GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance


Abstract
:1. Introduction
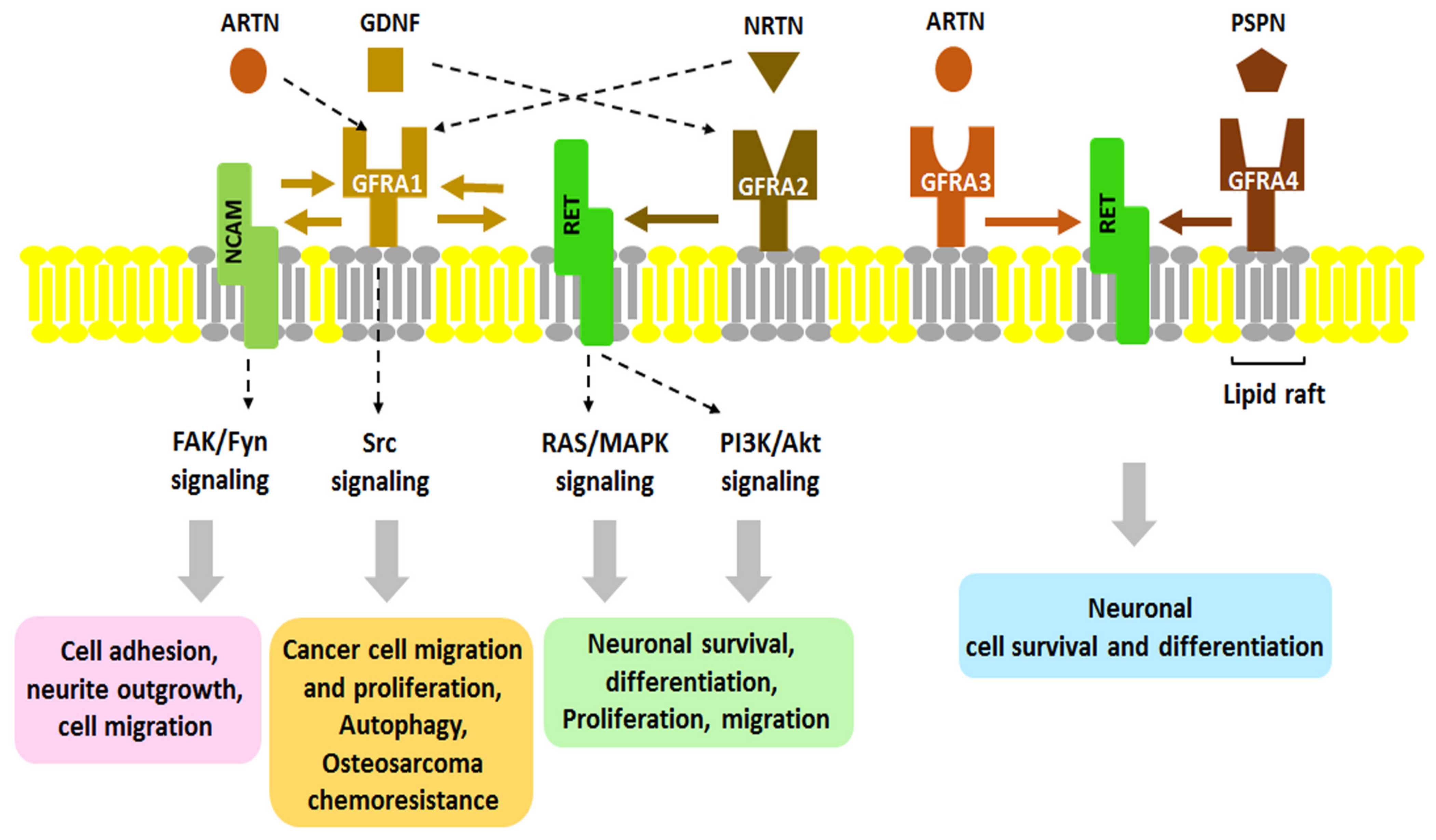
2. Regulatory Mechanisms of GDNF/GFRA1 Signaling
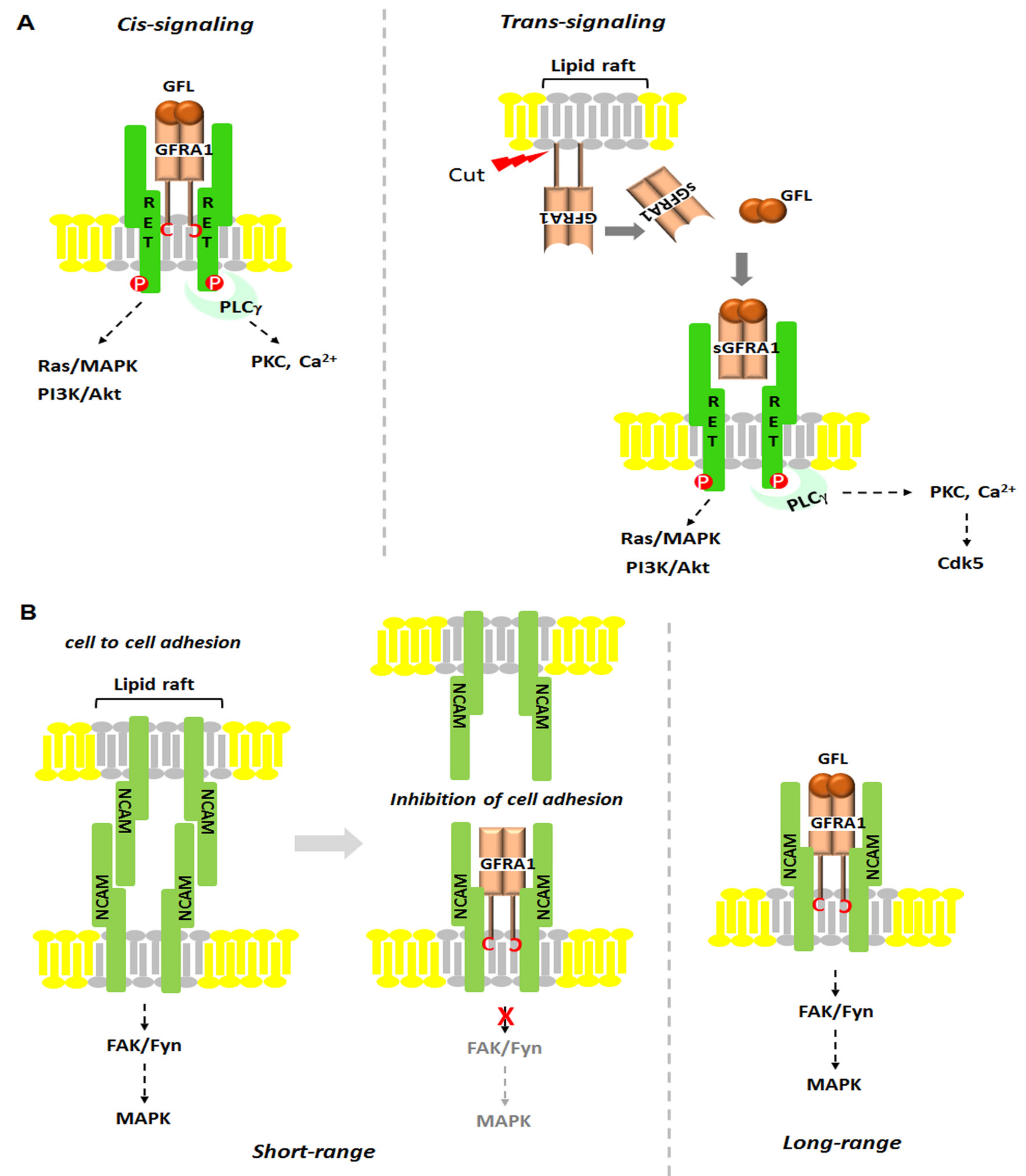
2.1. GFRA1-RET Signaling
2.2. GFRA1-NCAM Signaling
2.3. GDNF-GFRA1 Signaling: Ligand-Induced Cell Adhesion
2.4. GFRA1-SorLA Signaling
3. Physiological Roles of GFRA1 in Disease
4. GFRA1 in Cancer
5. Emerging Roles of GFRA1 in Chemoresistance
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jing, S.; Wen, D.; Yu, Y.; Holst, P.L.; Luo, Y.; Fang, M.; Tamir, R.; Antonio, L.; Hu, Z.; Cupples, R.; et al. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-α, a novel receptor for GDNF. Cell 1996, 85, 1113–1124. [Google Scholar] [CrossRef]
- Treanor, J.J.; Goodman, L.; de Sauvage, F.; Stone, D.M.; Poulsen, K.T.; Beck, C.D.; Gray, C.; Armanini, M.P.; Pollock, R.A.; Hefti, F.; et al. Characterization of a multicomponent receptor for GDNF. Nature 1996, 382, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H.; Tansey, M.G.; Golden, J.P.; Creedon, D.J.; Heuckeroth, R.O.; Keck, C.L.; Zimonjic, D.B.; Popescu, N.C.; Johnson, E.M., Jr.; Milbrandt, J. TRNR2, a novel receptor that mediates neurturin and GDNF signaling through Ret. Neuron 1997, 18, 793–802. [Google Scholar] [CrossRef]
- Baloh, R.H.; Gorodinsky, A.; Golden, J.P.; Tansey, M.G.; Keck, C.L.; Popescu, N.C.; Johnson, E.M., Jr.; Milbrandt, J. GFRα3 is an orphan member of the GDNF/neurturin/persephin receptor family. Proc. Natl. Acad. Sci. USA 1998, 95, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H.; Tansey, M.G.; Lampe, P.A.; Fahrner, T.J.; Enomoto, H.; Simburger, K.S.; Leitner, M.L.; Araki, T.; Johnson, E.M., Jr.; Milbrandt, J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3-RET receptor complex. Neuron 1998, 21, 1291–1302. [Google Scholar] [CrossRef]
- Naveilhan, P.; Baudet, C.; Mikaels, A.; Shen, L.; Westphal, H.; Ernfors, P. Expression and regulation of GFRα3, a glial cell line-derived neurotrophic factor family receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; de Sauvage, F.; Hongo, J.A.; Ninkina, N.; Rosenthal, A.; Buchman, V.L.; Davies, A.M. GFRα-4 and the tyrosine kinase Ret form a functional receptor complex for persephin. Curr. Biol. 1998, 8, 1019–1022. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, C.F.; Andressoo, J.O. Biology of GDNF and its receptors—Relevance for disorders of the central nervous system. Neurobiol. Dis. 2017, 97, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, C.F. Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Trupp, M.; Arenas, E.; Fainzilber, M.; Nilsson, A.S.; Sieber, B.A.; Grigoriou, M.; Kilkenny, C.; Salazar-Grueso, E.; Pachnis, V.; Arumae, U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature 1996, 381, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Saarma, M. GDNF—A stranger in the TGF-β superfamily? Eur. J. Biochem. 2000, 267, 6968–6971. [Google Scholar] [CrossRef] [PubMed]
