The DNA Methylome and Association of Differentially Methylated Regions with Differential Gene Expression during Heat Stress in Brassica rapa


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
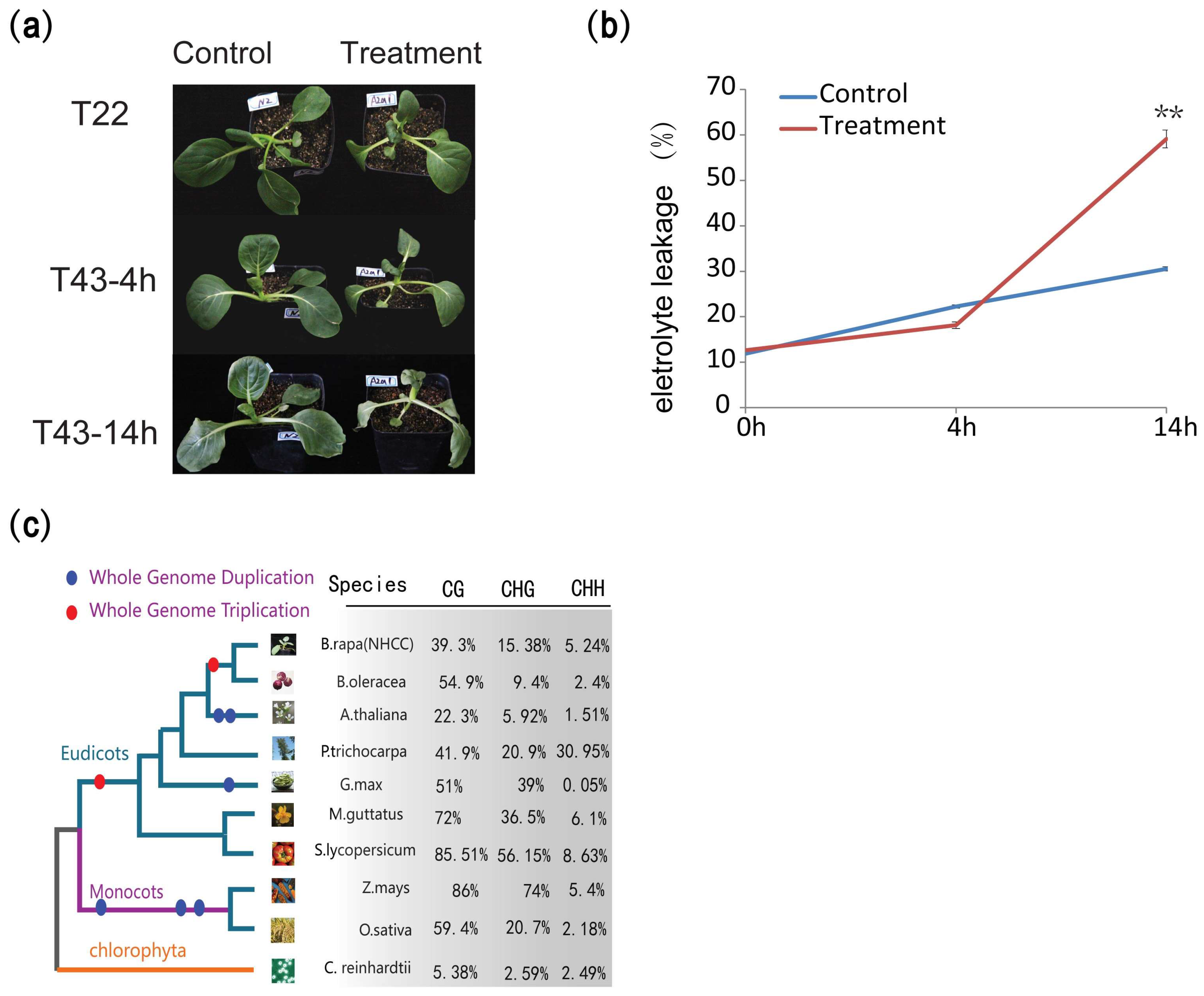
2.1. Methyltransferase Inhibitor Decreases the Heat Tolerance
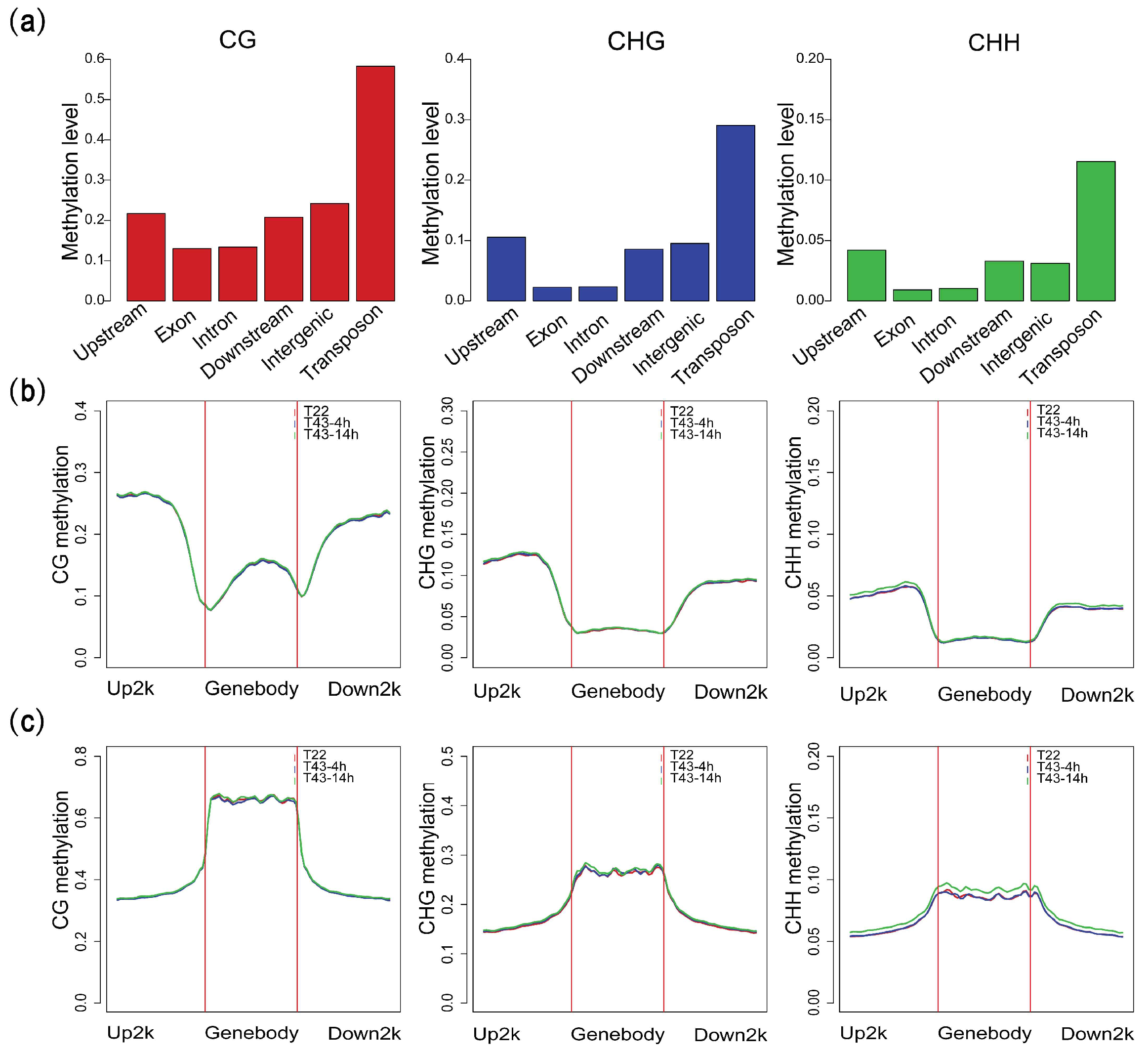
2.2. DNA Methylation Landscape of the NHCC Genome
2.3. Heat Stress Induces More Variation of mCG Site than the Other Two Contexts
2.4. Methylation Pathway Genes Are Conserved in B. rapa
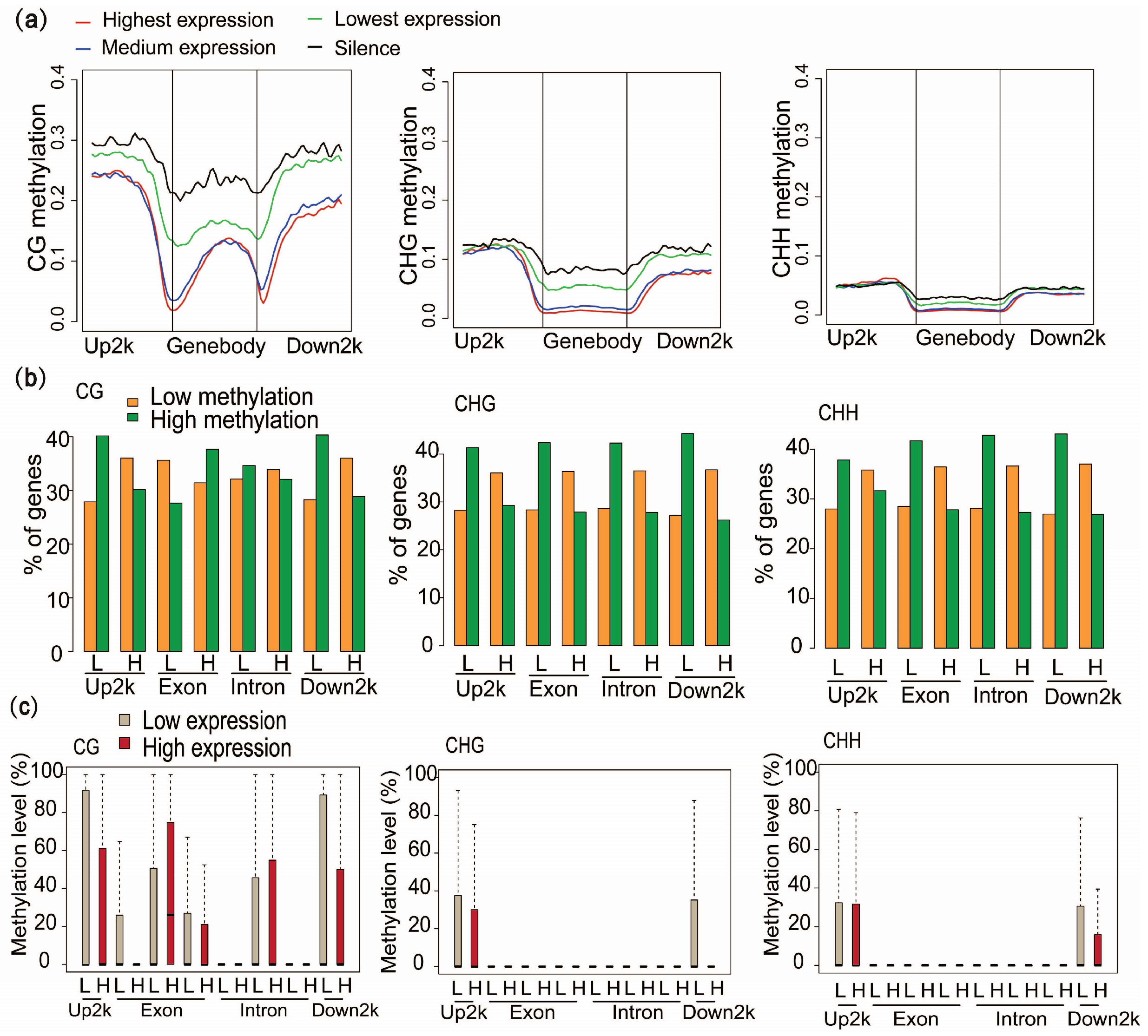
2.5. DNA Methylation at Different Genic Regions Differentially Correlates with Gene Expression
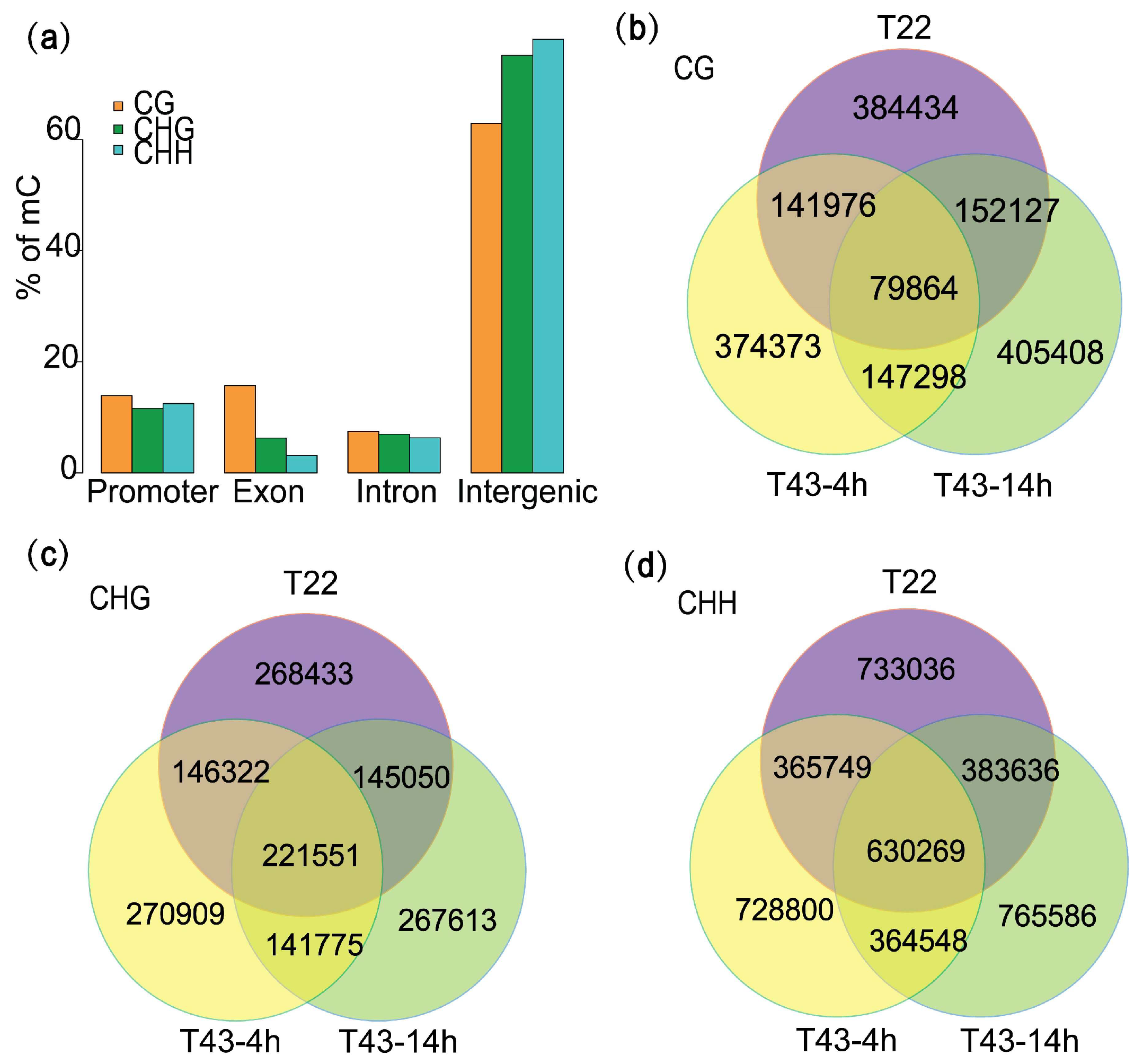
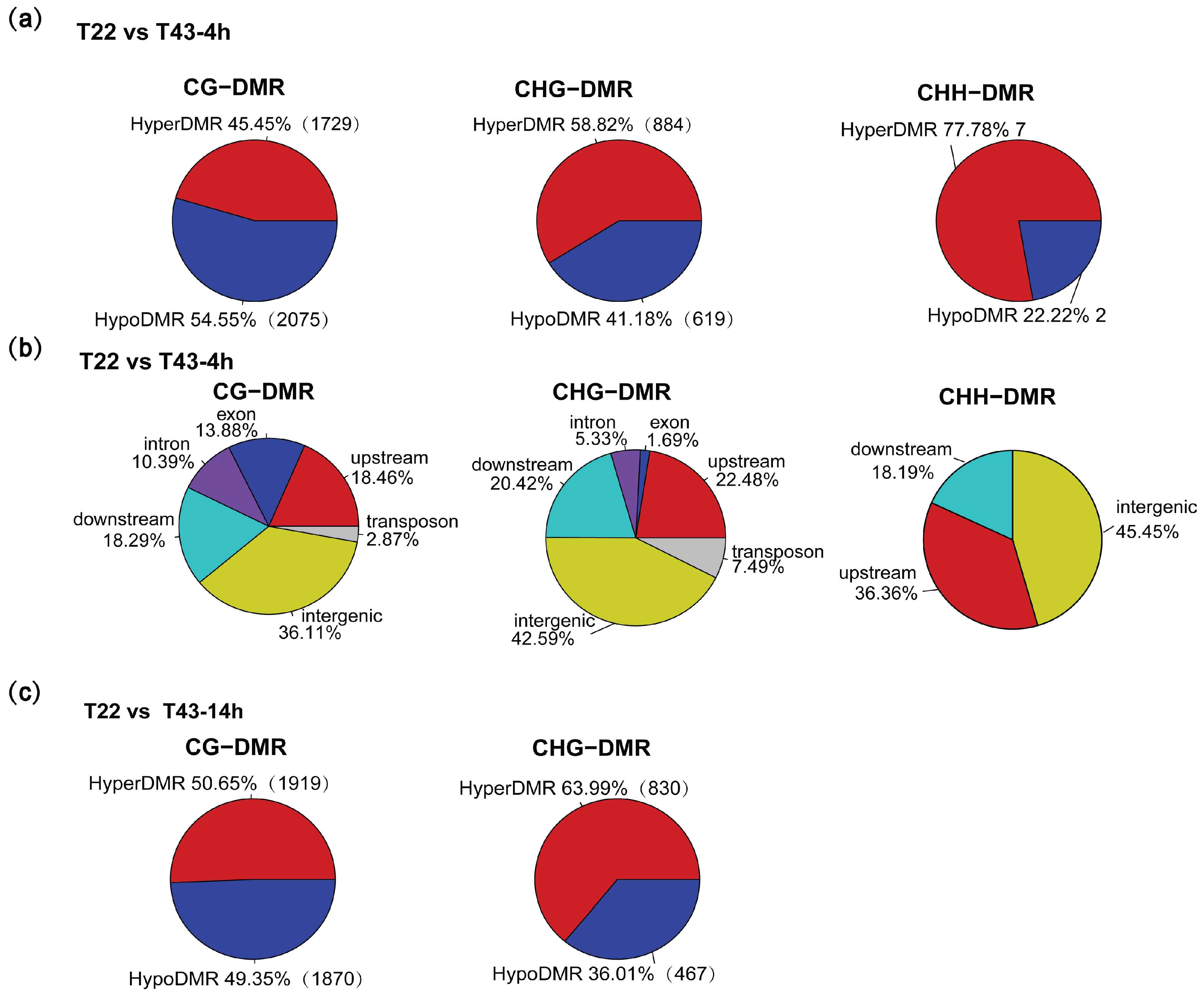
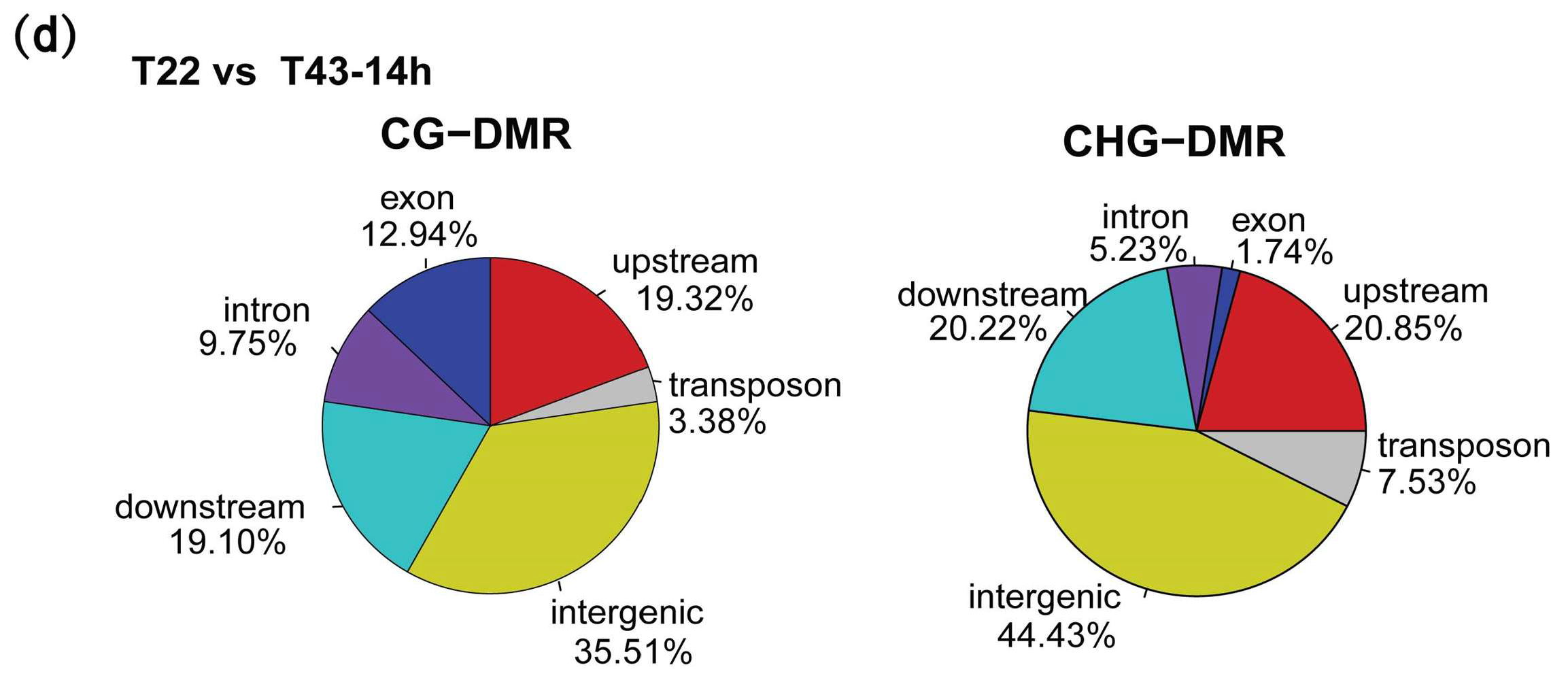
2.6. Heat-Stress Induced DMRs Are Mainly Located at Gene-Related Regions and Almost Absent in the CHH Context
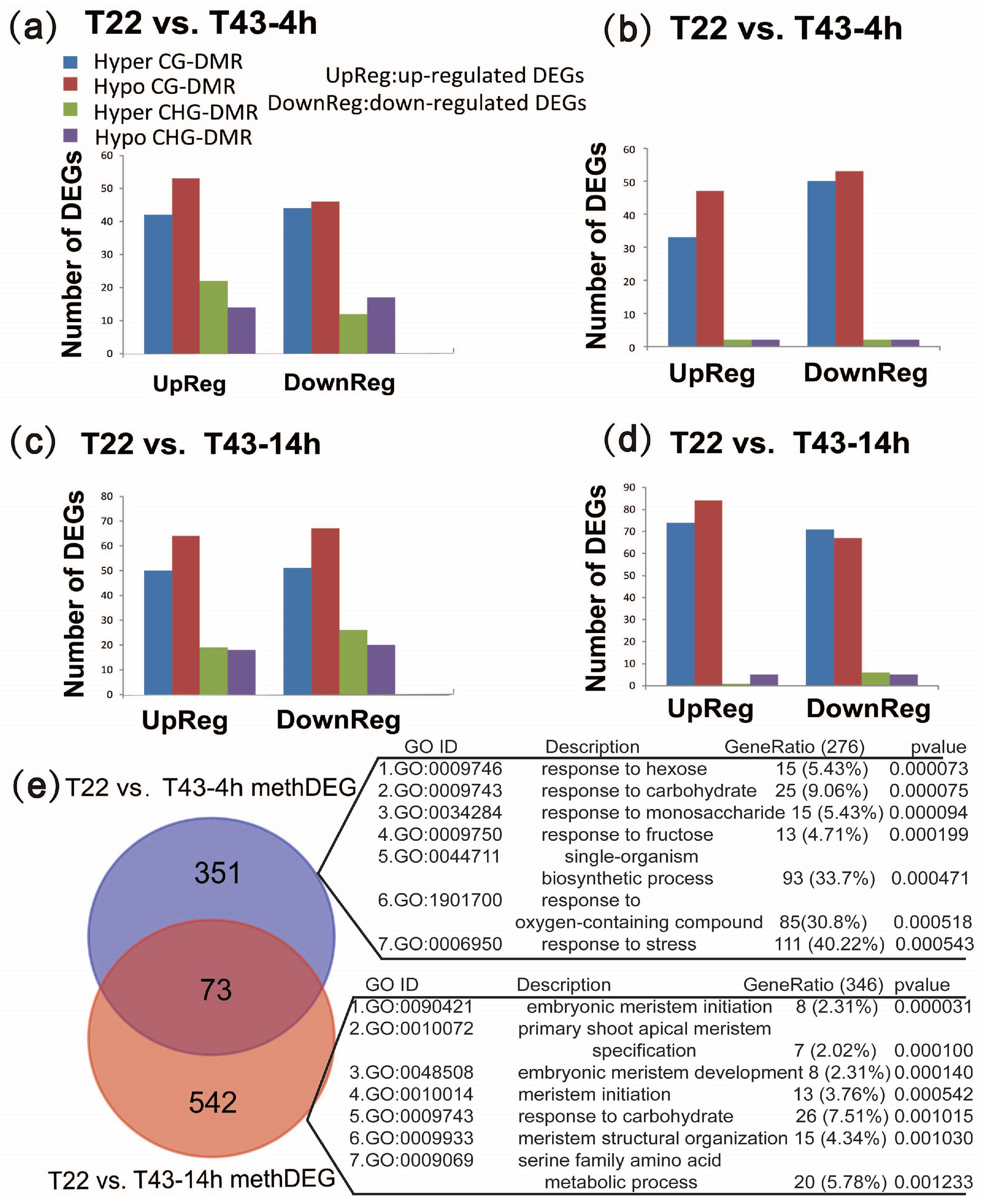
2.7. Biological Processes That Are Enriched in MethDEGs Are Different between 4 h and 14 h of Heat Stress
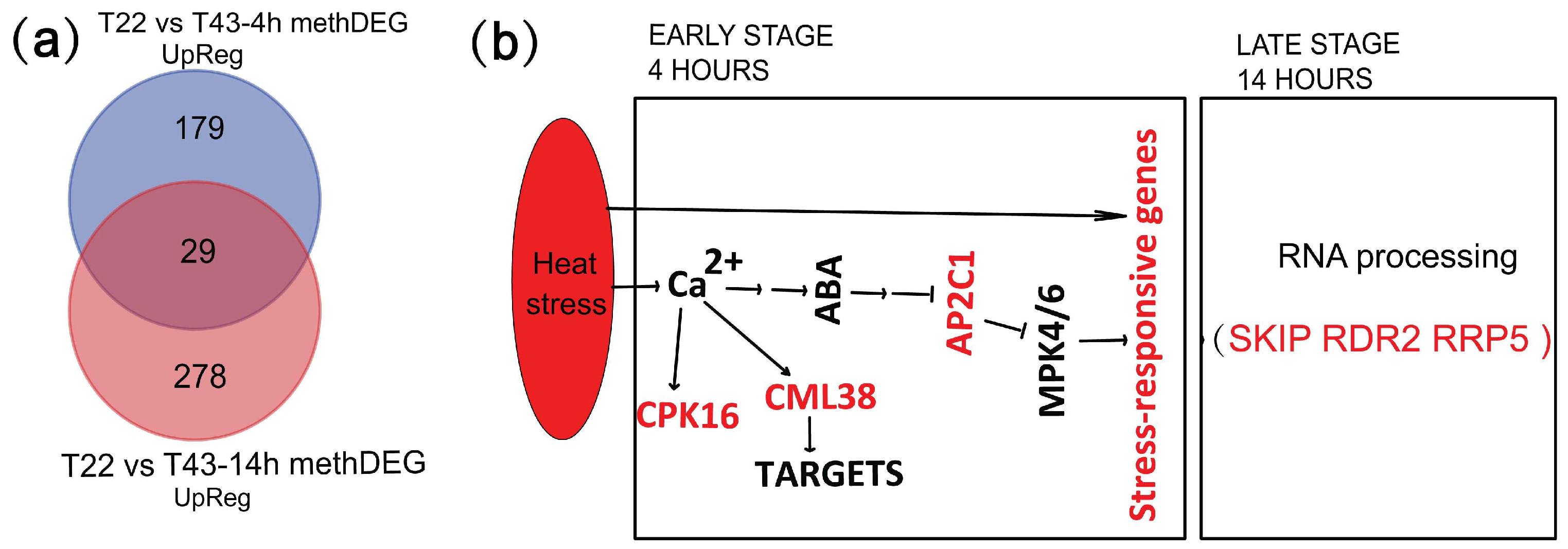
2.8. MethDEGs Are Significantly Involved in Signaling Transduction and RNA Metabolic Process under Heat Stress
3. Discussion
3.1. Dynamic DNA Methylation under Heat Treatment
3.2. Differences in Methylation Patterns of mC Sequence Contexts and Genic Regions during Heat Stress
3.3. DNA Methylation Is Potentially a Key Regulation Factor under Heat Stress
4. Materials and Methods
4.1. Plant Materials and Heat Treatment Conditions
4.2. Electrolyte Leakage Analysis
4.3. DNA Isolation and Whole-Genome Bisulfite Sequencing
4.4. Data Filtering and Identification of Methylated Cytosines
4.5. Identification of Differentially-Methylated Regions and Differentially-Methylated Genes
4.6. Total RNA Isolation and Transcriptome Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| WGBS | Whole-genome bisulfite sequencing |
| NHCC | Non-heading Chinese cabbage |
