GPCR Modulation in Breast Cancer



Abstract
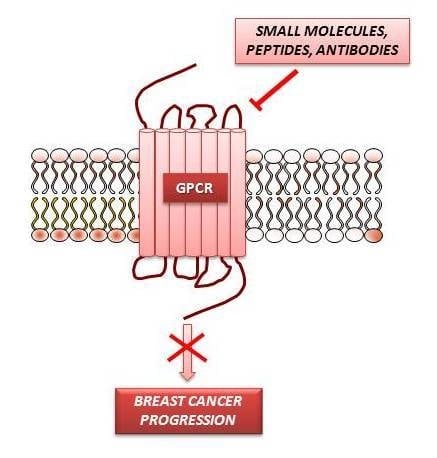
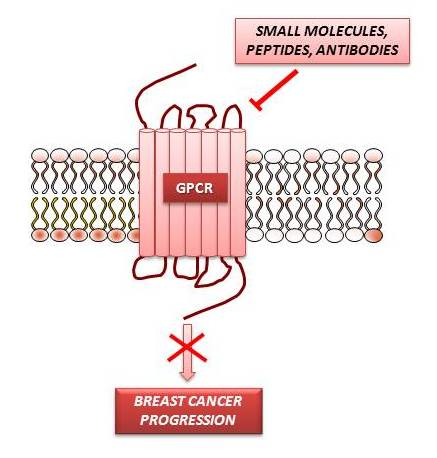
:
{kind=link}
{kind=link}
{kind=link}
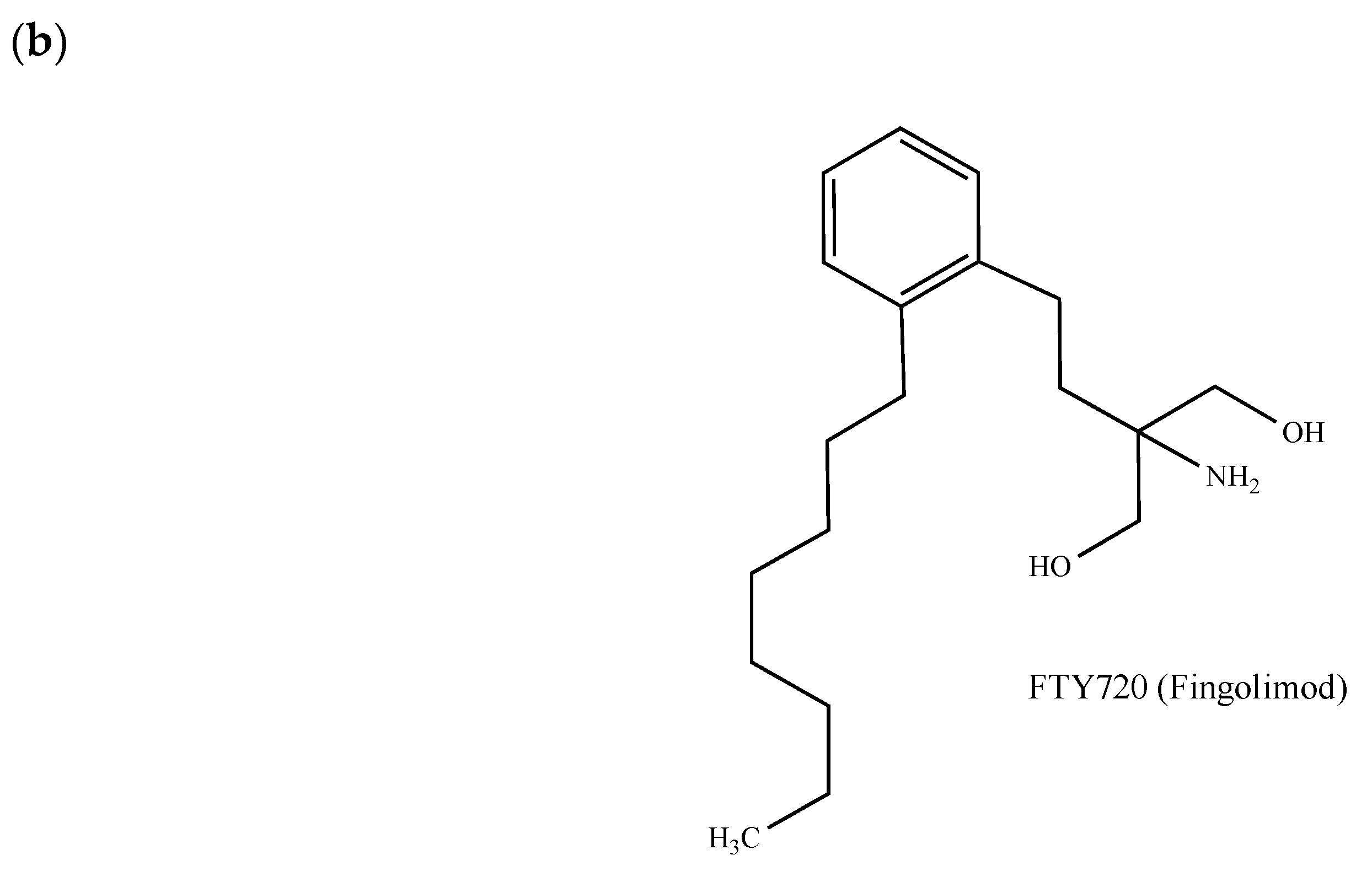{kind=link}
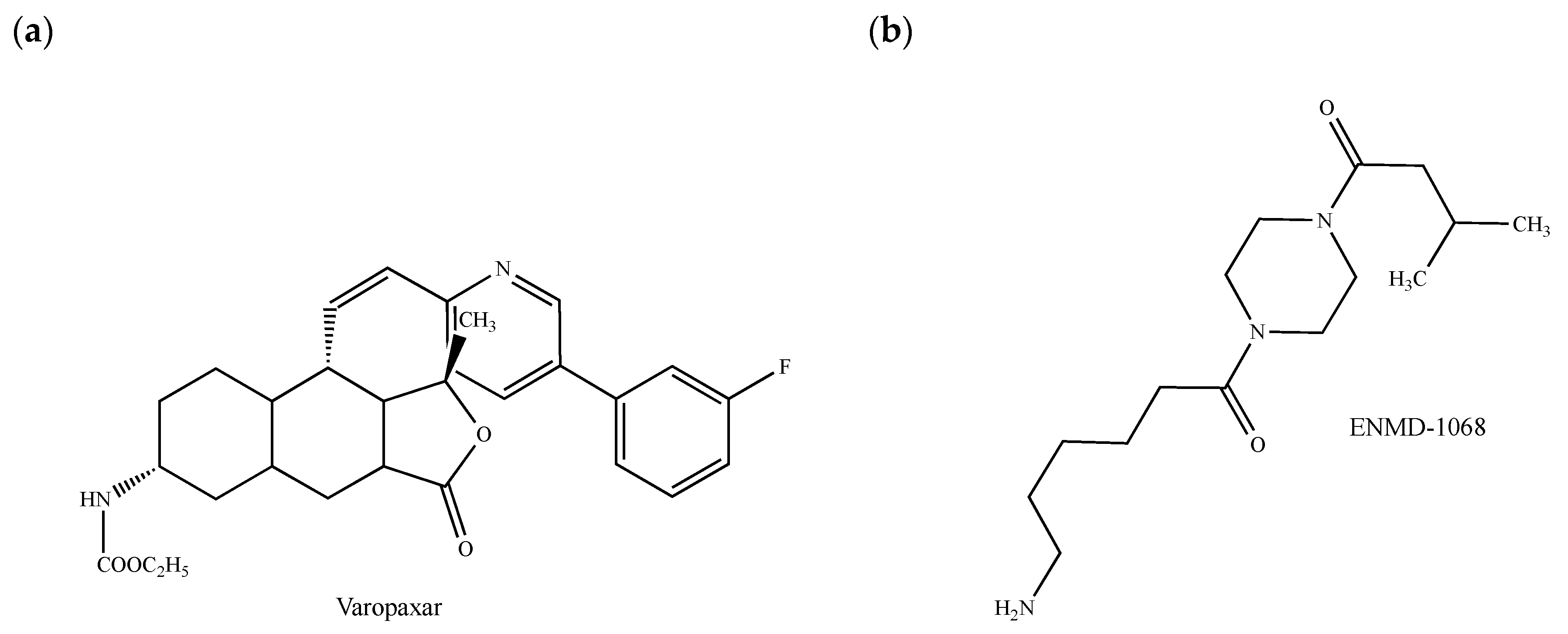
{kind=link}
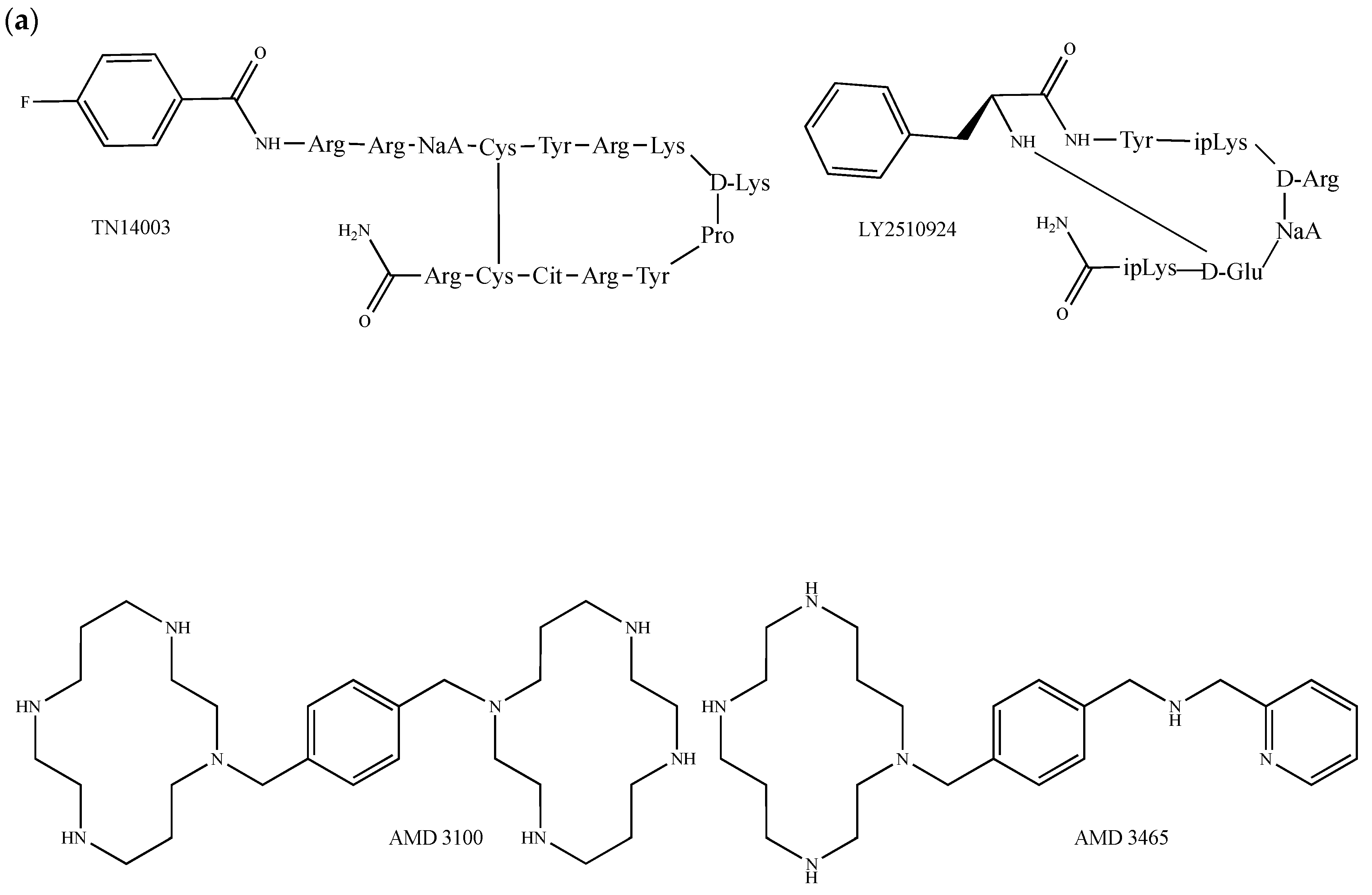
{kind=link}
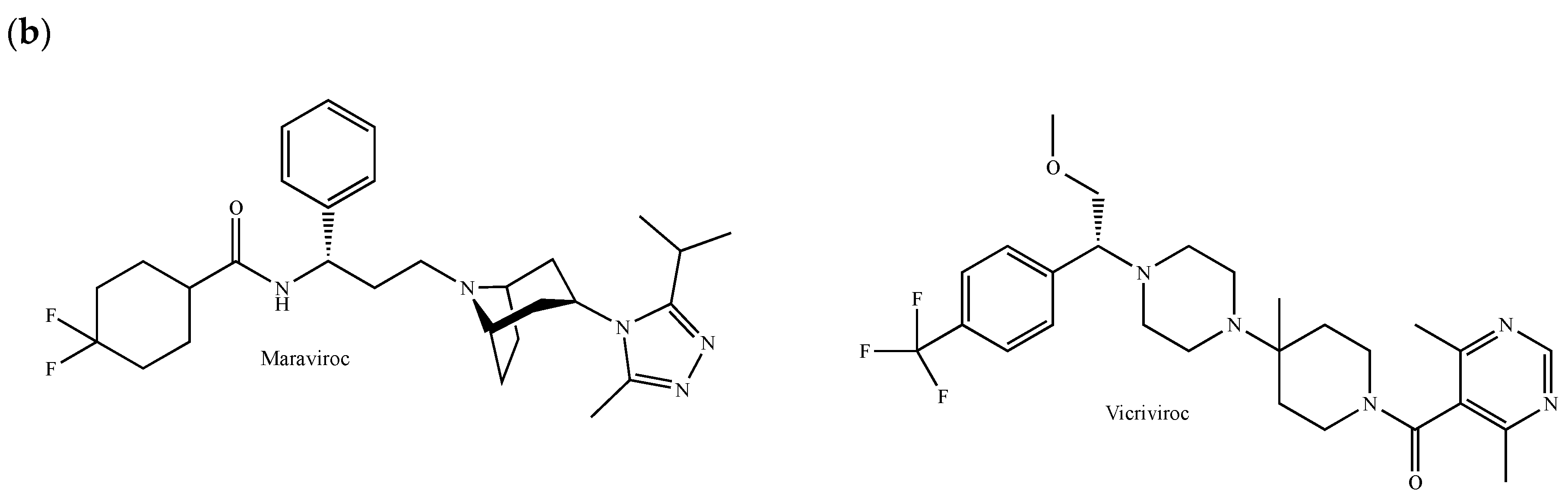
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
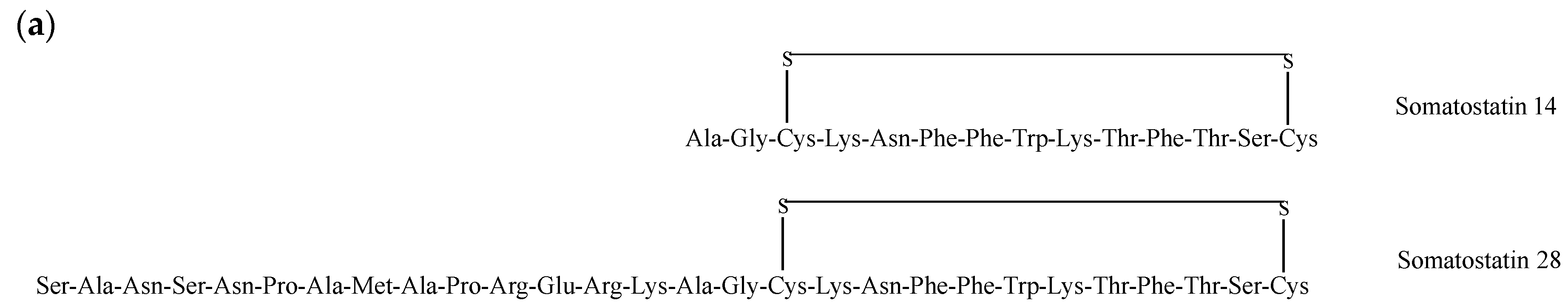{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
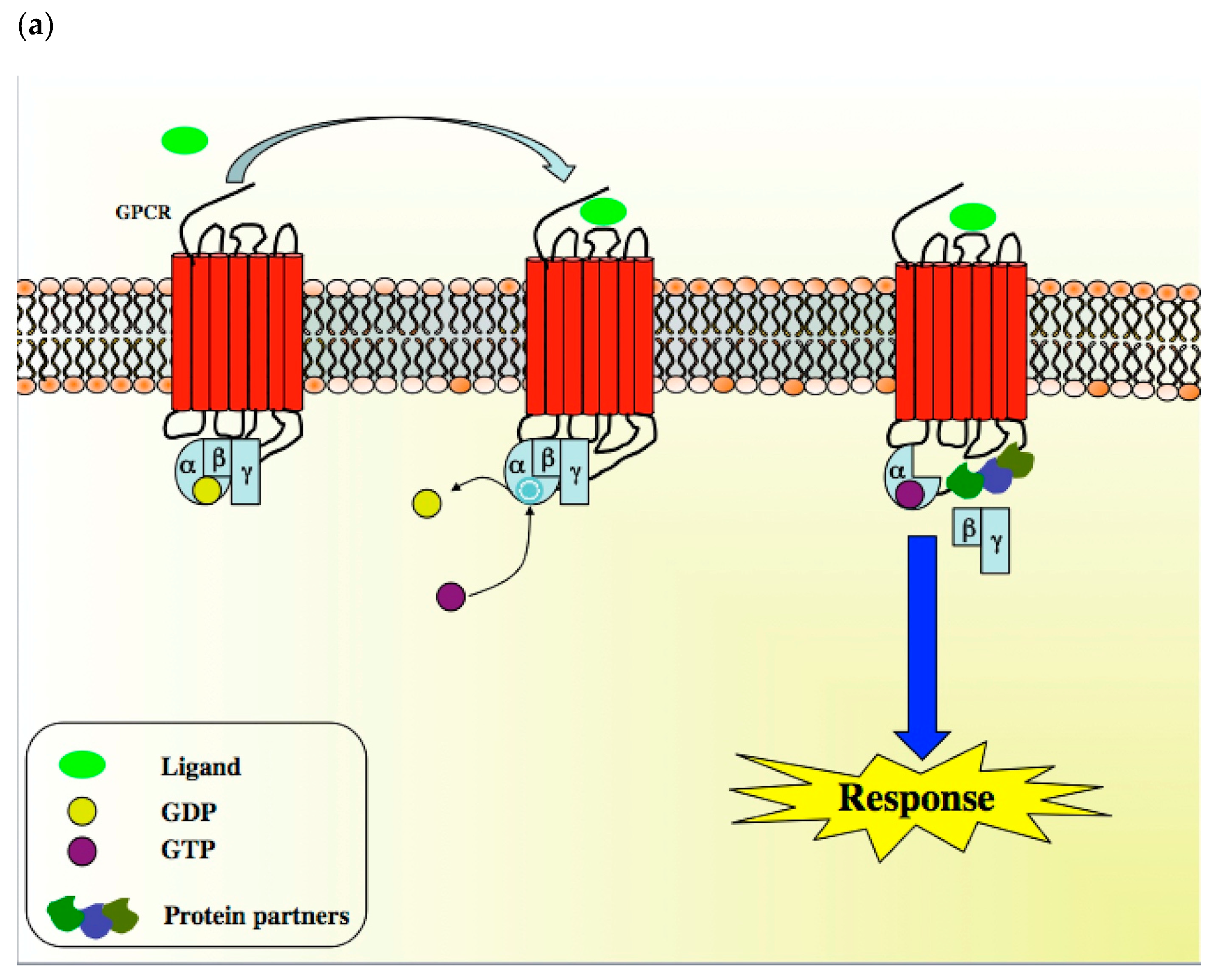{kind=link}
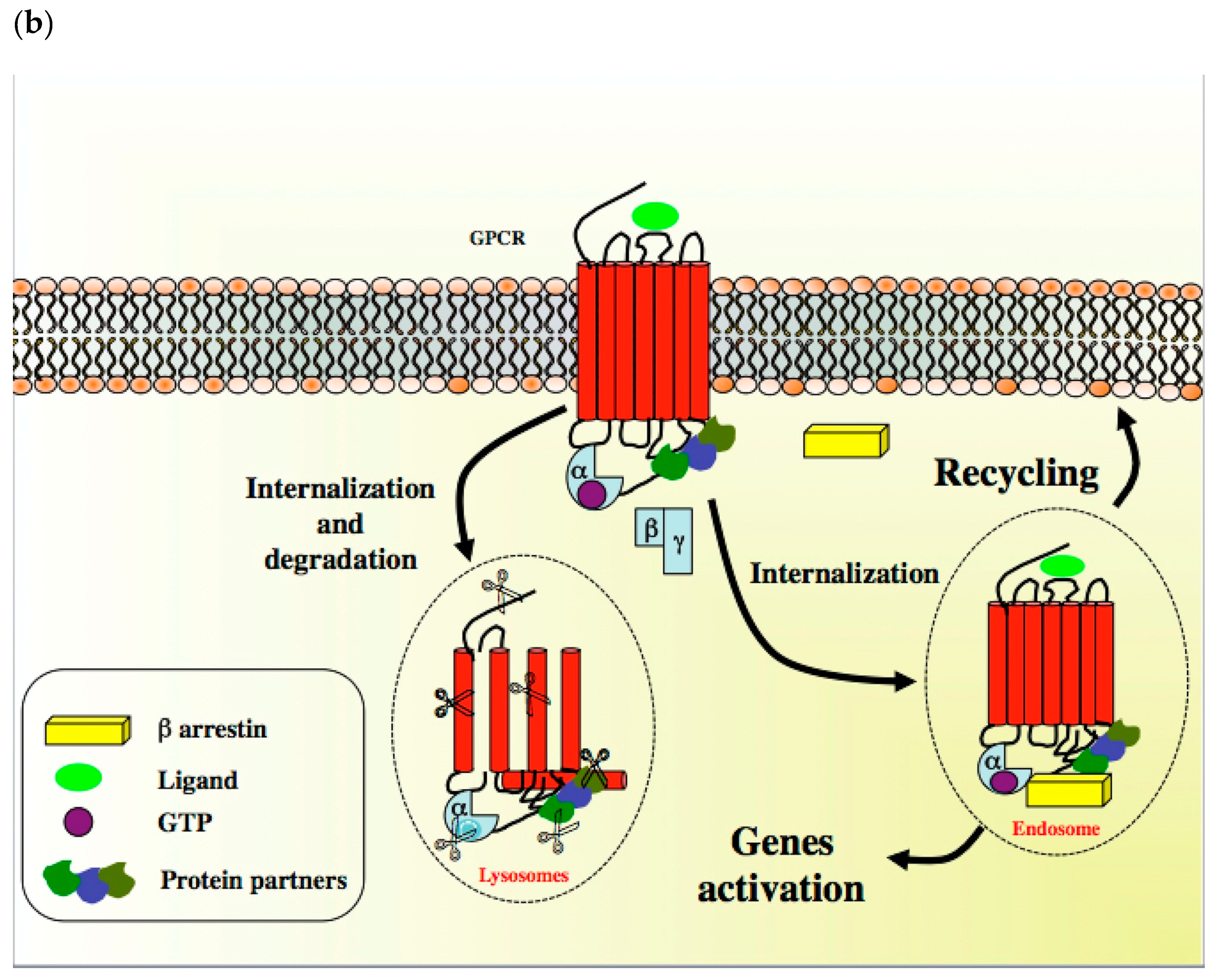{kind=link}
1. Generalities
2. GPCRs in Breast Cancer: Signaling, Biology and Modulators
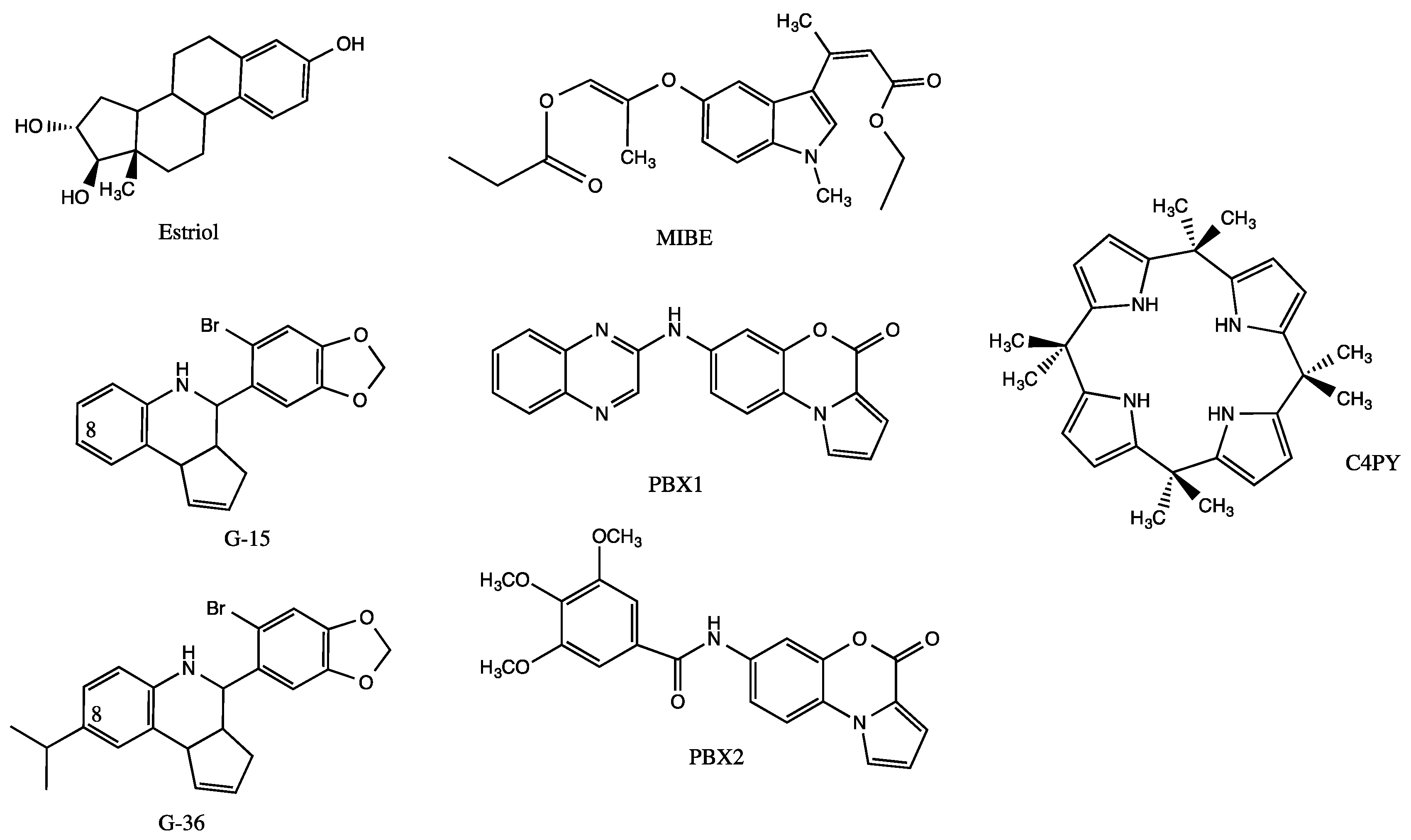
2.1. GPER
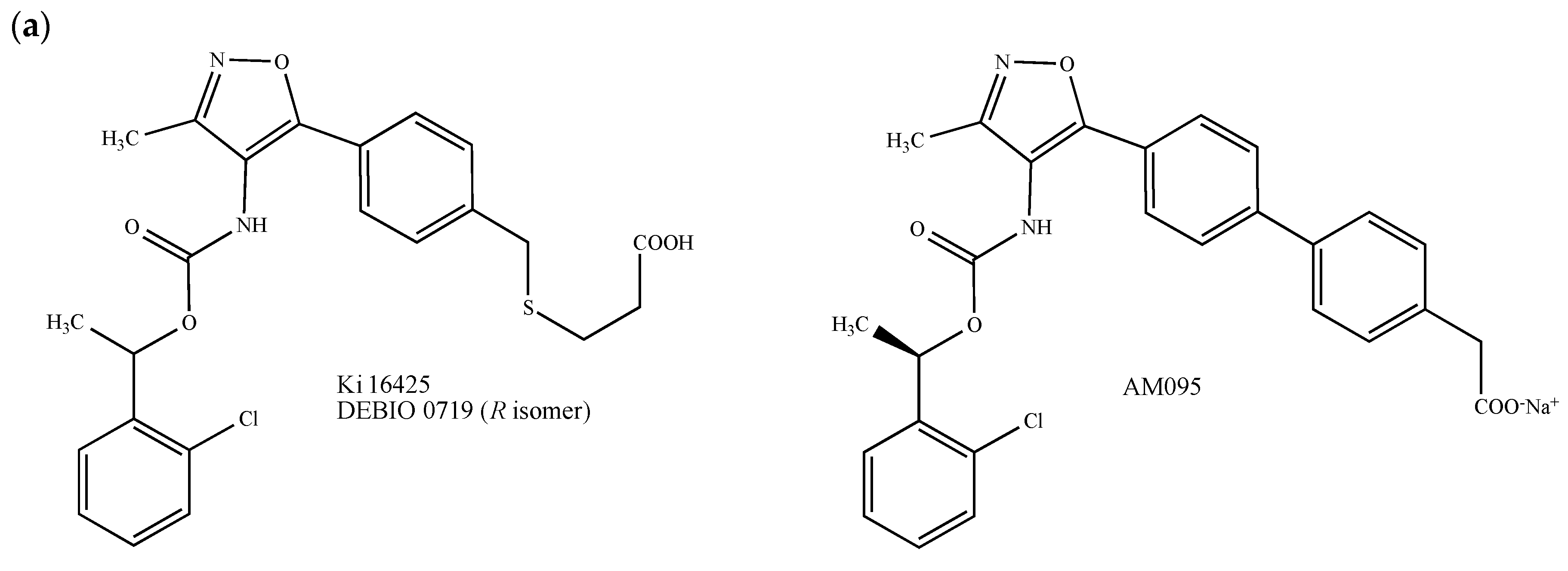
2.2. Lysophosphatidic Acid (LPA) and Sphingosine-1-Phosphate (S1P) Receptors
2.3. Prostaglandin E2 Receptors
2.4. Protease-Activated Receptors
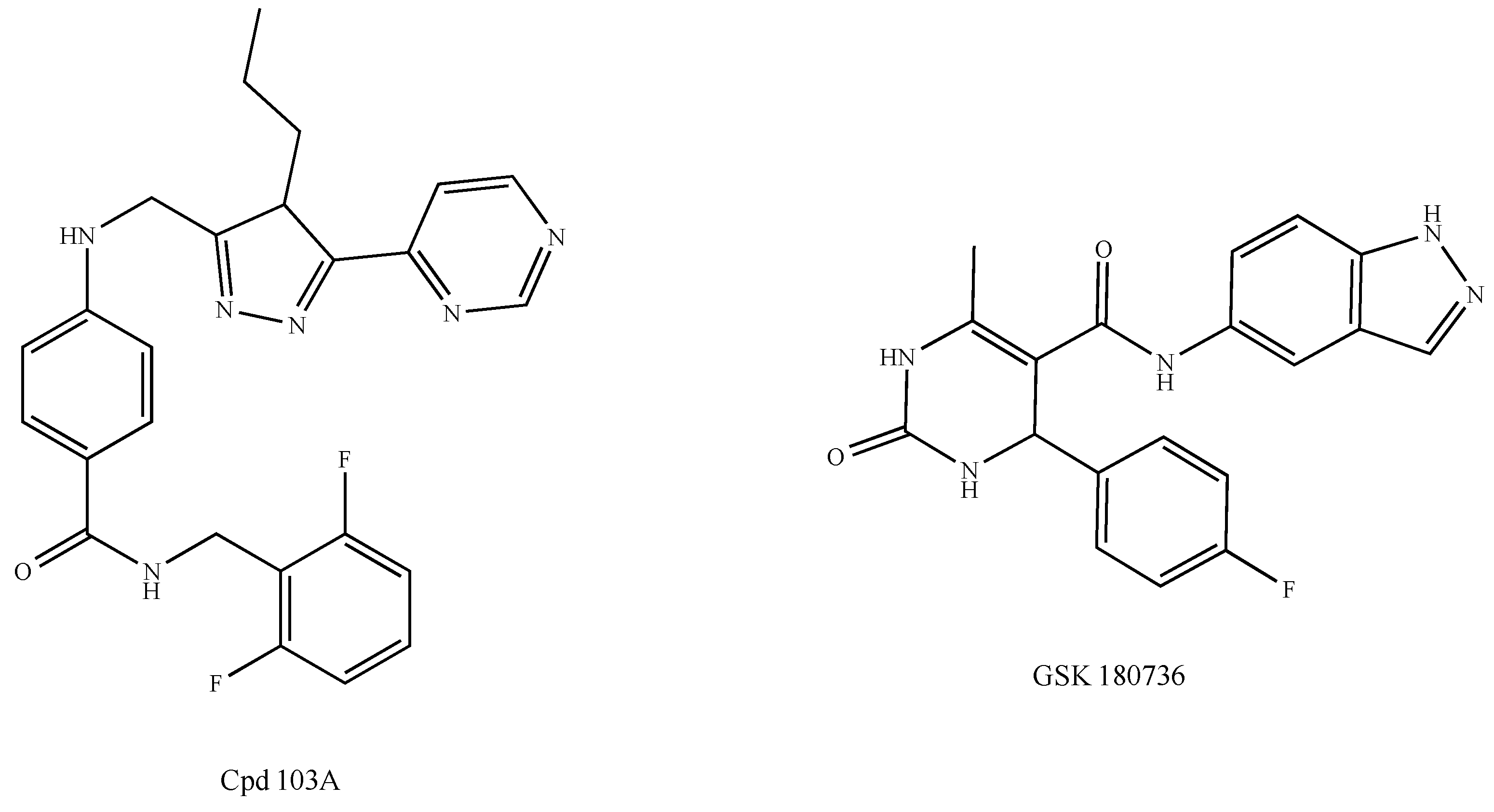
2.5. Chemokine Receptors
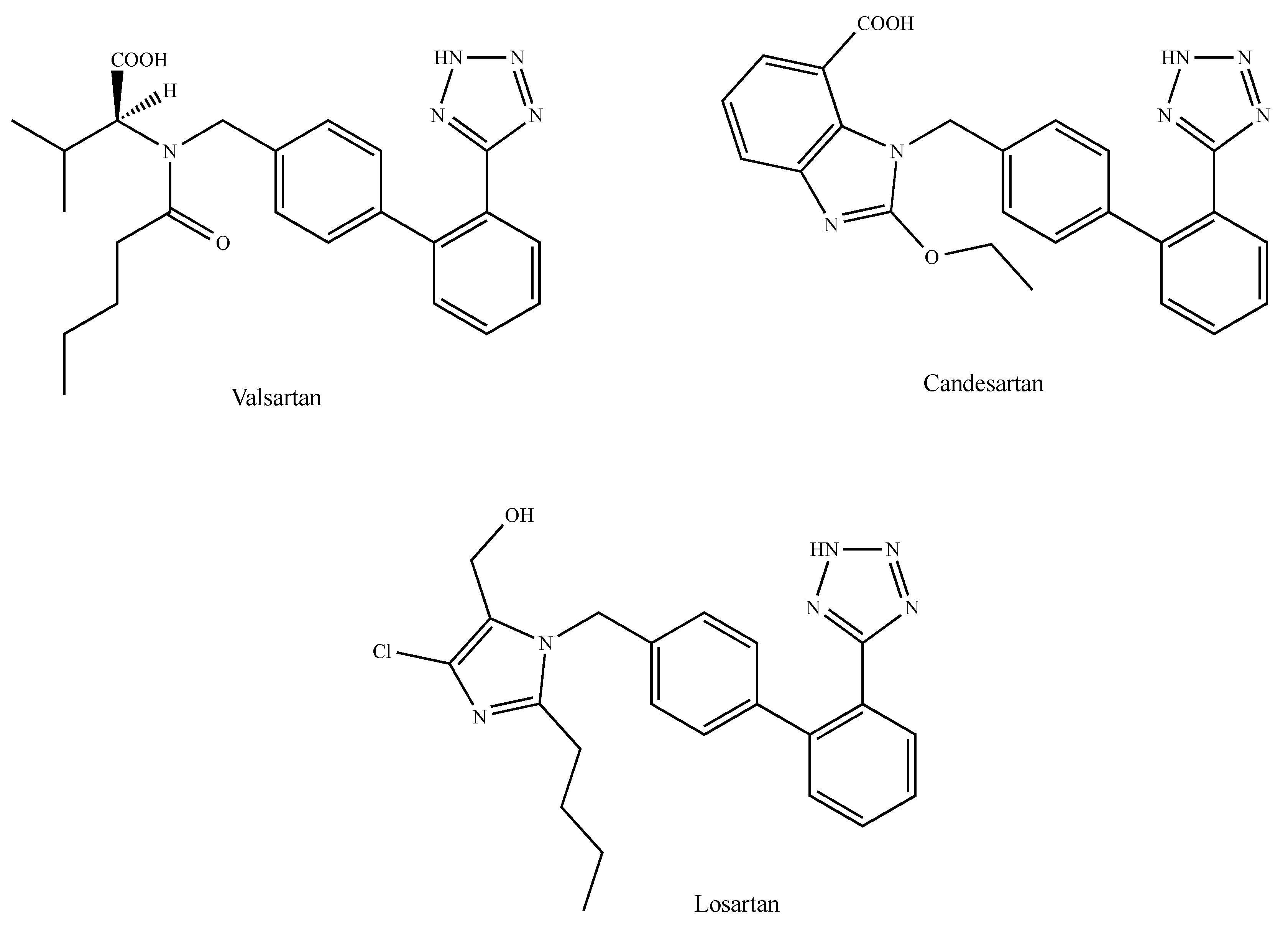
2.6. Angiotensin II Type 1 Receptor
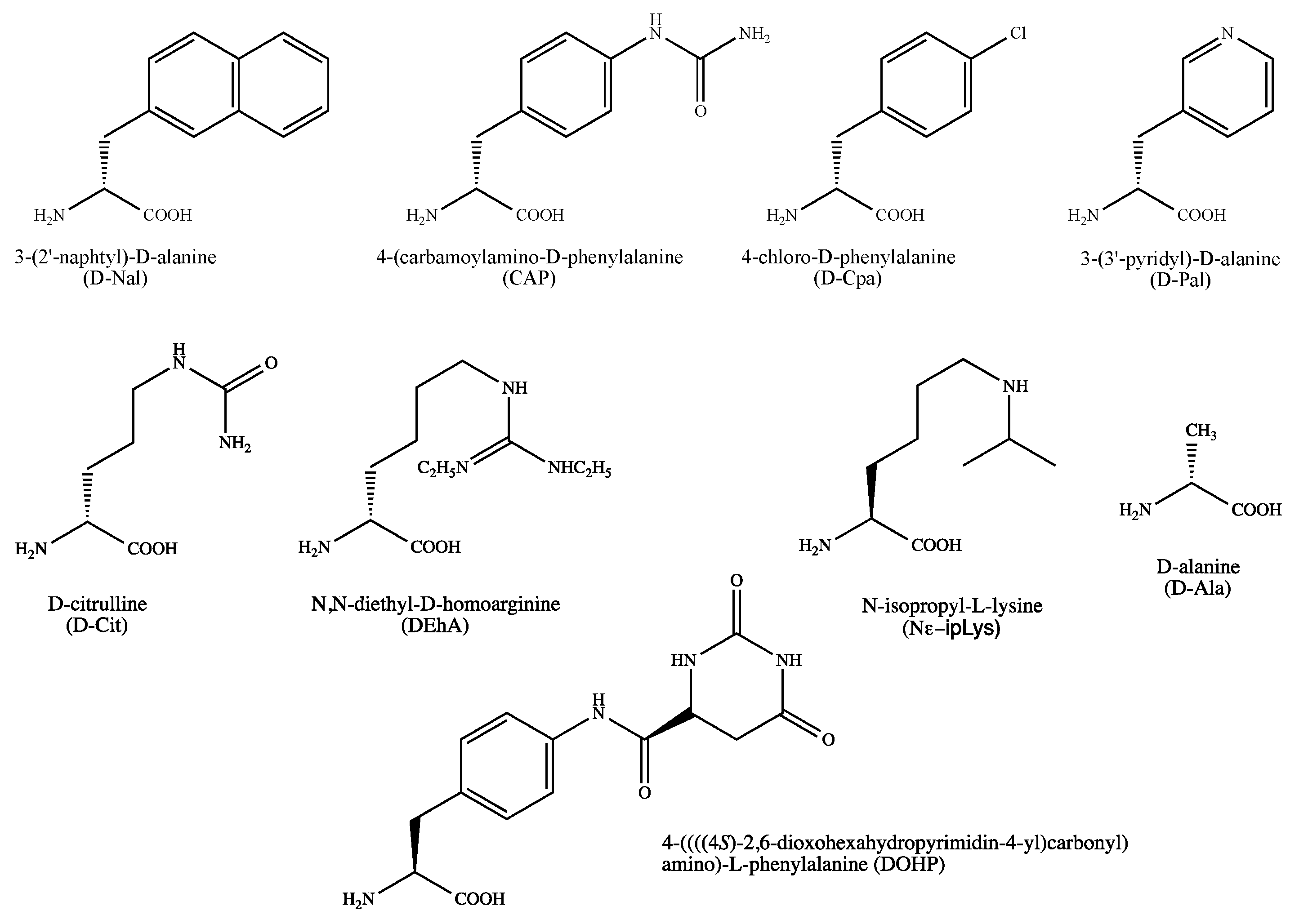
2.7. Gonadotropin-Releasing Hormone Receptors
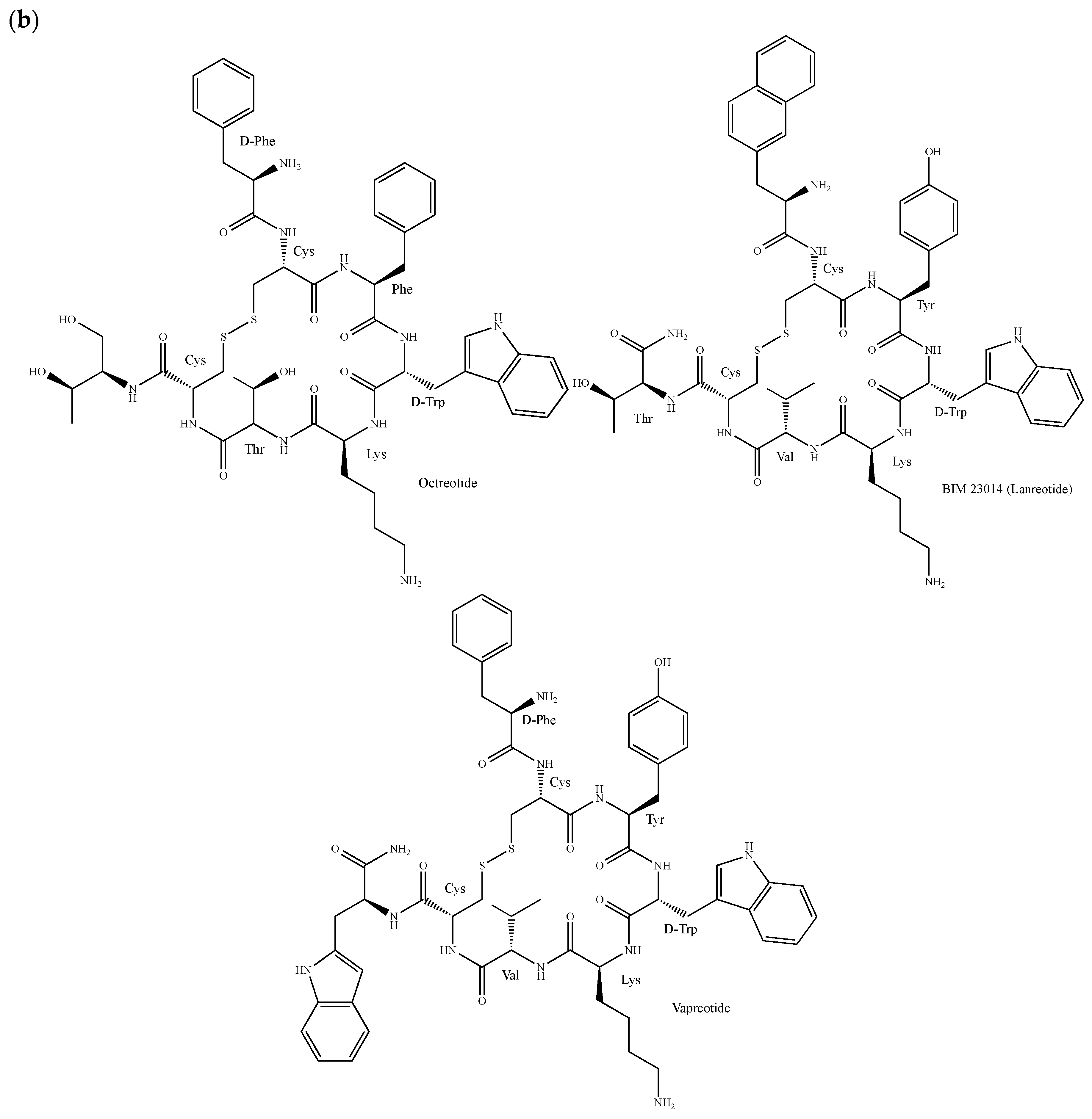
2.8. Somatostatin Receptors
2.9. Gastrin-Releasing Peptide Receptors
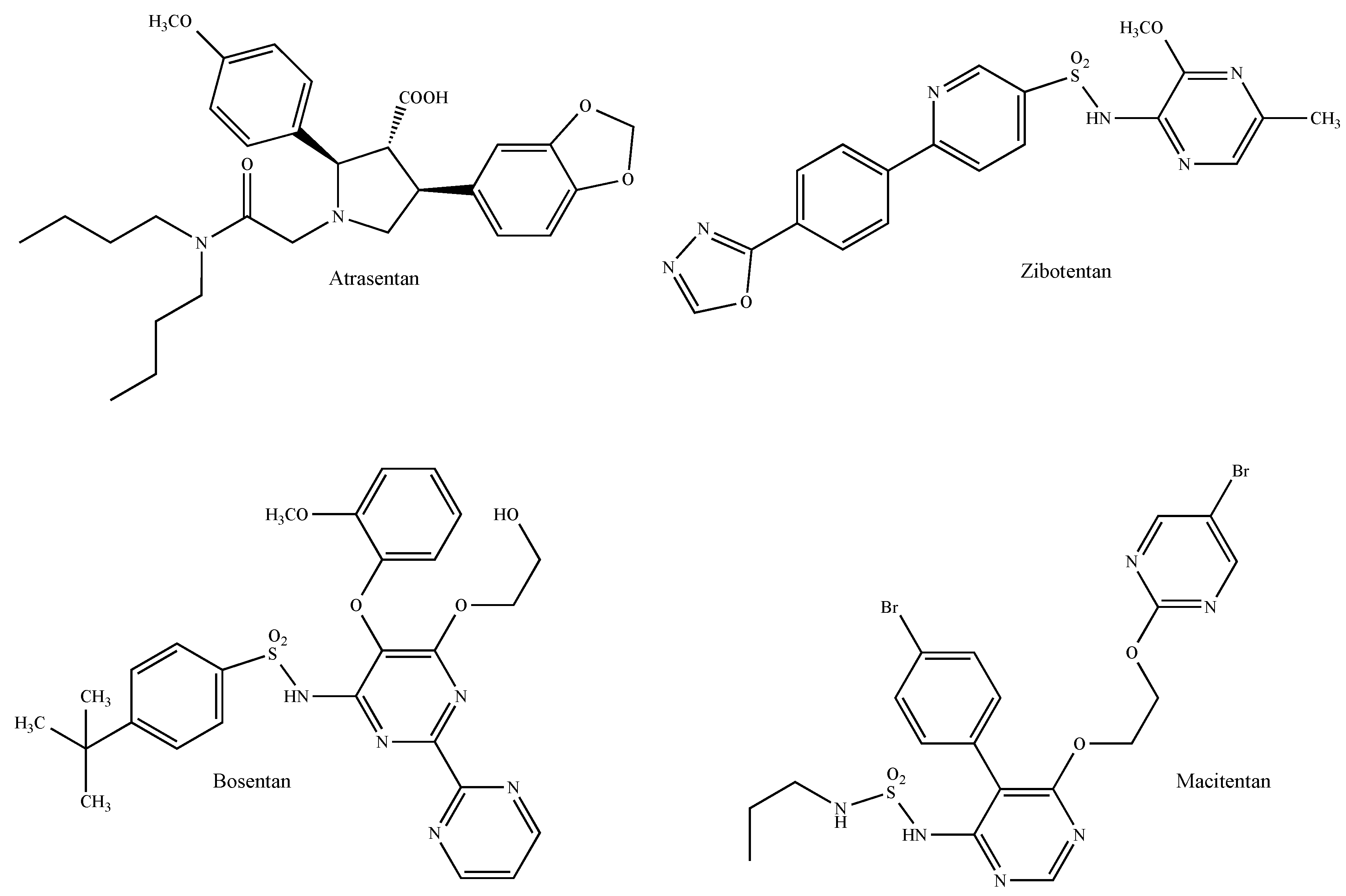
2.10. Endothelin Receptors
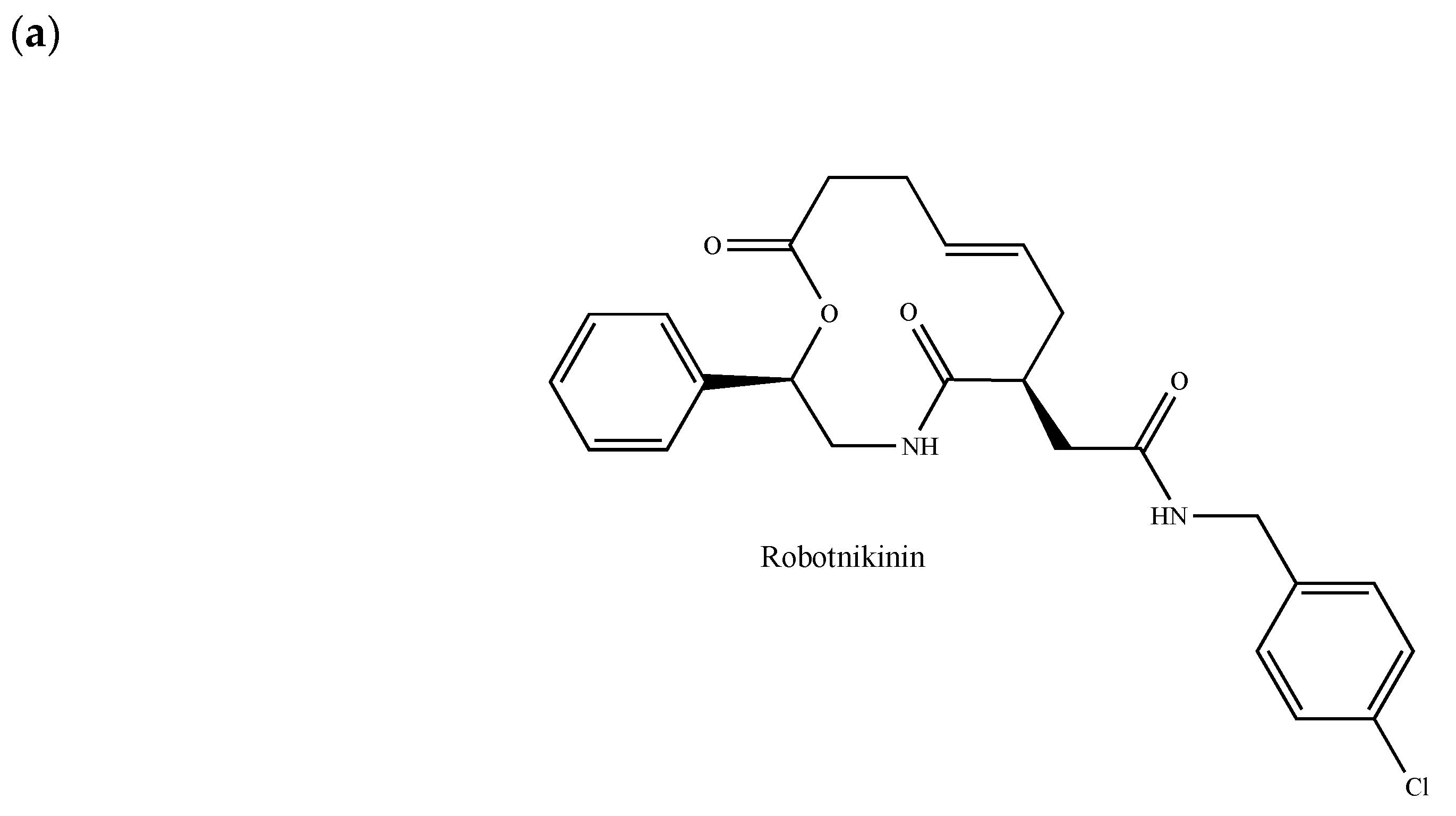
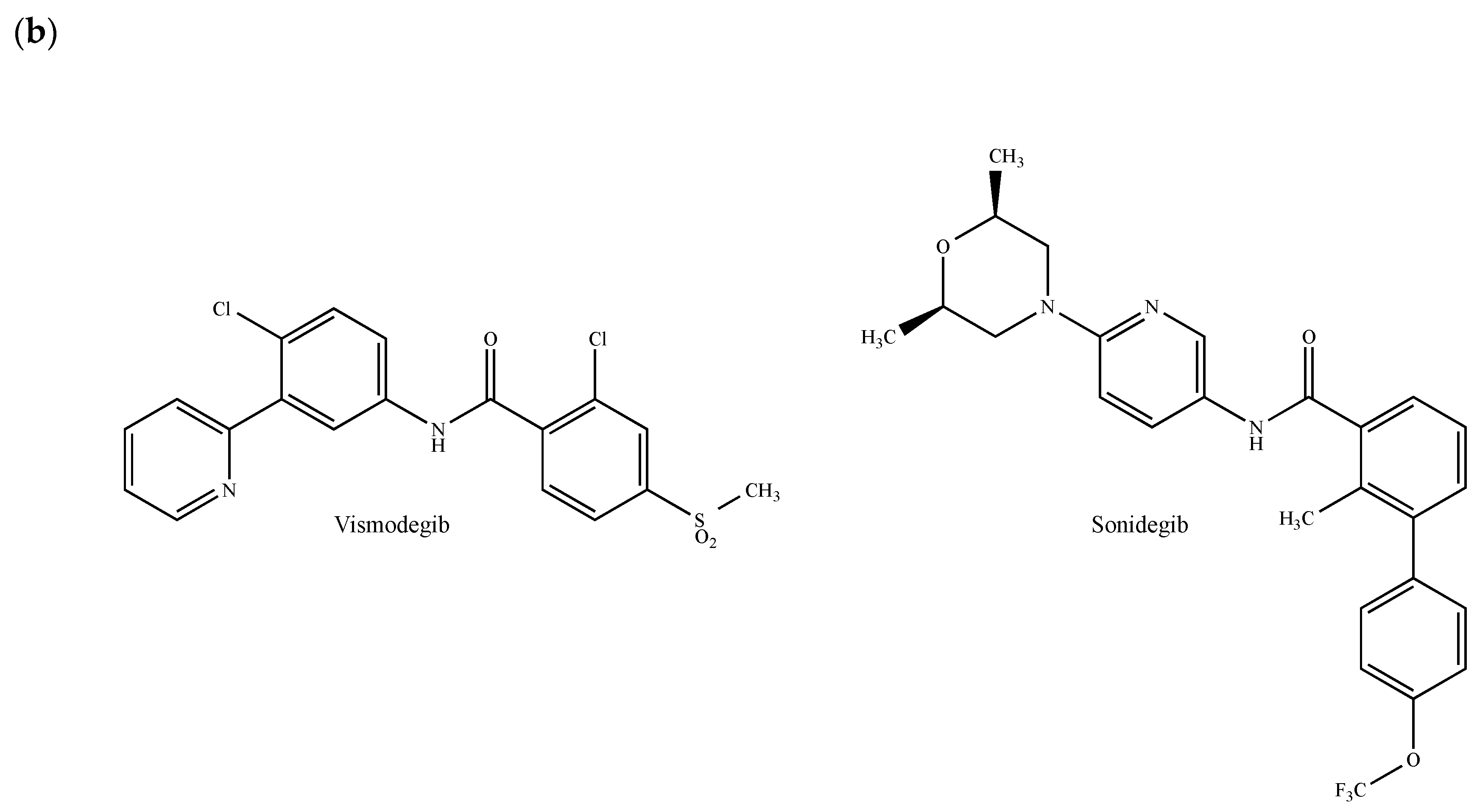
2.11. The Hedgehog and Wnt Signaling Pathways
3. Conclusions
Funding
Conflicts of Interest
References
- Wang, D.; Zhao, W.L.; Cai, M.J.; Wang, J.X.; Zaho, X.F. G-protein-coupled receptor controls hormone signaling in cell membrane. Sci. Rep. 2015, 5, 8675. [Google Scholar] [CrossRef]
- Wang, Z. Transactivation of epidermal growth factor receptor by G protein-coupled receptors: Recent progress, challenges and future research. Int. J. Mol. Sci. 2016, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Audet, M.; Bouvier, M. Restructuring G-protein-coupled receptor activation. Cell 2012, 151, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Flanagan, C.; Ballesteros, J.A.; Konvicka, K.; Davidson, J.S.; Weinstein, H.; Millar, R.P.; Sealfon, S.C. A reciprocal mutation supports helix 2 and helix 7 proximity in gonadotrophin-releasing hormone receptor. Mol. Pharmacol. 1994, 45, 165–170. [Google Scholar] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Neves, S.R.; Ram, P.T.; Lyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.; Viganó, M.; Newman, C.M.H.; Milligan, G.; Magee, A.I. A novel N-terminal motif for palmitoylation of G-protein α subunits. Biochem. J. 1993, 291, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Strange, P.G. Signaling mechanisms of GPCR ligands. Curr. Opin. Drug Discov. Dev. 2008, 11, 196–202. [Google Scholar]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Vilardaga, J.P.; Bünemann, M.; Feinstein, T.N.; Lambert, N.; Nikolaev, V.O.; Engelhardt, S.; Lohse, M.J.; Hoffmann, C. GPCR and G proteins: Drug efficacy and activation in live cells. Mol. Endocrinol. 2009, 23, 590–599. [Google Scholar] [CrossRef]
- Cvicek, V.; Godard, W.A., III; Abrol, R. Structure-based sequence alignment of the transmembrane domains of all human GPCRs: Phylogenic, structural and functional implications. PLoS Comput. Biol. 2016, 12, e1004805. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.S.; Tian, X.; Benovic, J.L. Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 2014, 27, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Leslie, M. Why endosomes recycle GPCRs. J. Cell Biol. 2016, 214, 785. [Google Scholar] [CrossRef]
- Bowman, S.L.; Shiwarski, D.J.; Puthenveedu, M.A. Distinct G protein-coupled receptor recycling pathways allow spatial control of downstream G protein signaling. J. Cell Biol. 2016, 214, 797–806. [Google Scholar] [CrossRef]
- Allaby, R.G.; Woodwark, M. Phylogenomic analysis reveals extensive phylogenetic mosaicism in the human GPCR superfamily. Evol. Bioinform. Online 2007, 3, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogen, D.J.; Meijer, H.J.G.; Seidl, M.F.; Govers, F. The ancient link between G-protein-coupled receptors and C-terminal phospholipid kinase domains. mBio 2018, 9, e02119-17. [Google Scholar] [CrossRef]
- Hu, G.M.; Mai, T.L.; Chen, C.M. Visualizing the GPCR network: Classification and evolution. Sci. Rep. 2017, 7, 15495. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Clarke, R.B.; Lisanti, M.P.; Maggiolini, M. G protein-coupled receptors at the crossroad between physiologic and pathologic angiogenesis: Old paradigms and emerging concepts. Int. J. Mol. Sci. 2017, 18, 2713. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. Pharmacotherapeutic targeting of G protein-coupled receptors in oncology: Examples of approved therapies and emerging concepts. Drugs 2017, 77, 951–965. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast cancer: Epidemiology and etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; de la Garza Salazar, J.; Pritchard, K.; Amadori, D.; Haidinger, R.; Hudis, C.A.; Khaled, H.; Liu, M.C.; Martin, M.; Namer, M.; et al. The global breast cancer burden: Variations in epidemiology and survival. Clin. Breast Cancer 2005, 6, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Warner, M.; Gustafsson, J.Å. Estrogen receptors in breast carcinogenesis and endocrine therapy. Mol. Cell. Endocrinol. 2015, 418, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, M.H.; Nevo, I.; Levy, R.; Vogel, Z. Role of the highly conserved Asp-Arg-Tyr motif in signal transduction of CB2 cannabinoid receptor. FEBS Lett. 2000, 466, 300–304. [Google Scholar] [CrossRef]
- Rovati, G.E.; Capra, V.; Neubig, R.R. The highly conserved DRY motif of class A G protein-coupled receptors: Beyond the ground state. Mol. Pharmacol. 2007, 71, 959–964. [Google Scholar] [CrossRef]
- Scheerer, P.; Park, J.H.; Hildebrand, P.W.; Kim, Y.J.; Krauss, N.; Choe, H.W.; Hofmann, K.P.; Ernst, O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Andò, S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17β-stradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Bonofiglio, D.; Recchia, A.G.; Musti, A.M.; Picard, D.; Andò, S.; Maggiolini, M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol. Endocrinol. 2006, 20, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Bonofiglio, D.; Albanito, L.; Madeo, A.; Rago, V.; Carpino, A.; Musti, A.M.; Picard, D.; Andò, S.; Maggiolini, M. 17β-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the G protein-coupled-receptor GPR30. Mol. Pharmacol. 2006, 70, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef]
- Leblanc, K.; Sexton, E.; Parent, S.; Bélanger, G.; Déry, M.C.; Boucher, V.; Asselin, E. Effects of 4-hydroxytamoxifen, raloxifen and ICI 182 780 on survival of uterine cancer cell lines in the presence and absence of exogenous estrogens. Int. J. Oncol. 2007, 30, 477–487. [Google Scholar]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; De Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-related fibroblasts. Environ. Health Perspect. 2012, 120, 1177–1182. [Google Scholar] [CrossRef]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER function in breast cancer: An overview. Front. Endocrinol. (Lausanne) 2014, 5, 66. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappano, R.; Rosano, C.; Santolla, M.F.; Pupo, M.; De Francesco, E.M.; De Marco, P.; Ponassi, M.; Spallarossa, A.; Ranise, A.; Maggiolini, M. Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr. Cancer Drug Targets 2012, 12, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Santolla, M.F.; Pupo, M.; Sinicropi, M.S.; Caruso, A.; Rosano, C.; Maggiolini, M. MIBE acts as antagonist ligand of both estrogen receptor α and GPER in breast cancer cells. Breast Cancer Res. 2012, 14, R12. [Google Scholar] [CrossRef] [PubMed]