- Sariola, H.; Saarma, M. Novel functions and signalling pathways for GDNF. J. Cell Sci. 2003, 116, 3855–3862. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Melillo, R.M.; Carlomagno, F.; Vecchio, G.; Fusco, A. Minireview: RET: Normal and abnormal functions. Endocrinology 2004, 145, 5448–5451. [Google Scholar] [CrossRef] [PubMed]
- Paratcha, G.; Ledda, F.; Ibanez, C.F. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 2003, 113, 867–879. [Google Scholar] [CrossRef]
- Meng, X.; Lindahl, M.; Hyvonen, M.E.; Parvinen, M.; de Rooij, D.G.; Hess, M.W.; Raatikainen-Ahokas, A.; Sainio, K.; Rauvala, H.; Lakso, M.; et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 2000, 287, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Viglietto, G.; Dolci, S.; Bruni, P.; Baldassarre, G.; Chiariotti, L.; Melillo, R.M.; Salvatore, G.; Chiappetta, G.; Sferratore, F.; Fusco, A.; et al. Glial cell line-derived neutrotrophic factor and neurturin can act as paracrine growth factors stimulating DNA synthesis of Ret-expressing spermatogonia. Int. J. Oncol. 2000, 16, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F.; Shakya, R. GDNF/Ret signaling and the development of the kidney. Bioessays 2006, 28, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Besset, V.; Scott, R.P.; Ibanez, C.F. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J. Biol. Chem. 2000, 275, 39159–39166. [Google Scholar] [CrossRef] [PubMed]
- Paratcha, G.; Ledda, F.; Baars, L.; Coulpier, M.; Besset, V.; Anders, J.; Scott, R.; Ibanez, C.F. Released GFRalpha1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron 2001, 29, 171–184. [Google Scholar] [CrossRef]
- Ledda, F.; Paratcha, G.; Ibanez, C.F. Target-derived GFRalpha1 as an attractive guidance signal for developing sensory and sympathetic axons via activation of Cdk5. Neuron 2002, 36, 387–401. [Google Scholar] [CrossRef]
- Grumet, M.; Rutishauser, U.; Edelman, G.M. Neural cell adhesion molecule is on embryonic muscle cells and mediates adhesion to nerve cells in vitro. Nature 1982, 295, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Covault, J.; Sanes, J.R. Neural cell adhesion molecule (N-CAM) accumulates in denervated and paralyzed skeletal muscles. Proc. Natl. Acad. Sci. USA 1985, 82, 4544–4548. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Duband, J.L.; Rutishauser, U.; Edelman, G.M. Cell adhesion molecules in early chicken embryogenesis. Proc. Natl. Acad. Sci. USA 1982, 79, 6737–6741. [Google Scholar] [CrossRef] [PubMed]