| TSS | Transcription start site |
| TES | Transcription end site |
| DMRs | Differentially-methylated regions |
| DEGs | Differentially-expressed gene |
| methDEGs | DEGs with DMR in the promoter or gene body |
References
- Christensen, J.H.; Christensen, O.B. A summary of the PRUDENCE model projections of changes in European climate by the end of this century. Clim. Chang. 2007, 81, 7–30. [Google Scholar] [CrossRef]
- Ahuja, I.; de Vos, R.C.; Bones, A.M.; Hall, R.D. Plant molecular stress responses face climate change. Trends Plant Sci. 2010, 15, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, X.; Chen, A.; Peterson, T.; Ma, K.; Bertzky, M.; Ciais, P.; Kapos, V.; Peng, C.; Poulter, B. Global priority conservation areas in the face of 21st century climate change. PLoS ONE 2013, 8, e54839. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Santoro, M.G. Stress-inducible responses and heat shock proteins: New pharmacologic targets for cytoprotection. Nat. Biotechnol. 1998, 16, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.-D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.-K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Chevala, V.N.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-S.; Pan, Y.-J.; Zhao, X.-Q.; Dwivedi, D.; Zhu, L.-H.; Ali, J.; Fu, B.-Y.; Li, Z.-K. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 2010, 62, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Wang, X.; Yang, G.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Yuan, Y. Analysis of the DNA methylation of maize (Zea mays L.) in response to cold stress based on methylation-sensitive amplified polymorphisms. J. Plant Biol. 2013, 56, 32–38. [Google Scholar] [CrossRef]
- Ding, H.; Gao, J.; Qin, C.; Ma, H.; Huang, H.; Song, P.; Luo, X.; Lin, H.; Shen, Y.O.; Pan, G. The dynamics of DNA methylation in maize roots under Pb stress. Int. J. Mol. Sci. 2014, 15, 23537–23554. [Google Scholar] [CrossRef] [PubMed]