- Sinicropi, M.S.; Lappano, R.; Caruso, A.; Santolla, M.F.; Pisano, A.; Rosano, C.; Capasso, A.; Panno, A.; Lancelot, J.C.; Rault, S.; et al. (6-bromo-1,4-dimethyl-9H-carbazol-3-yl-methylene)-hydrazine (carbhydraz) acts as a GPER agonist in breast cancer cells. Curr. Top. Med. Chem. 2015, 15, 1035–1042. [Google Scholar] [CrossRef]
- Lappano, R.; Rosano, C.; Pisano, A.; Santolla, M.F.; De Francesco, E.M.; De Marco, P.; Dolce, V.; Ponassi, M.; Felli, L.; Cafeo, G.; et al. A calixpyrrole derivative acts as an antagonist to GPER, a G-protein coupled receptor: Mechanisms and models. Dis. Model. Mech. 2015, 8, 1237–1246. [Google Scholar] [CrossRef]
- Maggiolini, M.; Santolla, M.F.; Avino, S.; Aiello, F.; Rosano, C.; Garofalo, A.; Grande, F. Identification of two benzopyrroloxazines acting as selective GPER antagonists in breast cancer cells and cancer-associated fibroblasts. Future Med. Chem. 2015, 7, 437–448. [Google Scholar] [CrossRef]
- Kampa, M.; Pelekanou, V.; Gallo, D.; Notas, G.; Troullinaki, M.; Pediaditakis, I.; Charalampopoulos, I.; Jacquot, Y.; Leclercq, G.; Castanas, E. ERα17p, an ERα P295-T311 fragment, modifies the migration of breast cancer cells, through actin cytoskeleton rearrangements. J. Cell. Biochem. 2011, 112, 3786–3796. [Google Scholar] [CrossRef]
- Leiber, D.; Burlina, F.; Byrne, C.; Robin, P.; Piesse, C.; Gonzalez, L.; Leclercq, G.; Tanfin, Z.; Jacquot, Y. The sequence Pro295-Thr311 of the hinge region of oestrogen receptor α is involved in ERK1/2 activation via GPR30 in leiomyoma cells. Biochem. J. 2015, 472, 97–109. [Google Scholar] [CrossRef]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marjon, N.A.; Hu, C.; Hathaway, H.J.; Prossnitz, E.R. G protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol. Cancer Res. 2014, 12, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Scaling, A.L.; Prossnitz, E.R.; Hathaway, H.J. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm. Cancer 2014, 5, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J. A role for G-protein coupled estrogen receptor (GPER) in estrogen-induced carcinogenesis: Dysregulated glandular homeostasis, survival and metastasis. J. Steroid Biochem. Mol. Biol. 2018, 176, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, T.; Eggemann, H.; Semczuk, A.; Smith, B.; Bischoff, J.; Roessner, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. Role of GPR30 in endometrial pathology after tamoxifen for breast cancer. Am. J. Obstet. Gynecol. 2010, 203, 595.e9–595.e16. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, A.; Ignatov, T.; Weissenborn, C.; Eggemann, H.; Bischoff, J.; Semczuk, A.; Roessner, A.; Costa, S.D.; Kalinski, T. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res. Treat. 2011, 128, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef] [PubMed]
- Rosano, C.; Lappano, R.; Santolla, M.F.; Ponassi, M.; Donadini, A.; Maggiolini, M. Recent advances in the rationale design of GPER ligands. Curr. Med. Chem. 2012, 19, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Luna, D.; Martínez-Archundia, M.; Maroun, R.C.; Ceballos-Reyes, G.; Fragoso-Vázquez, M.J.; González-Juárez, D.E.; Correa-Basurto, J. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J. Biomol. Struct. Dyn. 2015, 33, 2161–2172. [Google Scholar] [CrossRef]
- Rosano, C.; Ponassi, M.; Santolla, M.F.; Pisano, A.; Felli, L.; Vivacqua, A.; Maggiolini, M.; Lappano, R. Macromolecular modelling and docking simulations for the discovery of selective GPER ligands. AAPS J. 2016, 18, 41–46. [Google Scholar] [CrossRef]
- Méndez-Luna, D.; Bello, M.; Correa-Basurto, J. Understanding the molecular basis of agonist/antagonist mechanism of GPER1/GPR30 through structural and energetic analyses. J. Steroid Biochem. Mol. Biol. 2016, 158, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Rosano, C.; de Marco, P.; de Francesco, E.M.; Pezzi, V.; Maggiolini, M. Estriol acts as a GPR30 antagonist in estrogen receptor-negative breast cancer cells. Mol. Cell. Endocrinol. 2010, 320, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Cho, E.Y.; Min, C.; Park, J.H.; Kim, K.M. Characterization of functional roles of DRY motif in the 2nd intracellular loop of dopamine D2 and D3 receptors. Arch. Pharm. Res. 2008, 31, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Y.; Qu, J.; Yan, Y.; Yang, Y.; Cai, H. Roles of LPA receptor signaling in breast cancer. Expert Rev. Mol. Diagn. 2016, 16, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.A.; Lorincz, Z.; Bautista, D.L.; Liliom, K.; Tigyi, G.; Parrill, A.L. A single amino acid determines lysophospholipid specificity of the S1P1 (EDG1) and LPA1 (EDG2) phospholipid growth factor receptors. J. Biol. Chem. 2001, 276, 49213–49220. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Ghassemian, A.; Helleday, T. Lysophosphatidic acid receptor (LPAR) modulators: The current pharmacological toolbox. Prog. Lipid Res. 2015, 58, 51–75. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Sato, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T.; et al. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 2003, 64, 994–1005. [Google Scholar] [CrossRef]
- David, M.; Ribeiro, J.; Descotes, F.; Serre, C.M.; Barbier, M.; Murone, M.; Clézardin, P.; Peyruchaud, O. Targeting lysophosphatidic acid receptor type 1 with Debio 0719 inhibits spontaneous metastasis dissemination of breast cancer cells independently of cell proliferation and angiogenesis. Int. J. Oncol. 2012, 40, 1133–1141. [Google Scholar] [CrossRef]
- Sato, T.; Sugimoto, K.; Inoue, A.; Okudaira, S.; Aoki, J.; Tokuyama, H. Synthesis and biological evaluation of optically active Ki16425. Bioorg. Med. Chem. Lett. 2012, 22, 4323–4326. [Google Scholar] [CrossRef]
- Marshall, J.C.; Collins, J.W.; Nakayama, J.; Horak, C.E.; Liewehr, D.J.; Steinberg, S.M.; Albaugh, M.; Vidal-Vanaclocha, F.; Palmieri, D.; Barbier, M.; et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J. Natl. Cancer Inst. 2012, 104, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Brittain, J.E.; Seiders, T.J.; King, C.D. Preparation of Carbonylamino Isoxazolyl Biphenylacetic Acid Derivatives for Use as Lysophosphatidic Acid Receptor Antagonist; Amira Pharmaceuticals Inc.: San Diego, CA, USA, 2011; WO2011159550A2. [Google Scholar]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Ohotski, J.; Edwards, J.; Elsberger, B.; Watson, C.; Orange, C.; Mallon, E.; Pyne, S.; Pyne, N.J. Identification of novel functional and spatial associations between sphingosine kinase 1, sphingosine 1-phosphate receptors and other signaling proteins that affect prognostic outcome in estrogen receptor-positive breast cancer. Int. J. Cancer 2013, 132, 605–616. [Google Scholar] [CrossRef]
- Parrill, A.L.; Wang, D.A.; Bautista, D.L.; Van Brocklyn, J.R.; Lorinez, Z.; Fischer, D.J.; Baker, D.L.; Liliom, K.; Spiegel, S.; Tigyi, G. Identification of Edg1 receptor residues that recognize sphingosine 1-phosphate. J. Biol. Chem. 2000, 275, 39379–39384. [Google Scholar] [CrossRef]