- Rutishauser, U.; Thiery, J.P.; Brackenbury, R.; Edelman, G.M. Adhesion among neural cells of the chick embryo. III. Relationship of the surface molecule CAM to cell adhesion and the development of histotypic patterns. J. Cell Biol. 1978, 79, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.; Albrechtsen, M.; Moller, C.; Lyles, J.; Bock, E.; Goridis, C.; Watanabe, M.; Rutishauser, U. Glial cells express N-CAM/D2-CAM-like polypeptides in vitro. Nature 1985, 316, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Pollerberg, E.G.; Sadoul, R.; Goridis, C.; Schachner, M. Selective expression of the 180-kD component of the neural cell adhesion molecule N-CAM during development. J. Cell Biol. 1985, 101, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Van den Bergh, J.M.; Berneman, Z.N.; Lion, E.; Smits, E.L.; Van Tendeloo, V.F. Interleukin-15 enhances the proliferation, stimulatory phenotype, and antitumor effector functions of human gamma delta T cells. J. Hematol. Oncol. 2016, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Rogers, J.; Madrigal-Estebas, L.; O’Connor, T.; Doherty, D.G. Activation-induced expression of CD56 by T cells is associated with a reprogramming of cytolytic activity and cytokine secretion profile in vitro. Hum. Immunol. 2006, 67, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Roothans, D.; Smits, E.; Lion, E.; Tel, J.; Anguille, S. CD56 marks human dendritic cell subsets with cytotoxic potential. Oncoimmunology 2013, 2, e23037. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.A.; Small, S.J.; Akeson, R. At least 27 alternatively spliced forms of the neural cell adhesion molecule mRNA are expressed during rat heart development. Mol. Cell. Biol. 1991, 11, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Yang, F.; He, X.P.; Je, H.S.; Zhou, J.Z.; Eckermann, K.; Kawamura, D.; Feng, L.; Shen, L.; Lu, B. Regulation of neuromuscular synapse development by glial cell line-derived neurotrophic factor and neurturin. J. Biol. Chem. 2002, 277, 10614–10625. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Gotfryd, K.; Li, S.; Kulahin, N.; Soroka, V.; Rasmussen, K.K.; Bock, E.; Berezin, V. Role of glial cell line-derived neurotrophic factor (GDNF)-neural cell adhesion molecule (NCAM) interactions in induction of neurite outgrowth and identification of a binding site for NCAM in the heel region of GDNF. J. Neurosci. 2009, 29, 11360–11376. [Google Scholar] [CrossRef] [PubMed]
- Ledda, F. Ligand-induced cell adhesion as a new mechanism to promote synapse formation. Cell Adhes. Migr. 2007, 1, 137–139. [Google Scholar] [CrossRef]
- Glerup, S.; Lume, M.; Olsen, D.; Nyengaard, J.R.; Vaegter, C.B.; Gustafsen, C.; Christensen, E.I.; Kjolby, M.; Hay-Schmidt, A.; Bender, D.; et al. SorLA controls neurotrophic activity by sorting of GDNF and its receptors GFRα1 and RET. Cell Rep. 2013, 3, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E.; Trupp, M.; Akerud, P.; Ibanez, C.F. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron 1995, 15, 1465–1473. [Google Scholar] [CrossRef]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, H.B.; Acharya, S.; Sohn, H.M.; Jun, J.Y.; Chang, I.Y.; You, H.J. Ape1/Ref-1 induces glial cell-derived neurotropic factor (GDNF) responsiveness by upregulating GDNF receptor alpha1 expression. Mol. Cell. Biol. 2009, 29, 2264–2277. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.Y.; Kim, K.Y.; Yoon, Y.; Kang, Y.; Kim, H.B.; Youn, C.K.; Kim, D.H.; Kim, M.H. Ape1/Ref-1 Stimulates GDNF/GFRα1-mediated Downstream Signaling and Neuroblastoma Proliferation. Korean J. Physiol. Pharmacol. 2009, 13, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Newgreen, D.; Young, H.M. Enteric nervous system: Development and developmental disturbances—Part 2. Pediatr. Dev. Pathol. 2002, 5, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Pichel, J.G.; Mayeli, T.; Sariola, H.; Lu, B.; Westphal, H. Gdnf haploinsufficiency causes Hirschsprung-like intestinal obstruction and early-onset lethality in mice. Am. J. Hum. Genet. 2002, 70, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Borghini, S.; Bocciardi, R.; Bonardi, G.; Matera, I.; Santamaria, G.; Ravazzolo, R.; Ceccherini, I. Hirschsprung associated GDNF mutations do not prevent RET activation. Eur. J. Hum. Genet. 2002, 10, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Borrego, S.; Fernandez, R.M.; Dziema, H.; Niess, A.; Lopez-Alonso, M.; Antinolo, G.; Eng, C. Investigation of germline GFRA4 mutations and evaluation of the involvement of GFRA1, GFRA2, GFRA3, and GFRA4 sequence variants in Hirschsprung disease. J. Med. Genet. 2003, 40, e18. [Google Scholar] [CrossRef] [PubMed]
- Eketjall, S.; Ibanez, C.F. Functional characterization of mutations in the GDNF gene of patients with Hirschsprung disease. Hum. Mol. Genet. 2002, 11, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Bespalov, M.M.; Sidorova, Y.A.; Tumova, S.; Ahonen-Bishopp, A.; Magalhaes, A.C.; Kulesskiy, E.; Paveliev, M.; Rivera, C.; Rauvala, H.; Saarma, M. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J. Cell Biol. 2011, 192, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Pozas, E.; Ibanez, C.F. GDNF and GFRalpha1 promote differentiation and tangential migration of cortical GABAergic neurons. Neuron 2005, 45, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Perrinjaquet, M.; Sjostrand, D.; Moliner, A.; Zechel, S.; Lamballe, F.; Maina, F.; Ibanez, C.F. MET signaling in GABAergic neuronal precursors of the medial ganglionic eminence restricts GDNF activity in cells that express GFRα1 and a new transmembrane receptor partner. J. Cell Sci. 2011, 124, 2797–2805. [Google Scholar] [CrossRef] [PubMed]
- Charoy, C.; Nawabi, H.; Reynaud, F.; Derrington, E.; Bozon, M.; Wright, K.; Falk, J.; Helmbacher, F.; Kindbeiter, K.; Castellani, V. gdnf activates midline repulsion by Semaphorin3B via NCAM during commissural axon guidance. Neuron 2012, 75, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Duveau, V.; Fritschy, J.M. PSA-NCAM-dependent GDNF signaling limits neurodegeneration and epileptogenesis in temporal lobe epilepsy. Eur. J. Neurosci. 2010, 32, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Euteneuer, S.; Yang, K.H.; Chavez, E.; Leichtle, A.; Loers, G.; Olshansky, A.; Pak, K.; Schachner, M.; Ryan, A.F. Glial cell line-derived neurotrophic factor (GDNF) induces neuritogenesis in the cochlear spiral ganglion via neural cell adhesion molecule (NCAM). Mol. Cell. Neurosci. 