- Karan, R.; Deleon, T.; Biradar, H.; Subudhi, P.K. Salt Stress Induced Variation in DNA Methylation Pattern and Its Influence on Gene Expression in Contrasting Rice Genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, F.; Qin, Q.; Zhao, X.; Li, Z.; Fu, B. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem. Biophys. Res. Commun. 2015, 465, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Ji, D.; Li, S.; Wang, P.; Li, Q.; Xiang, F. The Dynamic Changes of DNA Methylation and Histone Modifications of Salt Responsive Transcription Factor Genes in Soybean. PLoS ONE 2012, 7, e41274. [Google Scholar] [CrossRef] [PubMed]
- Yaish, M.W.; Allawati, A.; Alharrasi, I.; Patankar, H.V. Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genom. 2018, 19, 78. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.P. Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol. Biochem. 2010, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tieman, D.M.; Jiao, C.; Xu, Y.; Chen, K.; Fe, Z.; Giovannoni, J.J.; Klee, H.J. Chilling-induced tomato flavor loss is associated with altered volatile synthesis and transient changes in DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.-L.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Springer, N.M.; Muszynski, M.G.; Phillips, R.L.; Kaeppler, S.; Jacobsen, S.E. Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc. Natl. Acad. Sci. USA 2000, 97, 4979–4984. [Google Scholar] [CrossRef] [PubMed]
- Ronemus, M.J.; Galbiati, M.; Ticknor, C.; Chen, J.; Dellaporta, S.L. Demethylation-induced developmental pleiotropy in Arabidopsis. Science 1996, 273, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhong, X.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 2012, 151, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.; Do Kim, K.; Page, J.T.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M. Widespread natural variation of DNA methylation within angiosperms. Biorxiv 2016, 045880. [Google Scholar] [CrossRef] [PubMed]