- Durgam, G.G.; Virag, T.; Walker, M.D.; Tsukahara, R.; Yasuda, S.; Liliom, K.; van Meeteren, L.A.; Moolenaar, W.H.; Wilke, N.; Siess, W.; et al. Synthesis, structure-activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARγ, and inhibitors of autotaxin. J. Med. Chem. 2005, 48, 4919–4930. [Google Scholar] [CrossRef]
- Cooper, A.; Singh, S.; Hook, S.; Tyndall, J.D.A.; Vernall, A.J. Chemical tools for studying lipid-binding class A G protein-coupled receptors. Pharmacol. Rev. 2017, 69, 316–353. [Google Scholar] [CrossRef]
- Sanchez, T.; Estrada-Hernandez, T.; Paik, J.H.; Wu, M.T.; Venkataraman, K.; Brinkmann, V.; Claffey, K.; Hla, T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J. Biol. Chem. 2003, 278, 47281–47290. [Google Scholar] [CrossRef]
- Nagaoka, Y.; Otsuki, K.; Fujita, T.; Uesato, S. Effects of phosphorylation of immunomodulatory agent FTY720 (fingolimod) on antiproliferative activity against breast and colon cancer cells. Biol. Pharm. Bull. 2008, 31, 1177–1181. [Google Scholar] [CrossRef]
- Mousseau, Y.; Mollard, S.; Faucher-Durand, K.; Richard, L.; Nizou, A.; Cook-Moreau, J.; Baaj, Y.; Qiu, H.; Plainard, X.; Fourcade, L.; et al. Fingolimod potentiales the effects of sunitinib malate in a rat breast cancer model. Breast Cancer Res. Treat. 2012, 134, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Yamada, A.; Miyazaki, H.; Allegood, J.C.; Tsuchida, J.; Aoyagi, T.; Huang, W.C.; Terracina, K.P.; Adams, B.J.; Rashid, O.M.; et al. Interstitial fluid sphingosine-1-phosphate in murine mammary gland and cancer and human breast tissue and cancer determined by novel methods. J. Mammary Gland Biol. Neoplasia 2016, 21, 9–17. [Google Scholar] [CrossRef]
- Martin, J.L.; Julovi, S.M.; Lin, M.Z.; de Silva, H.C.; Boyle, F.M.; Baxter, R.C. Inhibition of basal-like breast cancer growth by FTY720 in combination with epidermal growth factor receptor kinase blockade. Breast Cancer Res. 2017, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The emerging role of FTY720 (fingolimod) in cancer treatment. Oncotarget 2016, 7, 23106–23127. [Google Scholar] [CrossRef] [PubMed]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal structure of antagonist bound human lysophosphatidic receptor 1. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.; Holt, D.; Fulton, A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011, 30, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Kawamori, T.; Uchiya, N.; Nakatsugi, S.; Watanabe, K.; Ohuchida, S.; Yamamoto, H.; Maruyama, T.; Kondo, K.; Sugimura, T.; Wakabayashi, K. Chemopreventive effects of ONO-8711, a selective prostaglandin E receptor EP1 antagonist, on breast cancer development. Carcinogenesis 2001, 22, 2001–2004. [Google Scholar] [CrossRef]
- Ma, X.; Kundu, N.; Rifat, S.; Walser, T.; Fulton, A.M. Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res. 2006, 66, 2923–2927. [Google Scholar] [CrossRef]
- Timoshenko, A.V.; Chakraborty, C.; Wagner, G.F.; Lala, P.K. Cox-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br. J. Cancer 2006, 94, 1154–1163. [Google Scholar] [CrossRef]
- Yang, L.; Huang, Y.; Porta, R.; Yanagisawa, K.; Gonzalez, A.; Segi, E.; Johnson, D.H.; Narumiya, S.; Carbone, D.P. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006, 66, 9665–9672. [Google Scholar] [CrossRef]
- Robertson, F.M.; Simeone, A.M.; Mazumdar, A.; Shah, A.H.; McMurray, J.S.; Ghosh, S.; Cristofanilli, M. Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. J. Exp. Ther. Oncol. 2008, 7, 299–312. [Google Scholar] [PubMed]
- Xin, X.; Majumder, M.; Girish, G.V.; Mohindra, V.; Maruyama, T.; Lala, P.K. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab. Investig. 2012, 92, 1115–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Holt, D.; Kundu, N.; Reader, J.; Goloubeva, O.; Take, Y.; Fulton, A.M. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncolimmunology 2013, 2, e22647. [Google Scholar] [CrossRef]
- Kundu, N.; Ma, X.; Kochel, T.; Goloubeva, O.; Staats, P.; Thompson, K.; Martin, S.; Reader, J.; Take, Y.; Collin, P.; et al. Prostaglandin E receptor EP4 is a therapeutic target in breast cancer cells with stem-like properties. Breast Cancer Res. Treat. 2014, 143, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.M.; Hess, D.; Lala, P.K. COX-2 induces breast cancer stem cells via EP4/PI3K/AKT/NOTCH/WNT axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef]
- Okumura, Y.; Yamagishi, T.; Nukui, S.; Nakao, K. Discovery of AAT-008, a novel, potent, and selective prostaglandin EP4 receptor antagonist. Bioorg. Med. Chem. Lett. 2017, 27, 1186–1192. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)—Biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.J.; Salah, Z.; Grisaru-Granovsky, S.; Cohen, I.; Even-Ram, S.C.; Maoz, M.; Uziely, B.; Peretz, T.; Bar-Shavit, R. Human protease-activated receptor 1 expression in malignant epithelia: A role in invasiveness. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Li, Y.; Luo, Y.; Sheng, Y.; Su, Y.; Padia, R.N.; Pan, Z.K.; Dong, Z.; Huang, S. Proteinase-activated receptor 2 expression in breast cancer and its role in breast cancer cell migration. Oncogene 2009, 28, 3047–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, D.A.; Braunwald, E.; Bonaca, M.P.; Ameriso, S.F.; Dalby, A.J.; Fish, M.P.; Fox, K.A.; Lipka, L.J.; Liu, X.; Nicolau, J.C.; et al. TRA 2P-TIMI 50 Steering Commitee and Investigators. Varopaxar in the secondary prevention of atherothrombotic events. N. Engl. J. Med. 2012, 366, 1404–1413. [Google Scholar] [CrossRef]
- Chanakira, A.; Westmark, P.R.; Ong, I.M.; Sheehan, J.P. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol. Oncol. 2017, 145, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auvergne, R.; Wu, C.; Connell, A.; Au, S.; Cornwell, A.; Osipovitch, M.; Benraiss, A.; Dangelmajer, S.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; et al. PAR1 inhibition supresses the self-renewal and growth of A2B5-defined glioma progenitor cells and their derived gliomas in vivo. Oncogene 2016, 35, 3817–3828. [Google Scholar] [CrossRef] [PubMed]
- Covic, L.; Kuliopulos, A. Protease-activated receptor 1 as therapeutic target in breast, lung, and ovarian cancer: Pepducin approach. Int. J. Mol. Sci. 2018, 19, 2237. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Boire, A.; Agarwal, A.; Nguyen, N.; O’Callaghan, K.; Tu, P.; Kuliopulos, A.; Covic, L. Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res. 2009, 15, 6223–6231. [Google Scholar] [CrossRef]
- Cisowski, J.; O’Callaghan, K.; Kuliopulos, A.; Yang, J.; Nguyen, N.; Deng, Q.; Yang, E.; Fogel, M.; Tressel, S.; Foley, C.; et al. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am. J. Pathol. 2011, 179, 513–523. [Google Scholar] [CrossRef]
- Ray, T.; Kar, D.; Pal, A.; Mukherjee, S.; Das, C.; Pal, A. Molecular targeting of breast and colon cancer cells by PAR1 mediated apoptosis through a novel pro-apoptotic peptide. Apoptosis 2018, 23, 679–694. [Google Scholar] [CrossRef]
- Liu, Y.; Mueller, B.M. Protease-activated receptor-2 regulates vascular endothelial growth factor expression in MDA-MB-231 cells via MAPK pathways. Biochem. Biophys. Res. Commun. 2006, 344, 1263–1270. [Google Scholar] [CrossRef]
- Lidfeldt, J.; Bendahl, P.O.; Forsare, C.; Malmström, P.; Fernö, M.; Belting, M. Protease activated receptors 1 and 2 correlate differently with breast cancer aggressiveness depending on tumor ER status. PLoS ONE 2015, 10, e0134932. [Google Scholar] [CrossRef]
