. Predicting small RNAs in bacteria via sequence learning ensemble method. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Kansas City, MO, USA, 13–16 November 2017; pp. 643–647. [Google Scholar]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. Mlacp: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Chen, W.; Huang, Y.; Liu, X.; Jiang, Y. Identifying multi-functional enzyme by hierarchical multi-label classifier. J. Comput. Theor. Nanosci. 2013, 10, 1038–1043. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, X.; Fu, Y.; Tsuji, J.; Weng, Z. The prediction of human splicing branchpoints by multi-label learning. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 254–259. [Google Scholar]
- Zhang, W.; Zhu, X.; Fu, Y.; Tsuji, J.; Weng, Z. Predicting human splicing branchpoints by combining sequence-derived features and multi-label learning methods. BMC Bioinform. 2017, 18, 464. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, D.; Zeng, X.; Wu, Y.; Guo, L.; Zou, Q. nDNA-prot: Identification of DNA-binding proteins based on unbalanced classification. BMC Bioinform. 2014, 15, 298. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hu, L.; Guo, M.; Liu, X.; Zou, Q. Imdc: An ensemble learning method for imbalanced classification with miRNA data. Genet. Mol. Res. 2015, 14, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ju, Y.; Zou, Q. Protein folds prediction with hierarchical structured SVM. Curr. Proteom. 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Lin, C.; Zou, Y.; Qin, J.; Liu, X.; Jiang, Y.; Ke, C.; Zou, Q. Hierarchical classification of protein folds using a novel ensemble classifier. PLoS ONE 2013, 8, e56499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Wang, Z.; Liu, Y.; Deng, L. Ontological function annotation of long non-coding RNAs through hierarchical multi-label classification. Bioinformatics 2018, 34, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Burdukiewicz, M.; Sobczyk, P.; Rödiger, S.; Duda-Madej, A.; Mackiewicz, P.; Kotulska, M. Amyloidogenic motifs revealed by n-gram analysis. Sci. Rep. 2017, 7, 12961. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chen, W.; Qiu, C.; Wu, Y.; Krishnan, S.; Zou, Q. Libd3c: Ensemble classifiers with a clustering and dynamic selection strategy. Neurocomputing 2014, 123, 424–435. [Google Scholar] [CrossRef]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; De Baets, G.; Vranken, W.F.; Tompa, P.; Pancsa, R. Amypro: A database of proteins with validated amyloidogenic regions. Nucleic Acids Res. 2018, 46, D387–D392. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Zhang, N.; Huang, T.; Cai, Y.D. Discriminating between deleterious and neutral non-frameshifting indels based on protein interaction networks and hybrid properties. Mol. Genet. Genom. 2015, 290, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Li, X.; Jiang, Y.; Zhao, Y.; Wang, G. Binmempredict: A web server and software for predicting membrane protein types. Curr. Proteom. 2013, 10, 2–9. [Google Scholar] [CrossRef]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Söderquist, F.; Wallner, B. Proteus: A random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins. J. Comput. Aided Mol. Des. 2017, 31, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-P.; Wu, L.-Y.; Wang, Y.; Zhang, X.-S.; Chen, L. Prediction of protein–RNA binding sites by a random forest method with combined features. Bioinformatics 2010, 26, 1616–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Li, B.Q.; Gao, S.; Ruan, J.S.; Cai, Y.D. Computational prediction and analysis of protein γ-carboxylation sites based on a random forest method. Mol. Biosyst. 2012, 8, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Zhang, N.; Kong, X.; Huang, T.; Cai, Y.D. Predicting A-to-I RNA editing by feature selection and random forest. PLoS ONE 2014, 9, e110607. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J.; Lee, J. Random forest-based protein model quality assessment (RFMQA) using structural features and potential energy terms. PLoS ONE 2014, 9, e106542. [Google Scholar] [CrossRef] [PubMed]