- Ausin, I.; Feng, S.; Yu, C.; Liu, W.; Kuo, H.Y.; Jacobsen, E.L.; Zhai, J.; Gallego-Bartolome, J.; Wang, L.; Egertsdotter, U.; et al. DNA methylome of the 20-gigabase Norway spruce genome. Proc. Natl. Acad. Sci. USA 2016, 113, E8106–E8113. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Ragupathy, R.; You, F.M.; Cloutier, S. Arguments for standardizing transposable element annotation in plant genomes. Trends Plant Sci. 2013, 18, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Mirouze, M.; Vitte, C. Transposable elements, a treasure trove to decipher epigenetic variation: Insights from Arabidopsis and crop epigenomes. J. Exp. Bot. 2014, 65, 2801–2812. [Google Scholar] [CrossRef] [PubMed]
- Lippman, Z.; Gendrel, A.-V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Kubis, S.E.; Castilho, A.M.M.F.; Vershinin, A.V. Retroelements, transposons and methylation status in the genome of oil palm (Elaeis guineensis) and the relationship to somaclonal variation. Plant Mol. Biol. 2003, 52, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, S.; Kobayashi, A.; Kawabe, A.; Mathieu, O.; Miura, A.; Kakutani, T. Bursts of retrotransposition reproduced in Arabidopsis. Nature 2009, 461, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Bartee, L.; Bender, J. Two Arabidopsis methylation-deficiency mutations confer only partial effects on a methylated endogenous gene family. Nucleic Acids Res. 2001, 29, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.D.; Papp, I.; Moscone, E.; Iglesias, V.; Vaucheret, H.; Matzke, A.; Matzke, M. Gene silencing mediated by promoter homology occurs at the level of transcription and results in meiotically heritable alterations in methylation and gene activity. Plant J. 1996, 9, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Sakai, H.; Meyerowitz, E.M. Whorl-specific expression of the SUPERMAN gene of Arabidopsis is mediated by cis elements in the transcribed region. Curr. Biol. 2003, 13, 1524–1530. [Google Scholar] [CrossRef]