- Carneiro-Lobo, T.C.; Schaffner, F.; Disse, J.; Ostergaard, H.; Francischetti, I.M.; Monteiro, R.Q.; Ruf, W. The tick-derived inhibitor Ixolaris prevents tissue factor signaling on tumor cells. J. Thromb. Haemost. 2012, 10, 1849–1858. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Yau, M.K.; Lim, J.; Wu, K.C.; Xu, W.; Suen, J.Y.; Fairlie, D.P. A potent antagonist of protease-activated receptor 2 that inhibits multiple signaling functions in human cancer cells. J. Pharmacol. Exp. Ther. 2018, 364, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, É.; Hugo de Almeida, V.; Rondon, A.M.R.; Possik, P.A.; Viola, J.P.B.; Monteiro, R.Q. Protease-activated receptor 2 (PAR2) upregulates granulocyte colony stimulating factor (G-CSF) expression in breast cancer cells. Biochem. Biophys. Res. Commun. 2018, 504, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Hollmén, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage—Cancer cell crosstalk in estrogen receptor positive and triple negative breast cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.; Stahn, S.; Neudörfl, J.M.; Sperlich, J.; Schmalz, H.G.; Teusch, N. Synthetic indolactam V analogues as inhibitors of PAR2-induced calcium mobilization in triple-negative breast cancer cells. ChemMedChem 2018, 13, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.; Maoz, M.; Kancharla, A.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cell. Mol. Life Sci. 2014, 71, 2517–2533. [Google Scholar] [CrossRef] [PubMed]
- Kancharla, A.; Maoz, M.; Jaber, M.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. PH motifs in PAR1&2 endow breast cancer growth. Nat. Commun. 2015, 6, 8853. [Google Scholar] [Green Version]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Ahmed, S.; Iwasaki, Y.; Ohba, K.; Toi, M.; Yamamoto, N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed. Pharmacother. 2006, 60, 273–276. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: A systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Dev. Ther. 2015, 9, 4953–4964. [Google Scholar]
- Tamamura, H.; Omagari, A.; Hiramatsu, K.; Gotoh, K.; Kanamoto, T.; Xu, Y.; Kodama, E.; Matsuoka, M.; Hattori, T.; Yamamoto, N.; et al. Development of specific CXCR4 inhibitors possessing high selectivity indexes as well as complete stability in serum based on an anti-HIV peptide T140. Bioorg. Med. Chem. Lett. 2001, 11, 1897–1902. [Google Scholar] [CrossRef]
- Zhang, W.B.; Navenot, J.M.; Haribabu, B.; Tamamura, H.; Hiramatu, K.; Omagari, A.; Pei, G.; Manfredi, J.P.; Fujii, N.; Broach, J.R.; et al. A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40-4C are weak partial agonists. J. Biol. Chem. 2002, 277, 24515–24521. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Brooks, J.; Willard, M.; Liang, K.; Yoon, Y.; Kang, S.; Shim, H. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem. Biophys. Res. Commun. 2007, 359, 716–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamamura, H.; Hori, A.; Kanzaki, N.; Hiramatsu, K.; Mizumoto, M.; Nakashima, H.; Yamamoto, N.; Otaka, A.; Fujii, N. T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer. FEBS Lett. 2003, 550, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Wu, T.; Lou, H.; Yu, X.; Taichman, R.S.; Lau, S.K.; Nie, S.; Umbreit, J.; Shim, H. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004, 64, 4302–4308. [Google Scholar] [CrossRef]
- Lefort, S.; Thuleau, A.; Kieffer, Y.; Sirven, P.; Bieche, I.; Marangoni, E.; Vincent-Salomon, A.; Mechta-Grigoriou, F. CXCR4 inhibitors could benefit to HER2 but not to triple-negative breast cancer patients. Oncogene 2017, 36, 1211–1222. [Google Scholar] [CrossRef]
- Bumpers, H.; Huang, M.B.; Katkoori, V.; Manne, U.; Bond, V. Nef-M1, a CXCR4 peptide antagonist, enhances apoptosis and inhibits primary tumor growth and metastasis in breast cancer. J. Cancer Ther. 2013, 4, 898–906. [Google Scholar] [CrossRef]
- Huang, M.B.; Giesler, K.E.; Katzman, B.M.; Prosser, A.R.; Truax, V.; Liotta, D.C.; Wilson, L.J.; Bond, V.C. Small molecule CXCR4 antagonists block the HIV-1 Nef/CXCR4 axis and selectively initiate the apoptotic program in breast cancer cells. Oncotarget 2018, 9, 16996–17013. [Google Scholar] [CrossRef]
- Katkoori, V.R.; Basson, M.D.; Bond, V.C.; Manne, U.; Bumpers, H.L. Nef-M1, a peptide antagonist of CXCR4, inhibits tumor angiogenesis and epithelial-to-mesenchymal transition in colon and breast cancers. Oncotarget 2015, 6, 27763–27777. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, C.; Yang, Z.; Gao, Y.; Tang, J. Suppression of breast cancer proliferation and induction of apoptosis via AKT and ERK1/2 signal transduction pathways by synthetic polypeptide derived from viral macrophage inflammatory protein II. J. Huazhong Univ. Sci. Technol. 2011, 31, 497–503. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, F.; Ding, Y.; Huang, J.; Chen, S.; Wu, Q.; Wang, Z.; Wang, Z.; Chen, C. Antitumour activity of the recombination polypeptide GST-NT21MP is mediated by inhibition of CXCR4 pathway in breast cancer. Br. J. Cancer 2014, 110, 1288–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.H.; Singh, B.; Cristofanilli, M.; Gelovani, J.; Wei, C.; Vincent, L.; Cook, K.R.; Lucci, A. A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of breast cancer. J. Surg. Res. 2009, 155, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Richert, M.M.; Vaidya, K.S.; Mills, C.N.; Wong, D.; Korz, W.; Hurst, D.R.; Welch, D.R. Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol. Rep. 2009, 21, 761–767. [Google Scholar] [PubMed]
- Hassan, S.; Buchanan, M.; Jahan, K.; Aguilar-Mahecha, A.; Gaboury, L.; Muller, W.J.; Alsawafi, Y.; Mourskaia, A.A.; Siegel, P.M.; Salvucci, O.; et al. CXCR4 peptide antagonist inhibits primary breast tumor growth, metastasis and enhances the efficacy of anti-VEGF treatment or docetaxel in a transgenic mouse model. Int. J. Cancer 2011, 129, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Bachelder, R.E.; Wendt, M.A.; Mercurio, A.M. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002, 62, 7203–7206. [Google Scholar]
- Peng, S.B.; Zhang, X.; Paul, D.; Kays, L.M.; Gough, W.; Stewart, J.; Uhlik, M.T.; Chen, Q.; Hui, Y.H.; Zamek-Gliszczynski, M.J.; et al. Identification of LY2510924, a novel cyclic peptide CXCR4 antagonist that exhibits antitumor activities in solid tumor and breast cancer metastatic models. Mol. Cancer Ther. 2015, 14, 480–490. [Google Scholar] [CrossRef]
- Ling, X.; Spaeth, E.; Chen, Y.; Shi, Y.; Zhang, W.; Schober, W.; Hail, N., Jr.; Konopleva, M.; Andreeff, M. The CXCR4 antagonist AMD3465 regulates oncogenic signaling and invasiveness in vitro and prevents breast cancer growth and metastasis in vivo. PLoS ONE 2013, 8, e58426. [Google Scholar] [CrossRef]
- Pusic, I.; DiPersio, J.F. Update on clinical experience with AMD3100, an SDF-1/CXCL12-CXCR4 inhibitor, in mobilization of hematopoietic stem and progenitor cells. Curr. Opin. Hematol. 2010, 17, 319–326. [Google Scholar] [CrossRef]
- Rosenkilde, M.M.; Gerlach, L.O.; Jakobsen, J.S.; Skerlj, R.T.; Bridgers, G.J.; Schwartz, T.W. Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: Transfer of binding site to the CXCR3 receptor. J. Biol. Chem. 2004, 279, 3033–3041. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, Q.; Bai, X.; Yu, Z.; Sun, M.; Zhao, H.; Mi, X.; Wang, E.; Yao, W.; Jin, F.; et al. Activation of STAT3 is involved in malignancy mediated by CXCL12-CXCR4 signaling in human breast cancer. Oncol. Rep. 2014, 32, 2760–2768. [Google Scholar] [CrossRef]