- Dao, F.-Y.; Yang, H.; Su, Z.-D.; Yang, W.; Wu, Y.; Hui, D.; Chen, W.; Tang, H.; Lin, H. Recent advances in conotoxin classification by using machine learning methods. Molecules 2017, 22, 1057. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. Aippred: Sequence-based prediction of anti-inflammatory peptides using random forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J. Svmqa: Support–vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ding, C.; Song, Q.; Yang, P.; Ding, H.; Deng, K.-J.; Chen, W. The prediction of protein structural class using averaged chemical shifts. J. Biomol. Struct. Dyn. 2012, 29, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Lee, G. PVP-SVM: Sequence-based prediction of phage virion proteins using a support vector machine. Front Microbiol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.-Y.; Chen, X.-X.; Chen, W.; Tang, H.; Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 2017, 8, 28169–28175. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Lee, G. DHSpred: Support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget 2018, 9, 1944–1956. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | ACC (%) | MCC | SE | SP | F-Measure |
|---|---|---|---|---|---|
| 188-D+Pse-in-One | 89.1941 | 0.739 | 0.781 | 0.927 | 0.891 |
| 188-D | 84.8482 | 0.626 | 0.655 | 0.932 | 0.626 |
| Pse-in-one | 81.31 | 0.5626 | 0.6374 | 0.8989 | 0.792 |
| 400-D | 84.1105 | 0.634 | 0.691 | 0.917 | 0.838 |
| n-gram (n = 1) | 81.3187 | 0.522 | 0.534 | 0.930 | 0.802 |
| Method | ACC (%) | MCC | SE | SP | F-Measure |
|---|---|---|---|---|---|
| 188-D+Pse-in-One | 89.7196 | 0.757 | 0.818 | 0.932 | 0.897 |
| 188-D | 73.1841 | 0.524 | 0.512 | 0.960 | 0.678 |
| Pse-in-one | 78.7037 | 0.679 | 0.676 | 0.880 | 0.782 |
| 400-D | 71.2963 | 0.543 | 0.503 | 0.893 | 0.684 |
| n-gram (n = 1) | 69.4444 | 0.522 | 0.534 | 0.893 | 0.657 |
| Classifier | ACC (%) | MCC | SE | SP | F-Measure |
|---|---|---|---|---|---|
| Random Forest | 89.19 | 0.739 | 0.781 | 0.927 | 0.891 |
| Naive Bayes | 75.50 | 0.3791 | 0.4606 | 0.8822 | 0.8721 |
| SGD | 77.51 | 0.4451 | 0.5515 | 0.8717 | 0.6533 |
| Nearest Neighbors | 77.70 | 0.4293 | 0.2970 | 0.9843 | 0.8818 |
| Decision Tree | 67.28 | 0.2567 | 0.5333 | 0.7330 | 0.7461 |
| LinearSVC | 77.51 | 0.4654 | 0.6242 | 0.8403 | 0.8658 |
| Logistic Regression | 79.52 | 0.5123 | 0.6545 | 0.8560 | 0.8694 |
| LibSVM | 70.02 | 0.0651 | 0.0061 | 1.0000 | 0.8239 |
| ExtraTrees | 74.95 | 0.4128 | 0.6061 | 0.8115 | 0.8087 |
| Bagging | 74.95 | 0.4128 | 0.6061 | 0.8115 | 0.7727 |
| AdaBoost | 76.78 | 0.4700 | 0.6788 | 0.8063 | 0.8763 |
| GradientBoosting | 80.26 | 0.5298 | 0.6667 | 0.8613 | 0.8668 |
| LibD3C | 86.99 | 0.683 | 0.732 | 0.929 | 0.868 |
| Method | ACC (%) | MCC | SE | SP |
|---|---|---|---|---|
| RFAmy | 89.1941 | 0.739 | 0.781 | 0.927 |
| BioSeq-SVM | 76.86 | 0.4419 | 0.4953 | 0.9006 |
| BioSeq-RF | 81.31 | 0.5626 | 0.6374 | 0.8989 |
| Method | ACC (%) | MCC | SE | SP | F-Measure |
|---|---|---|---|---|---|
| unbalanced | 89.1941 | 0.739 | 0.781 | 0.927 | 0.891 |
| balanced | 83.4962 | 0.757 | 0.847 | 0.823 | 0.865 |
| Physical-Chemical Property | Dimensions |
|---|---|
| Amino acid composition | 20 |
| Hydrophobicity | 21 |
| Normalized van der Waals volume | 21 |
| Polarity | 21 |
| Polarizability | 21 |
| Charge | 21 |
| Surface tension | 21 |
| Secondary structure | 21 |
| Solvent accessibility | 21 |
| Category | Method |
|---|---|
| Amino acid composition | K-mer |
| DR | |
| Distance Pair | |
| Autocorrelation | AC |
| CC | |
| ACC | |
| PDT | |
| Pseudo amino acid composotion | PC-PseAAC |
| SC-PseAAC | |
| PC-PseAAC-General | |
| SC-PseAAC-General | |
| Profile-based features | Top-n-gram |
| PDT-Profile | |
| DT | |
| AC-PSSM | |
| CC-PSSM | |
| ACC-PSSM | |
| PSSM-DT | |
| PSSM-RT | |
| CS | |
| Predicted structure features | SS |
| SASA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, M.; Li, Y.; Wang, C.; Han, K. RFAmyloid: A Web Server for Predicting Amyloid Proteins. Int. J. Mol. Sci. 2018, 19, 2071. https://doi.org/10.3390/ijms19072071
Niu M, Li Y, Wang C, Han K. RFAmyloid: A Web Server for Predicting Amyloid Proteins. International Journal of Molecular Sciences. 2018; 19(7):2071. https://doi.org/10.3390/ijms19072071
Chicago/Turabian StyleNiu, Mengting, Yanjuan Li, Chunyu Wang, and Ke Han. 2018. "RFAmyloid: A Web Server for Predicting Amyloid Proteins" International Journal of Molecular Sciences 19, no. 7: 2071. https://doi.org/10.3390/ijms19072071