- Xun, C.; Ge, X.; Jing, W.; Chen, T.; King, G.J.; Liu, K. Genome-wide DNA methylation profiling by modified reduced representation bisulfite sequencing in Brassica rapa suggests that epigenetic modifications play a key role in polyploid genome evolution. Front. Plant Sci. 2015, 6, 836. [Google Scholar]
- Liu, T.; Li, Y.; Duan, W.; Huang, F.; Hou, X. Cold acclimation alters DNA methylation patterns and confers tolerance to heat and increases growth rate in Brassica rapa. J. Exp. Bot. 2017, 68, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.W.; Kendall, T.; Baten, A.; Burgess, D.; Freeling, M.; King, G.J.; Mosher, R.A. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 19, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Parkin, I.A.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.L. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef] [PubMed]
- Pecinka, A.; Dinh, H.Q.; Baubec, T.; Rosa, M.; Lettner, N.; Scheid, O.M. Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 2010, 22, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, J.; Li, H.; Li, F.; Xu, K.; Yan, G.; Chen, B.; Qiao, J.; Wu, X. Comparison of the heat stress induced variations in DNA methylation between heat-tolerant and heat-sensitive rapeseed seedlings. Breed. Sci. 2014, 64, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, Q.; Sun, M.; Zhang, T.; Li, H.; Chen, B.; Xu, K.; Gao, G.; Li, F.; Yan, G. Global DNA methylation variations after short-term heat shock treatment in cultured microspores of Brassica napus cv. Topas. Sci. Rep. 2016, 6, 38401. [Google Scholar] [CrossRef] [PubMed]