- Greco, S.J.; Patel, S.A.; Bryan, M.; Pliner, L.F.; Banerjee, D.; Rameshwar, P. AMD3100-mediated production of interleukin-1 from mesenchymal stem cells is key to chemosensitivity of breast cancer cells. Am. J. Cancer Res. 2011, 1, 701–715. [Google Scholar] [PubMed]
- Tverdal, A.; Stensvold, I.; Solvoll, K.; Foss, O.P.; Lund-Larsen, P.; Bjartveit, K. Coffee consumption and death from coronary heart disease in middle aged Norwegian men and women. BMJ 1990, 300, 566–569. [Google Scholar] [CrossRef]
- Cronin, P.A.; Wang, J.H.; Redmond, H.P. Hypoxia increases the metastatic ability of breast cancer cells via upregulation of CXCR4. BMC Cancer 2010, 10, 225. [Google Scholar] [CrossRef]
- Smith, M.C.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Cervoni-Curet, F.N.; Busch, J.I.; Sutton, E.; Webster, J.D.; Kavalukas, S.L.; Wakefield, L.M.; Barbul, A.; Niederhuber, J.E. SDF-1α mediates wound-promoted tumor growth in a syngeneic orthotopic mouse model of breast cancer. PLoS ONE 2013, 8, e60919. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.X.; Xie, L.H.; Peng, X.; Guo, Q.M.; Wu, Q.Y.; Wang, W.H.; Zhang, G.L.; Wu, J.F.; Zhang, G.J.; Du, C.W. CXCR4 antagonist AMD3100 enhances the response of MDA-MB-231 triple-negative breast cancer cells to ionizing radiation. Cancer Lett. 2018, 418, 196–203. [Google Scholar] [CrossRef]
- Xiang, J.; Hurchla, M.A.; Fontana, F.; Su, X.; Amend, S.R.; Esser, A.K.; Douglas, G.J.; Mudalagiriyappa, C.; Luker, K.E.; Pluard, T.; et al. CXCR4 protein epitope mimetic antagonist POL5551 disrupts metastasis and enhances chemotherapy effect in triple-negative breast cancer. Mol. Cancer Ther. 2015, 14, 2473–2485. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, M.K.; Kumar, D.; Jones, H.; Amaya-Chanaga, C.I.; Choi, M.Y.; Melo-Cardenas, J.; Ale-Ali, A.; Kuhne, M.R.; Sabbatini, P.; Cohen, L.J.; et al. Ulocuplumab (BMS-936564/MDX1338): A fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway. Oncotarget 2016, 7, 2809–2822. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velázquez, M.; Xolalpa, W.; Pestell, R.G. The potential to target CCL5/CCR5 in breast cancer. Expert Opin. Ther. Targets 2014, 18, 1265–1275. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef] [PubMed]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Devel. Ther. 2015, 9, 5447–5468. [Google Scholar] [PubMed]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor marivoc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, O.M. Vicriviroc, a CCR5 receptor antagonist for the potential treatment of HIV infection. Curr. Opin. Investig. Drugs 2009, 10, 845–859. [Google Scholar] [PubMed]
- Sax, M.J.; Gasch, C.; Athota, V.R.; Freeman, R.; Rasighaemi, P.; Westcott, D.E.; Day, C.J.; Nikolic, I.; Elsworth, B.; Wei, M.; et al. Cancer cell CCL5 mediates bone marrow independent angiogenesis in breast cancer. Oncotarget 2016, 7, 85437–85449. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.; Velasco-Velázquez, M.A.; Wang, M.; Li, Z.; Rui, H.; Peck, A.R.; Korkola, J.E.; Chen, X.; Xu, S.; DuHadaway, J.B.; et al. CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer Res. 2018, 78, 1657–1671. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Sun, L.; Jiao, Y.; Lee, L.T.O. The role of G protein-coupled receptor kinases in cancer. Int. J. Biol. Sci. 2018, 14, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Rajbhandari, P.; Schalper, K.A.; Solodin, N.M.; Ellison-Zelski, S.J.; Ping Lu, K.; Rim, D.L.; Alarid, E.T. Pin1 modulates ERα levels in breast cancer through inhibition of phosphorylation-dependent ubiquitination and degradation. Oncogene 2014, 33, 1438–1447. [Google Scholar] [CrossRef]
- Nogués, L.; Reglero, C.; Rivas, V.; Salcedo, A.; Lafarga, V.; Neves, M.; Ramos, P.; Mendiola, M.; Berjón, A.; Stamatakis, K.; et al. G protein-coupled receptor kinase 2 (GRK2) promotes breast tumorigenesis through a HDAC6-Pin1 axis. EBioMedicine 2016, 13, 132–145. [Google Scholar] [CrossRef]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef]
- Homan, K.T.; Larimore, K.M.; Elkins, J.M.; Szklarz, M.; Knapp, S.; Tesmer, J.J.G. Identification and structure-function analysis of subfamily selective G protein coupled receptor kinase inhibitors. ACS Chem. Biol. 2015, 10, 310–319. [Google Scholar] [CrossRef]
- Billard, M.J.; Fitzhugh, D.J.; Parker, J.S.; Brozowski, J.M.; McGinnis, M.W.; Timoshchenko, R.G.; Serafin, D.S.; Lininger, R.; Klauber-Demore, N.; Sahagian, G.; et al. G protein coupled receptor kinase 3 regulates breast cancer migration, invasion, and metastasis. PLoS ONE 2016, 11, e0152856. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The renin-angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Nouet, S.; Nahmias, C. Signal transduction from the angiotensin II AT2 receptor. Trends Endocrinol. Metab. 2000, 11, 1–6. [Google Scholar] [CrossRef]
- Deshayes, F.; Nahmias, C. Angiotensin receptors: A new role in cancer? Trends Endocrinol. Metab. 2005, 16, 293–299. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Ateeq, B.; Cao, Q.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Kalyana-Sundaram, S.; Lonigro, R.J.; Helgeson, B.E.; Bhojani, M.S.; et al. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc. Natl. Acad. Sci. USA 2009, 106, 10284–10289. [Google Scholar] [CrossRef] [Green Version]
- Ishikane, S.; Takahashi-Yanaga, F. The role of angiotensin II in cancer metastasis: Potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochem. Pharmacol. 2018, 151, 96–103. [Google Scholar] [CrossRef]
- Stergiou, G.S.; Skeva, I.I. Renin-angiotensin system blockade at the level of the angiotensin converting enzyme or the angiotensin type-1 receptor: Similarities and differences. Curr. Top. Med. Chem. 2004, 4, 473–481. [Google Scholar] [CrossRef]
- Bhuiyan, M.A.; Ishiguro, M.; Hossain, M.; Nakamura, T.; Ozaki, M.; Miura, S.; Nagatomo, T. Binding sites of valsartan, candesartan and losartan with angiotensin II receptor 1 subtype by molecular modeling. Life Sci. 2009, 85, 136–140. [Google Scholar] [CrossRef]
- Bhuiyan, M.A.; Shahriar, M.; Nagatomo, T. Binding Affinity of Candesartan, Losartan, Telmisartan and Valsartan with Angiotensin II Receptor 1 Subtype. Bangladesh Pharm. J. 2013, 16, 10–14. [Google Scholar] [CrossRef]
- Chen, X.; Meng, Q.; Zhao, Y.; Liu, M.; Li, D.; Yang, Y.; Sun, L.; Sui, G.; Cai, L.; Dong, X. Angiotensin II type 1 receptor antagonists inhibit cell proliferation and angiogenesis in breast cancer. Cancer Lett. 2013, 328, 318–324. [Google Scholar] [CrossRef]
- Muscella, A.; Greco, S.; Elia, M.G.; Storelli, C.; Marsigliante, S. Angiotensin II stimulation of Na+/K+ATPase activity and cell growth by calcium-independent pathway in MCF-7 breast cancer cells. J. Endocrinol. 2002, 173, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulson, R.; Liew, S.H.; Connelly, A.A.; Yee, N.S.; Deb, S.; Kumar, B.; Vargas, A.C.; O’Toole, S.A.; Parslow, A.C.; Poh, A.; et al. The angiotensin receptor blocker, Losartan, inhibits mammary tumor development and progression to invasive carcinoma. Oncotarget 2017, 8, 18640–18656. [Google Scholar] [CrossRef] [PubMed]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Liu, X.; Ran, W.; Meng, J.; Zhai, Y.; Zhang, P.; Yin, Q.; Yu, H.; Zhang, Z.; Li, Y. Regulating cancer associated fibroblasts with losartan-loaded injectable peptide hydrogel to potentiate chemotherapy in inhibiting growth and lung metastasis of triple negative breast cancer. Biomaterials 2017, 144, 60–72. [Google Scholar] [CrossRef]