2013, 54, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Paratcha, G.; Ledda, F. GDNF and GFRalpha: A versatile molecular complex for developing neurons. Trends Neurosci. 2008, 31, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Paratcha, G.; Ibanez, C.F. Lipid rafts and the control of neurotrophic factor signaling in the nervous system: Variations on a theme. Curr. Opin. Neurobiol. 2002, 12, 542–549. [Google Scholar] [CrossRef]
- Krakora, D.; Mulcrone, P.; Meyer, M.; Lewis, C.; Bernau, K.; Gowing, G.; Zimprich, C.; Aebischer, P.; Svendsen, C.N.; Suzuki, M. Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model. Mol. Ther. 2013, 21, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Meissner, W.G.; Frasier, M.; Gasser, T.; Goetz, C.G.; Lozano, A.; Piccini, P.; Obeso, J.A.; Rascol, O.; Schapira, A.; Voon, V.; et al. Priorities in Parkinson’s disease research. Nat. Rev. Drug Discov. 2011, 10, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Trupp, M.; Belluardo, N.; Funakoshi, H.; Ibanez, C.F. Complementary and overlapping expression of glial cell line-derived neurotrophic factor (GDNF), c-ret proto-oncogene, and GDNF receptor-alpha indicates multiple mechanisms of trophic actions in the adult rat CNS. J. Neurosci. 1997, 17, 3554–3567. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Kholodilov, N.; Kim, S.R.; Yarygina, O.; Kareva, T.; Cho, J.W.; Baohan, A.; Burke, R.E. Glial cell line-derived neurotrophic factor receptor-α1 expressed in striatum in trans regulates development and injury response of dopamine neurons of the substantia nigra. J. Neurochem. 2011, 116, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Pruett, B.S.; Salvatore, M.F. Nigral GFRalpha1 infusion in aged rats increases locomotor activity, nigral tyrosine hydroxylase, and dopamine content in synchronicity. Mol. Neurobiol. 2013, 47, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Ledda, F.; Paratcha, G.; Sandoval-Guzman, T.; Ibanez, C.F. GDNF and GFRα1 promote formation of neuronal synapses by ligand-induced cell adhesion. Nat. Neurosci. 2007, 10, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Marks, C.; Belluscio, L.; Ibanez, C.F. Critical role of GFRalpha1 in the development and function of the main olfactory system. J. Neurosci. 2012, 32, 17306–17320. [Google Scholar] [CrossRef] [PubMed]
- Duarte, E.P.; Curcio, M.; Canzoniero, L.M.; Duarte, C.B. Neuroprotection by GDNF in the ischemic brain. Growth Factors 2012, 30, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Reeben, M.; Laurikainen, A.; Hiltunen, J.O.; Castren, E.; Saarma, M. The messenger RNAs for both glial cell line-derived neurotrophic factor receptors, c-ret and GDNFRalpha, are induced in the rat brain in response to kainate-induced excitation. Neuroscience 1998, 83, 151–159. [Google Scholar] [CrossRef]