- Campo, C.G. Biology of Brassica Coenospecies; Elsevier: New York, NY, USA, 1999. [Google Scholar]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.-H.; Bancroft, I.; Cheng, F. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Marz, M.; Ihling, C.; Sinz, A.; Stadler, P.F.; Krauss, V. The correlation of genome size and DNA methylation rate in metazoans. Theory Biosci. 2013, 132, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P. Epigenetic variation and environmental change. J. Exp. Bot. 2015, 66, 3541–3548. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Schweighofer, A.; Kazanaviciute, V.; Scheikl, E.; Teige, M.; Doczi, R.; Hirt, H.; Schwanninger, M.; Kant, M.; Schuurink, R.; Mauch, F. The PP2C-type phosphatase AP2C1, which negatively regulates MPK4 and MPK6, modulates innate immunity, jasmonic acid, and ethylene levels in Arabidopsis. Plant Cell 2007, 19, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.-L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Perochon, A.; Aldon, D.; Galaud, J.-P.; Ranty, B. Calmodulin and calmodulin-like proteins in plant calcium signaling. Biochimie 2011, 93, 2048–2053. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, J.; Gao, Z.; Lu, Y.; Yu, J.; Zheng, Q.; Yan, S.; Zhang, W.; He, H.; Ma, L. SKIP confers osmotic tolerance during salt stress by controlling alternative gene splicing in Arabidopsis. Mol. Plant 2015, 8, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.; Kovalchuk, I. Epigenetic control of plant stress response. Environ. Mol. Mutagen. 2008, 49, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.-Y.; Tang, S.; Wang, Y.; Yang, L.; Wang, J. Single-base-resolution methylomes of populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15, S9. [Google Scholar] [CrossRef] [PubMed]
- Kou, H.; Li, Y.; Song, X.; Ou, X.; Xing, S.; Ma, J.; Von Wettstein, D.; Liu, B. Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhanced tolerance by progenies to the stress in rice (Oryza sativa L.). J. Plant Physiol. 