- Xia, T.; He, Q.; Shi, K.; Wang, Y.; Yu, Q.; Zhang, L.; Zhang, Q.; Gao, H.; Ma, L.; Liu, J. Losartan loaded liposomes improve the antitumor efficacy of liposomal paclitaxel modified with pH sensitive peptides by inhibition of collagen in breast cancer. Pharm. Dev. Technol. 2018, 1, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Namazi, S.; Rostami-Yalmeh, J.; Sahebi, E.; Jaberipour, M.; Razmkhah, M.; Hosseini, A. The role of captopril and losartan in prevention and regression of tamoxifen-induced resistance of breast cancer cell line MCF-7: An in vitro study. Biomed. Pharmacother. 2014, 68, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Kociecka, B.; Surazynski, A.; Miltyk, W.; Palka, J. The effect of telmisartan on collagen biosynthesis depends on the status of estrogen activation in breast cancer cells. Eur. J. Pharmacol. 2010, 628, 51–56. [Google Scholar] [CrossRef]
- Du, N.; Feng, J.; Hu, L.J.; Sun, X.; Sun, H.B.; Zhao, Y.; Yang, Y.P.; Ren, H. Angiotensin II receptor type 1 blockers suppress the cell proliferation effects of angiotensin II in breast cancer cells by inhibiting AT1R signaling. Oncol. Rep. 2012, 27, 1893–1903. [Google Scholar] [Green Version]
- Millar, R.P.; Lu, Z.L.; Pawson, A.J.; Flanagan, C.A.; Morgan, K.; Maudsley, S.R. Gonadotropin-releasing hormone receptors. Endocr. Rev. 2004, 25, 235–275. [Google Scholar] [CrossRef]
- Schally, A.V. Luteinizing hormone-releasing hormone analogs: Their impact on the control of tumorigenesis. Peptides 1999, 20, 1247–1262. [Google Scholar] [CrossRef]
- Cheng, C.K.; Leung, P.C. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr Rev. 2005, 26, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, M.M.; Moretti, R.M.; Januszkiewicz-Caulier, J.; Motta, M.; Limonta, P. Gonadotropin-releasing hormone (GnRH) receptors in tumors: A new rationale for the therapeutical application of GnRH analogs in cancer patients? Curr. Cancer Drug Targets 2006, 6, 257–269. [Google Scholar]
- Tan, S.H.; Wolff, A.C. Luteinizing hormone-releasing hormone agonists in premenopausal hormone receptor-positive breast cancer. Clin. Breast Cancer 2007, 76, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Klijn, J.G.; de Jong, F.H.; Lamberts, S.W.; Blankenstein, M.A. LHRH-agonist treatment in clinical and experimental human breast cancer. J. Steroid Biochem. 1985, 23, 867–873. [Google Scholar] [CrossRef]
- Lissoni, P.; Barni, S.; Crispino, S.; Cattaneo, G.; Tancini, G. Endocrine and clinical effects of an LHRH analogue in pretreated advanced breast cancer. Tumori 1988, 74, 303–308. [Google Scholar] [CrossRef]
- Dowsett, M.; Mehta, A.; Mansi, J.; Smith, I.E. A dose-comparative endocrine-clinical study of leuprorelin in premenopausal breast cancer patients. Br. J. Cancer 1990, 62, 834–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.W.; Green, S.; Dalton, W.S.; Martino, S.; Rector, D.; Ingle, J.N.; Robert, N.J.; Budd, G.T.; Paradelo, J.C.; Natale, R.B.; et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: An intergroup study. J. Clin. Oncol. 1998, 16, 994–999. [Google Scholar] [CrossRef]
- Klijn, J.G.; Blamey, R.W.; Boccardo, F.; Tominaga, T.; Duchateau, L.; Sylvester, R. Combined Hormone Agents Trialists’ Group and the European Organization for Research and Treatment of Cancer. Combined tamoxifen and luteinizing hormone-releasing hormone (LHRH) agonist versus LHRH agonist alone in premenopausal advanced breast cancer: A meta-analysis of four randomized trials. J. Clin. Oncol. 2001, 19, 343–353. [Google Scholar]
- Flanagan, C.A.; Millar, R.P.; Illing, N. Advances in understanding gonadotrophin-releasing hormone receptor structure and ligand interaction. Rev. Reprod. 1997, 2, 113–120. [Google Scholar] [CrossRef]
- Pappa, E.V.; Zompra, A.A.; Spyranti, Z.; Diamantopoulou, Z.; Pairas, G.; Lamari, F.N.; Katsoris, P.; Spyroulias, G.A.; Cordopatis, P. Enzymatic stability, solution structure, and antiproliferative effect on prostate cancer cells of leuprolide and new gonadotrophin-releasing hormone peptide analogs. Pept. Sci. 2011, 96, 260–272. [Google Scholar] [CrossRef]
- Stein, R.C.; Dowsett, M.; Hedley, A.; Gazet, J.C.; Ford, H.T.; Coombes, R.C. The clinical and endocrine effects of 4-hydroxyandrostenedione alone and in combination with goserelin in premenopausal women with advanced breast cancer. Br. J. Cancer 1990, 62, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Celio, L.; Martinetti, A.; Ferrari, L.; Buzzoni, R.; Mariani, L.; Miceli, R.; Seregni, E.; Procopio, G.; Cassata, A.; Bombardieri, E.; et al. Premenopausal breast cancer patients treated with a gonadotropin-releasing hormone analog alone or in combination with an aromatase inhibitor: A comparative endocrine study. Anticancer Res. 1999, 19, 2261–2268. [Google Scholar]
- Rody, A.; Loibl, S.; von Minckwitz, G.; Kaufmann, M. Use of goserelin in the treatment of breast cancer. Expert Rev. Anticancer Ther. 2005, 5, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, H. Goserelin (‘Zoladex’)—Offering patients more choice in early breast cancer. Eur. J. Oncol. Nurs. 2004, 2, S95–S103. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.B.; Schally, A.V. Drug Insight: Clinical use of agonists and antagonists of luteinizing-hormone-releasing hormone. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Tan, O.; Bukulmez, O. Biochemistry, molecular biology and cell biology of gonadotropin-releasing hormone antagonists. Curr. Opin. Obstet. Gynecol. 2011, 23, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Patrick, E.; Whitson, M.; Smith, A.; Parnell, J.; Thomas, S.E.; Blankenship, C.C. GnRH receptor antagonists for prostate cancer. JAAPA 2013, 26, 51–52. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhu, Y.F.; Gross, T.D.; Tucci, F.C.; Gao, Y.; Moorjani, M.; Connors, P.J., Jr.; Rowbottom, M.W.; Chen, Y.; Struthers, S.; et al. Synthesis and structure-activity relationships of 1-arylmethyl-5-aryl-6-methyluracils as potent gonadotrophin-releasing hormone receptor antagonists. J. Med. Chem. 2004, 47, 1259–1271. [Google Scholar] [CrossRef]
- Bonger, K.M.; van den Berg, R.J.B.H.N.; Knijnenburg, A.D.; Heitman, L.H.; IJzerman, A.P.; Oosterom, J.; Timmers, C.M.; Overkleeft, H.S.; van der Marel, G.A. Synthesis and evaluation of homodimeric GnRHR antagonists having a rigid bis-propargylated benzene core. Bioorg. Med. Chem. 2008, 16, 3744–3758. [Google Scholar] [CrossRef]
- Tukun, F.L.; Olberg, D.E.; Riss, P.J.; Haraldsen, I.; Kaass, A.; Klaveness, J. Recent development of non-peptide GnRH antagonists. Molecules 2017, 22, 2188. [Google Scholar] [CrossRef]
- Rosenkulde, M.M.; Cahir, M.; Gether, U.; Hjorth, S.A.; Schwartz, T.W. Mutations along transmembrane segment II of the NK-1 receptor affect substance P competition with non-peptide antagonists but not substance P binding. J. Biol. Chem. 1994, 269, 28160–28164. [Google Scholar]
- Zhou, W.; Rodic, V.; Kitanovic, S.; Flanagan, C.A.; Chi, L.; Weinstein, H.; Maayani, S.; Millar, R.P.; Sealfon, S.C. A locus of the gonadotropin-releasing hormone receptor that differentiates agonist and antagonist binding sites. J. Biol. Chem. 1995, 270, 18853–18857. [Google Scholar] [CrossRef]
- Patel, Y.C. Somatostatin and its receptor family. Front. Neuroendocrinol. 1999, 20, 157–198. [Google Scholar] [CrossRef]