- Scholz, D.; Chernyshova, Y.; Leist, M. Control of Aβ release from human neurons by differentiation status and RET signaling. Neurobiol. Aging 2013, 34, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Britschgi, M.; Herbert, C.; Takeda-Uchimura, Y.; Boxer, A.; Blennow, K.; Friedman, L.F.; Galasko, D.R.; Jutel, M.; Karydas, A.; et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 2007, 13, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Messer, C.J.; Eisch, A.J.; Carlezon, W.A., Jr.; Whisler, K.; Shen, L.; Wolf, D.H.; Westphal, H.; Collins, F.; Russell, D.S.; Nestler, E.J. Role for GDNF in biochemical and behavioral adaptations to drugs of abuse. Neuron 2000, 26, 247–257. [Google Scholar] [CrossRef]
- Ubhi, K.; Inglis, C.; Mante, M.; Patrick, C.; Adame, A.; Spencer, B.; Rockenstein, E.; May, V.; Winkler, J.; Masliah, E. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of α-synucleinopathy. Exp. Neurol. 2012, 234, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Tunca, Z.; Kivircik Akdede, B.; Ozerdem, A.; Alkin, T.; Polat, S.; Ceylan, D.; Bayin, M.; Cengizcetin Kocuk, N.; Simsek, S.; Resmi, H.; et al. Diverse glial cell line-derived neurotrophic factor (GDNF) support between mania and schizophrenia: A comparative study in four major psychiatric disorders. Eur. Psychiatry 2015, 30, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Fontenelle, L.F.; Barbosa, I.G.; Luna, J.V.; Rocha, N.P.; Silva Miranda, A.; Teixeira, A.L. Neurotrophic factors in obsessive-compulsive disorder. Psychiatry Res. 2012, 199, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Moises, H.W.; Zoega, T.; Gottesman, I.I. The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry 2002, 2, 8. [Google Scholar] [CrossRef]
- Maheu, M.; Lopez, J.P.; Crapper, L.; Davoli, M.A.; Turecki, G.; Mechawar, N. MicroRNA regulation of central glial cell line-derived neurotrophic factor (GDNF) signalling in depression. Transl. Psychiatry 2015, 5, e511. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jung, J.Y.; Choi, S.; Lee, H.; Morales, L.D.; Koh, J.T.; Kim, S.H.; Choi, Y.D.; Choi, C.; Slaga, T.J.; et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy 2017, 13, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Esseghir, S.; Todd, S.K.; Hunt, T.; Poulsom, R.; Plaza-Menacho, I.; Reis-Filho, J.S.; Isacke, C.M. A role for glial cell derived neurotrophic factor induced expression by inflammatory cytokines and RET/GFRα1 receptor up-regulation in breast cancer. Cancer Res. 2007, 67, 11732–11741. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Liu, P.; Xie, X.; Zhou, Y.; Liao, Q.; Xiong, W.; Li, X.; Li, G.; Zeng, Z.; Tang, H. circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J. Exp. Clin. Cancer Res. 2017, 36, 145. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.S.; Pandey, V.; Wu, W.Y.; Ye, S.; Zhu, T.; Lobie, P.E. Prognostic significance of the expression of GFRα1, GFRalpha3 and syndecan-3, proteins binding ARTEMIN, in mammary carcinoma. BMC Cancer 2013, 13, 34. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, Y.; Yajima, I.; Takeda, K.; Iida, M.; Kumasaka, M.; Matsumoto, Y.; Kato, M. c-RET molecule in malignant melanoma from oncogenic RET-carrying transgenic mice and human cell lines. PLoS ONE 2010, 5, e10279. [Google Scholar] [CrossRef] [PubMed]