2011, 168, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Chwialkowska, K.; Nowakowska, U.; Mroziewicz, A.; Szarejko, I.; Kwasniewski, M. Water-deficiency conditions differently modulate the methylome of roots and leaves in barley (Hordeum vulgare L.). J. Exp. Bot. 2016, 67, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollander, J.; Meins, F., Jr.; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef]
- Ferreira, L.J.; Azevedo, V.; Maroco, J.; Oliveira, M.M.; Santos, A.P. Salt tolerant and sensitive rice varieties display differential methylome flexibility under salt stress. PLoS ONE 2015, 10, e0124060. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chang, F.; You, C.; Cui, J.; Zhu, G.; Wang, L.; Zheng, Y.; Qi, J.; Ma, H. Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J. Cell Mol. Biol. 2015, 81, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; De Jonge, J.; Forsberg, S.K.; Pettersson, M.E.; Sheng, Z.; Hennig, L.; Carlborg, Ö. Natural CMT2 variation is associated with genome-wide methylation changes and temperature seasonality. PLoS Genet. 2014, 10, e1004842. [Google Scholar] [CrossRef] [PubMed]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.-S.; Osborne, E.J.; Casale, F.P.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjálmsson, B. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife 2015, 4, e05255. [Google Scholar] [CrossRef] [PubMed]
- Amoah, S.; Kurup, S.; Lopez, C.M.R.; Welham, S.J.; Powers, S.J.; Hopkins, C.J.; Wilkinson, M.J.; King, G.J. A Hypomethylated population of Brassica rapa for forward and reverse Epi-genetics. BMC Plant Biol. 2012, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.-J.; Claussen, K.; Zimmerman, J.L. Genotypic differences in the heat-shock response and thermotolerance in four potato cultivars. Plant Sci. 2004, 166, 901–911. [Google Scholar] [CrossRef]
- Saelim, S.; Zwiazek, J.J. Preservation of thermal stability of cell membranes and gas exchange in high temperature acclimated Xylia xylocarpa seedlings. J. Plant Physiol. 2000, 156, 380–385. [Google Scholar] [CrossRef]