- Bousquet, C.; Puente, E.; Buscail, L.; Vaysse, N.; Susini, C. Antiproliferative effect of somatostatin and analogs. Chemotherapy 2001, 47, 30–39. [Google Scholar] [CrossRef]
- Watt, H.L.; Kharmate, G.; Kumar, U. Biology of somatostatin in breast cancer. Mol. Cell. Endocrinol. 2008, 286, 251–261. [Google Scholar] [CrossRef]
- Fani, M.; Nicolas, G.P.; Wild, D. Somatostatin receptor antagonists for imaging and therapy. J. Nucl. Med. 2017, 58, 61S–66S. [Google Scholar] [CrossRef]
- Ruscica, M.; Arvigo, M.; Steffani, L.; Ferone, D.; Magni, P. Somatostatin, somatostatin analogs and somatostatin receptor dynamics in the biology of cancer progression. Curr. Mol. Med. 2013, 13, 555–571. [Google Scholar] [CrossRef]
- Díez, M.; Teulé, A.; Salazar, R. Gastroenteropancreatic neuroendocrine tumors: Diagnosis and treatment. Ann. Gastroenterol. 2013, 26, 29–36. [Google Scholar]
- Costa, F.; Gumz, B. Octreotide—A Review of its use in treating neuroendocrine tumours. Eur. Endocrinol. 2014, 10, 70–74. [Google Scholar] [CrossRef]
- Gadelha, M.R.; Wildemberg, L.E.; Bronstein, M.D.; Gatto, F.; Ferone, D. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary 2017, 20, 100–108. [Google Scholar] [CrossRef]
- Weckbecker, G.; Liu, R.; Tolcsvai, L.; Bruns, C. Antiproliferative effects of the somatostatin analogue octreotide (SMS 201-995) on ZR-75-1 human breast cancer cells in vivo and in vitro. Cancer Res. 1992, 52, 4973–4978. [Google Scholar]
- Huang, C.M.; Wu, Y.T.; Chen, S.T. Targeting delivery of paclitaxel into tumor cells via somatostatin receptor endocytosis. Chem. Biol. 2000, 7, 453–461. [Google Scholar] [CrossRef]
- Hruby, V.J. Designing peptide receptor agonists and antagonists. Nat. Rev. Drug Discov. 2002, 1, 847–858. [Google Scholar] [CrossRef]
- Prévost, G.; Foehrlé, E.; Thomas, F.; Pihan, I.; Veber, N.; Starzec, A.; Israël, L. Growth of human breast cancer cell lines is inhibited by the somatostatin analog BIM23014. Endocrinology 1991, 129, 323–329. [Google Scholar] [CrossRef]
- Kahán, Z.; Nagy, A.; Schally, A.V.; Hebert, F.; Sun, B.; Groot, K.; Halmos, G. Inhibition of growth of MX-1, MCF-7-MIII and MDA-MB-231 human breast cancer xenografts after administration of a targeted cytotoxic analog of somatostatin, AN-238. Int. J. Cancer 1999, 82, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Seitz, S.; Schally, A.V.; Treszl, A.; Papadia, A.; Rick, F.; Szalontay, L.; Szepeshazi, K.; Ortmann, O.; Halmos, G.; Hohla, F.; et al. Preclinical evaluation of properties of a new targeted cytotoxic somatostatin analog, AN-162 (AEZS-124), and its effects on tumor growth inhibition. Anticancer Drugs 2009, 20, 553–558. [Google Scholar] [CrossRef]
- Feng, Q.; Yu, M.Z.; Wang, J.C.; Hou, W.J.; Gao, L.Y.; Ma, X.F.; Pei, X.W.; Niu, Y.J.; Liu, X.Y.; Qiu, C.; et al. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core-shell nanoparticles. Biomaterials 2014, 35, 5028–5038. [Google Scholar] [CrossRef]
- O’Byrne, K.J.; Dobbs, N.; Propper, D.J.; Braybrooke, J.P.; Koukourakis, M.I.; Mitchell, K.; Woodhull, J.; Talbot, D.C.; Schally, A.V.; Harris, A.L. Phase II study of RC-160 (vapreotide), an octapeptide analogue of somatostatin, in the treatment of metastatic breast cancer. Br. J. Cancer 1999, 79, 1413–1418. [Google Scholar] [CrossRef]
- Dolan, J.T.; Miltenburg, D.M.; Granchi, T.S.; Miller, C.C., 3rd; Brunicardi, F.C. Treatment of metastatic breast cancer with somatostatin analogues—A meta-analysis. Ann. Surg. Oncol. 2001, 8, 227–233. [Google Scholar]
- Van Essen, M.; Krenning, E.P.; De Jong, M.; Valkema, R.; Kwekkeboom, D.J. Peptide Receptor Radionuclide Therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol. 2007, 46, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Laverman, P.; Sosabowski, J.K.; Boerman, O.C.; Oyen, W.J. Radiolabelled peptides for oncological diagnosis. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, S78–S92. [Google Scholar] [CrossRef]
- Smith-Jones, P.M.; Bischof, C.; Leimer, M.; Gludovacz, D.; Angelberger, P.; Pangerl, T.; Peck-Radosavljevic, M.; Hamilton, G.; Kaserer, K.; Kofler, A.; et al. DOTA-lanreotide: A novel somatostatin analog for tumor diagnosis and therapy. Endocrinology 1999, 140, 5136–5148. [Google Scholar] [CrossRef]
- Ischia, J.; Patel, O.; Shulkes, A.; Baldwin, G.S. Gastrin-releasing peptide: Different forms, different functions. Biofactors 2009, 35, 69–75. [Google Scholar] [CrossRef]
- Van de Wiele, C.; Dumont, F.; van Belle, S.; Slegers, G.; Peers, S.H.; Dierckx, R.A. Is there a role for agonist gastrin-releasing peptide receptor radioligands in tumour imaging? Nucl. Med. Commun. 2001, 22, 5–15. [Google Scholar] [CrossRef]
- Smith, C.J.; Volkert, W.A.; Hoffman, T.J. Gastrin releasing peptide (GRP) receptor targeted radiopharmaceuticals: A concise update. Nucl. Med. Biol. 2003, 30, 861–868. [Google Scholar] [CrossRef]
- Parry, J.J.; Andrews, R.; Rogers, B.E. MicroPET imaging of breast cancer using radiolabeled bombesin analogs targeting the gastrin-releasing peptide receptor. Breast Cancer Res. Treat. 2007, 101, 175–183. [Google Scholar] [CrossRef]
- Dalm, S.U.; Martens, J.W.; Sieuwerts, A.M.; van Deurzen, C.H.; Koelewijn, S.J.; de Blois, E.; Maina, T.; Nock, B.A.; Brunel, L.; Fehrentz, J.A.; et al. In vitro and in vivo application of radiolabeled gastrin-releasing peptide receptor ligands in breast cancer. J. Nucl. Med. 2015, 56, 752–757. [Google Scholar] [CrossRef]
- Prignon, A.; Nataf, V.; Provost, C.; Cagnolini, A.; Montravers, F.; Gruaz-Guyon, A.; Lantry, L.E.; Talbot, J.N.; Nunn, A.D. (68)Ga-AMBA and (18)F-FDG for preclinical PET imaging of breast cancer: Effect of tamoxifen treatment on tracer uptake by tumor. Nucl. Med. Biol. 2015, 42, 92–98. [Google Scholar] [CrossRef]
- Maina, T.; Bergsma, H.; Kulkarni, H.R.; Mueller, D.; Charalambidis, D.; Krenning, E.P.; Nock, B.A.; de Jong, M.; Baum, R.P. Preclinical and first clinical experience with the gastrin-releasing peptide receptor-antagonist [⁶⁸Ga]SB3 and PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 964–973. [Google Scholar] [CrossRef]
- Morgat, C.; MacGrogan, G.; Brouste, V.; Vélasco, V.; Sévenet, N.; Bonnefoi, H.; Fernandez, P.; Debled, M.; Hindié, E. Expression of gastrin-releasing peptide receptor in breast cancer and its association with pathologic, biologic, and clinical parameters: A study of 1432 primary tumors. J. Nucl. Med. 2017, 58, 1401–1407. [Google Scholar] [CrossRef]
- Vangsted, A.J.; Andersen, E.V.; Nedergaard, L.; Zeuthen, J. Gastrin releasing peptide GRP(14-27) in human breast cancer cells and in small cell lung cancer. Breast Cancer Res. Treat. 1991, 19, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Shulkes, A.; Baldwin, G.S. Gastrin-releasing peptide and cancer. Biochim. Biophys. Acta 2006, 1766, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Shirahige, Y.; Cai, R.Z.; Szepeshazi, K.; Halmos, G.; Pinski, J.; Groot, K.; Schally, A.V. Inhibitory effect of bombesin/gastrin-releasing peptide (GRP) antagonists RC-3950-II and RC-3095 on MCF-7 MIII human breast cancer xenografts in nude mice. Biomed. Pharmacother. 1994, 48, 465–472. [Google Scholar] [CrossRef]
- Miyazaki, M.; Lamharzi, N.; Schally, A.V.; Halmos, G.; Szepeshazi, K.; Groot, K.; Cai, R.Z. Inhibition of growth of MDA-MB-231 human breast cancer xenografts in nude mice by bombesin/gastrin-releasing peptide (GRP) antagonists RC-3940-II and RC-3095. Eur. J. Cancer 1998, 34, 710–717. [Google Scholar] [CrossRef]