- Morandi, A.; Martin, L.A.; Gao, Q.; Pancholi, S.; Mackay, A.; Robertson, D.; Zvelebil, M.; Dowsett, M.; Plaza-Menacho, I.; Isacke, C.M. GDNF-RET signaling in ER-positive breast cancers is a key determinant of response and resistance to aromatase inhibitors. Cancer Res. 2013, 73, 3783–3795. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, S.; Crocker, L.M.; Chen, Y.; Hazen, M.; Schutten, M.M.; Li, D.; Kuijl, C.; Ohri, R.; Zhong, F.; Poon, K.A.; et al. An anti-GDNF Family Receptor α 1(GFRA1) Antibody-Drug Conjugate for the Treatment of Hormone Receptor-Positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [PubMed]
- Luetke, A.; Meyers, P.A.; Lewis, I.; Juergens, H. Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.; Kleinerman, E.S.; Betcher, D.; Bernstein, M.L.; Conrad, E.; Ferguson, W.; Gebhardt, M.; Goorin, A.M.; et al. Osteosarcoma: A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J. Clin. Oncol. 2005, 23, 2004–2011. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; DeBerardinis, R.J.; Thompson, C.B. Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell. Biol. 2005, 6, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Levine, B. Unraveling the role of autophagy in cancer. Autophagy 2006, 2, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta 2009, 1793, 1516–1523. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Ligand | Type of Receptor | Biological Functions | Type of Cancer | References |
|---|---|---|---|---|
| ARTN | GFRA1/GFRA3 | ND | mammary carcinoma | [76] |
| GDNF | RET/GFRA1 | ND | Breast cancer | [74] |
| GDNF | RET/GFRA1 | Oncogenicity | malignant melanoma | [77] |
| GDNF | RET/GFRA1 | Aromatase inhibitor resistance | Breast cancer | [78] |
| ND | GFRA1 | Cisplatin resistance by autophagy | Osteosarcoma | [73] |
| ND | GFRA1 | ND | Breast cancer | [79] |
| ND | GFRA1 | ND | Breast cancer | [75] |
| GDNF | GFRA1 | Cell migration and proliferation | Pancreatic cancer | [40] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Kim, D.J. GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance. Int. J. Mol. Sci. 2018, 19, 1078. https://doi.org/10.3390/ijms19041078
Kim M, Kim DJ. GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance. International Journal of Molecular Sciences. 2018; 19(4):1078. https://doi.org/10.3390/ijms19041078
Chicago/Turabian StyleKim, Mihwa, and Dae Joon Kim. 2018. "GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance" International Journal of Molecular Sciences 19, no. 4: 1078. https://doi.org/10.3390/ijms19041078