- Ziller, M.J.; Hansen, K.D.; Meissner, A.; Aryee, M.J. Coverage recommendations for methylation analysis by whole genome bisulfite sequencing. Nat. Methods 2015, 12, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrated Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.E.; Yao, Y.; Zhang, X.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Colicchio, J.M.; Miura, F.; Kelly, J.K.; Ito, T.; Hileman, L.C. DNA methylation and gene expression in Mimulus guttatus. BMC Genom. 2015, 16, 507. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010, 8, e1000533. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Coreteam, R. R: A language and environment for statistical computing. Computing 2015, 1, 12–21. [Google Scholar]
- Song, X.; Liu, G.; Huang, Z.; Duan, W.; Tan, H.; Li, Y.; Hou, X. Temperature expression patterns of genes and their coexpression with LncRNAs revealed by RNA-Seq in non-heading Chinese cabbage. BMC Genom. 2016, 17, 297. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Xia, Y.; Liu, T.; Dai, S.; Hou, X. The DNA Methylome and Association of Differentially Methylated Regions with Differential Gene Expression during Heat Stress in Brassica rapa. Int. J. Mol. Sci. 2018, 19, 1414. https://doi.org/10.3390/ijms19051414
Liu G, Xia Y, Liu T, Dai S, Hou X. The DNA Methylome and Association of Differentially Methylated Regions with Differential Gene Expression during Heat Stress in Brassica rapa. International Journal of Molecular Sciences. 2018; 19(5):1414. https://doi.org/10.3390/ijms19051414
Chicago/Turabian StyleLiu, Gaofeng, Yudong Xia, Tongkun Liu, Shaojun Dai, and Xilin Hou. 2018. "The DNA Methylome and Association of Differentially Methylated Regions with Differential Gene Expression during Heat Stress in Brassica rapa" International Journal of Molecular Sciences 19, no. 5: 1414. https://doi.org/10.3390/ijms19051414