- Kahán, Z.; Sun, B.; Schally, A.V.; Arencibia, J.M.; Cai, R.Z.; Groot, K.; Halmos, G. Inhibition of growth of MDA-MB-468 estrogen-independent human breast carcinoma by bombesin/gastrin-releasing peptide antagonists RC-3095 and RC-3940-II. Cancer 2000, 88, 1384–1392. [Google Scholar] [CrossRef]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosanò, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2013, 13, 637–651. [Google Scholar] [CrossRef]
- Wülfing, P.; Diallo, R.; Kersting, C.; Wülfing, C.; Poremba, C.; Rody, A.; Greb, R.R.; Böcker, W.; Kiesel, L. Expression of endothelin-1, endothelin-A, and endothelin-B receptor in human breast cancer and correlation with long-term follow-up. Clin. Cancer Res. 2003, 9, 4125–4131. [Google Scholar]
- Wülfing, P.; Kersting, C.; Tio, J.; Fischer, R.J.; Wülfing, C.; Poremba, C.; Diallo, R.; Böcker, W.; Kiesel, L. Endothelin-1-, endothelin-A-, and endothelin-B-receptor expression is correlated with vascular endothelial growth factor expression and angiogenesis in breast cancer. Clin. Cancer Res. 2004, 10, 2393–2400. [Google Scholar] [CrossRef]
- Wülfing, P.; Tio, J.; Kersting, C.; Sonntag, B.; Buerger, H.; Wülfing, C.; Euler, U.; Boecker, W.; Tulusan, A.H.; Kiesel, L. Expression of endothelin-A-receptor predicts unfavourable response to neoadjuvant chemotherapy in locally advanced breast cancer. Br. J. Cancer 2004, 91, 434–440. [Google Scholar] [CrossRef]
- Bagnato, A.; Rosanò, L. The endothelin axis in cancer. Int. J. Biochem. Cell Biol. 2008, 40, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Smollich, M.; Götte, M.; Kersting, C.; Fischgräbe, J.; Kiesel, L.; Wülfing, P. Selective ETAR antagonist atrasentan inhibits hypoxia-induced breast cancer cell invasion. Breast Cancer Res. Treat. 2008, 108, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Fischgräbe, J.; Götte, M.; Michels, K.; Kiesel, L.; Wülfing, P. Targeting endothelin A receptor enhances anti-proliferative and anti-invasive effects of the HER2 antibody trastuzumab in HER2-overexpressing breast cancer cells. Int. J. Cancer 2010, 127, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smollich, M.; Götte, M.; Fischgräbe, J.; Macedo, L.F.; Brodie, A.; Chen, S.; Radke, I.; Kiesel, L.; Wülfing, P. ETAR antagonist ZD4054 exhibits additive effects with aromatase inhibitors and fulvestrant in breast cancer therapy, and improves in vivo efficacy of anastrozole. Breast Cancer Res. Treat. 2010, 123, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Jewell, A.N.; Swamydas, M.; Castillo, C.I.; Wyan, H.; Allen, L.D.; McDermott, K.A.; Eddy, J.M.; Dréau, D. The endothelin axis stimulates the expression of pro-inflammatory cytokines and pro-migratory molecules in breast cancer. Cancer Investig. 2010, 28, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Bolli, M.H.; Boss, C.; Binkert, C.; Buchmann, S.; Bur, D.; Hess, P.; Iglarz, M.; Meyer, S.; Rein, J.; Rey, M.; et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (Macitentan), an orally active, potent dual endothelin receptor antagonist. J. Med. Chem. 2012, 55, 7849–7861. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Hanibuchi, M.; Kim, S.J.; Yu, H.; Kim, M.S.; He, J.; Langley, R.R.; Lehembre, F.; Regenass, U.; Fidler, I.J. Treatment of experimental human breast cancer and lung cancer brain metastases in mice by macitentan, a dual antagonist of endothelin receptors, combined with paclitaxel. Neuro-Oncol. 2016, 18, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhang, Y.; Sun, B.; McMahon, A.P.; Wang, Y. Hedgehog signaling: From basic biology to cancer therapy. Cell Chem. Biol. 2017, 24, 252–280. [Google Scholar] [CrossRef]
- Harris, P.J.; Speranza, G.; Dansky Ullmann, C. Targeting embryonic signaling pathways in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 131–145. [Google Scholar] [CrossRef]
- Hui, M.; Cazet, A.; Nair, R.; Watkins, D.N.; O’Toole, S.A.; Swarbrick, A. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy. Breast Cancer Res. 2013, 15, 203. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mauro, C.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Formisano, L.; De Falco, S.; Cicatiello, V.; et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br. J. Cancer 2017, 116, 1425–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, M.; Ruiz-Borrego, M.; Trigo Perez, J.M.; Antolín, S.; García-Sánez, J.A.; Corral, J.; Jerez, Y.; Urruticoechea, A.; Colom, H.; Gonzalo, N.; et al. A phase I study of sonidegib (S) in combination with docetaxel (D) in patients (pts) with triple negative (TN) advanced breast cancer (ABC): GEICAM/2012-12 (EDALINE study). Ann. Oncol. 2016, 27, 281P. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Hess, D.; von Moos, R.; Homicsko, K.; Griguolo, G.; Joerger, M.; Mark, M.; Ackermann, C.J.; Allegrini, S.; Catapano, C.V.; et al. Swiss Group for Clinical Cancer Research (SAKK). Phase I trial of the oral smoothened inhibitor sonidegib in combination with paclitaxel in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 766–772. [Google Scholar] [CrossRef]
- Ruiz-Borrego, M.; Jimenez, B.; Antolín, S.; García-Saenz, J.A.; Corral, J.; Jerez, Y.; Trigo, J.; Urruticoechea, A.; Colom, H.; Gonzalo, N.; et al. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012-12 (EDALINE) study. Investig. New Drugs 2018. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Acebron, S.P.; Niehrs, C. β-Catenin-Independent Roles of Wnt/LRP6 Signaling. Trends Cell Biol. 2016, 26, 956–967. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Lin, S.Y.; Xia, W.; Wang, J.C.; Kwong, K.Y.; Spohn, B.; Wen, Y.; Pestell, R.G.; Hung, M.C. Beta-catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci. USA 2000, 97, 4262–4266. [Google Scholar] [CrossRef]
- De, P.; Carlson, J.H.; Wu, H.; Marcus, A.; Leyland-Jones, B.; Dey, N. Wnt-beta-catenin pathway signals metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget 2016, 7, 43124–43149. [Google Scholar] [CrossRef] [Green Version]
- Klarmann, G.J.; Decker, A.; Farrar, W.L. Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics 2008, 3, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Prior, J.; Piwnica-Worms, D.; Bu, G. LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 5136–5141. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.C.; Deng, X.; Chen, L.; Kim, C.C.; Lau, S.; et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.Y.; Soloviev, I.; Huang, X.; Chang, P.; Ernst, J.A.; Polakis, P.; Sakanaka, C. Mammary tumor regression elicited by Wnt signaling inhibitor requires IGFBP5. Cancer Res. 2012, 72, 1568–1578. [Google Scholar] [CrossRef]
- Säfholm, A.; Tuomela, J.; Rosenkvist, J.; Dejmek, J.; Härkönen, P.; Andersson, T. The Wnt-5a-derived hexapeptide Foxy-5 inhibits breast cancer metastasis in vivo by targeting cell motility. Clin. Cancer Res. 2008, 14, 6556–6563. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed] [Green Version]



















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lappano, R.; Jacquot, Y.; Maggiolini, M. GPCR Modulation in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 3840. https://doi.org/10.3390/ijms19123840
Lappano R, Jacquot Y, Maggiolini M. GPCR Modulation in Breast Cancer. International Journal of Molecular Sciences. 2018; 19(12):3840. https://doi.org/10.3390/ijms19123840
Chicago/Turabian StyleLappano, Rosamaria, Yves Jacquot, and Marcello Maggiolini. 2018. "GPCR Modulation in Breast Cancer" International Journal of Molecular Sciences 19, no. 12: 3840. https://doi.org/10.3390/ijms19123840